
Computer-Aided and AILDE Approaches to Design Novel 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors



Abstract
1. Introduction
2. Results and Discussion
2.1. Topomer CoMFA Analysis
2.2. Topomer Search
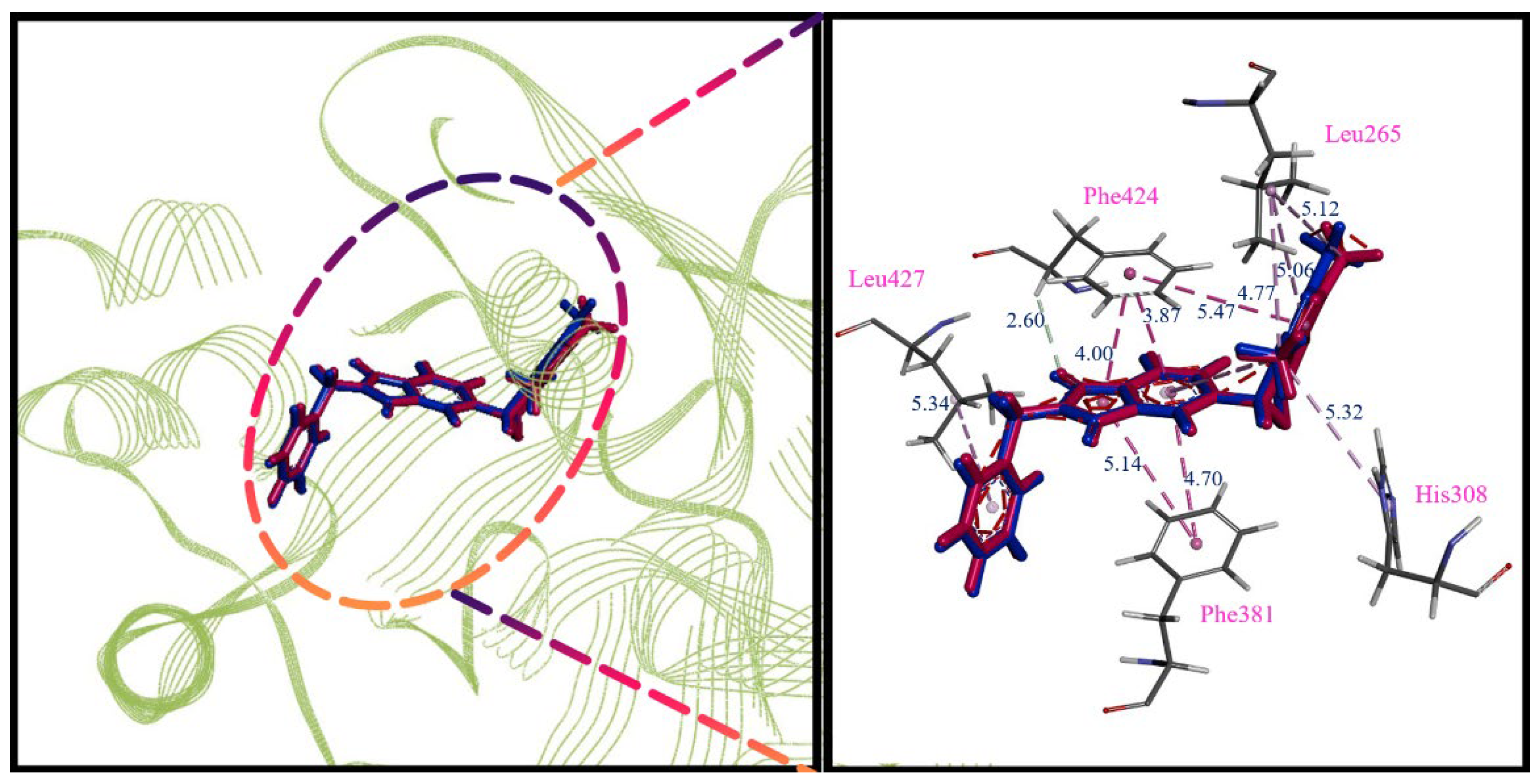
2.3. Molecular Docking Analysis
2.4. ADMET Prediction
2.5. MD Simulations
2.6. AILDE Optimized Hit Compounds and Physicochemical Properties
3. Methods and Materials
3.1. Information Collection
3.2. Topomer CoMFA Modeling Construction and Virtual Screening
3.3. Molecular Docking
3.4. ADMET Prediction
3.5. MD Simulation
3.6. AILDE and Physicochemical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ahrens, H.; Lange, G.; Muller, T.; Rosinger, C.; Willms, L.; van Almsick, A. 4-Hydroxyphenylpyruvate dioxygenase inhibitors in combination with safeners: Solutions for modern and sustainable agriculture. Angew. Chem. Int. Ed. Engl. 2014, 52, 9388–9398. [Google Scholar] [CrossRef]
- He, B.; Wang, D.W.; Yang, W.C.; Chen, Q.; Yang, G.F. Advances in research on 4-hydroxyphenylpyruvate dioxygenase (HPPD) structure and pyrazole-containing herbicides. Chin. J. Org. Chem. 2017, 37, 2895–2904. [Google Scholar] [CrossRef]
- Li, H.B.; Li, L.; Li, J.X.; Han, T.F.; He, J.L.; Zhu, Y.Q. Novel HPPD inhibitors: Triketone 2H-benzo[b] [1,4] oxazin-3(4H)-one analogs. Pest Manag. Sci. 2018, 74, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, S.Q.; Liu, Y.X.; Wang, J.Y.; Gao, S.; Zhao, L.X.; Ye, F. Design, synthesis, SAR and molecular docking of novel green niacin-triketone HPPD inhibitor. Ind. Crops Prod. 2019, 137, 566–575. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, D.; Zhang, S.Q.; Liu, Y.X.; Guo, Y.Y.; Wang, M.X.; Gao, S.; Zhao, L.X.; Ye, F. Discovery of n-aroyl diketone/triketone derivatives as novel 4-hydroxyphenylpyruvatedioxygenase inhibiting based herbicide. J. Agric. Food Chem. 2019, 67, 11839–11847. [Google Scholar] [CrossRef]
- Neidig, M.L.; Kavana, M.; Moran, G.R.; Solomon, E.I. CD and MCD studies of the non-heme ferrous active site in (4-hydroxyphenyl) pyruvate dioxygenase: Correlation between oxygen activation in the extradiol and alpha-dependent dioxygenases. J. Am. Chem. Soc. 2004, 126, 4486–4487. [Google Scholar] [CrossRef]
- Cho, J.E.; Kim, J.T.; Kim, E.; Ko, Y.K.; Kang, N.S. The structure-based three-dimensional pharmacophore models for arabidopsis thaliana HPPD inhibitors as herbicide. Bull. Korean Chem. Soc. 2013, 34, 2909–2914. [Google Scholar] [CrossRef]
- Ndikuryayo, F.; Moosavi, B.; Yang, W.C.; Yang, G.F. 4-Hydroxyphenylpyruvate dioxygenase inhibitors: From chemical biology to agrochemicals. J. Agric. Food Chem. 2017, 65, 8523–8537. [Google Scholar] [CrossRef]
- Zhang, F.W.; Qiao, Z.H.; Yao, C.T.; Sun, S.; Liu, W.T.; Wang, J.X. Effects of the novel HPPD-inhibitor herbicide QYM201 on enzyme activity and microorganisms, and its degradation in soil. Ecotoxicology 2021, 30, 80–90. [Google Scholar] [CrossRef]
- Hu, W.; Gao, S.; Zhao, L.X.; Guo, K.L.; Wang, J.Y.; Gao, Y.C.; Shao, X.X.; Fu, Y.; Ye, F. Design, synthesis and biological activity of novel triketone-containing quinoxaline as HPPD inhibitor. Pest Manag. Sci. 2022, 78, 938–946. [Google Scholar] [CrossRef]
- Wang, X.N.; Lin, H.Y.; Liu, J.J.; Zhao, X.Y.; Chen, X.; Yang, W.C.; Yang, G.F.; Zhan, C.G. The structure of 4-hydroxylphenylpyruvate dioxygenase complexed with 4-hydroxylphenylpyruvic acid reveals an unexpected inhibition mechanism. Chin. Chem. Lett. 2021, 32, 1920–1924. [Google Scholar] [CrossRef]
- Santucci, A.; Bernardini, G.; Braconi, D.; Petricci, E.; Manetti, F. 4 Hydroxyphenylpyruvate dioxygenase and its inhibition in plants and animals: Small molecules as herbicides and agents for the treatment of human inherited diseases. J. Med. Chem. 2017, 60, 4101–4125. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.K.; Ovejero, R.F.L.; Belchior, G.G.; Maymone, G.P.L.; Dayan, F.E. ACCase-inhibiting herbicides: Mechanism of action, resistance evolution and stewardship. Plant Physiol. Biochem. 2021, 78, 176–187. [Google Scholar] [CrossRef]
- Wang, Z.W.; Zhao, L.X.; Ma, P.; Ye, T.; Fu, Y.; Ye, F. Fragments recombination, design, synthesis, safener activity and CoMFA model of novel substituted dichloroacetylphenyl sulfonamide derivatives. Pest Manag. Sci. 2021, 77, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Song, H.M.; Zhao, L.X.; Zhang, S.Q.; Ye, T.; Fu, Y.; Ye, F. Design, synthesis, structure-activity relationship, molecular docking and herbicidal evaluation of 2-cinnamoyl-3-hydroxycyclohex-2-en-1-one as novel 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem. 2021, 69, 12621–12633. [Google Scholar] [CrossRef]
- Huang, D.D.; Liu, Y.L.; Shi, B.B.; Li, Y.T.; Wang, G.X.; Liang, G.Z. Comprehensive 3D-QSAR and binding mode of BACE-1 inhibitors using R-group search and molecular docking. J. Mol. Graph. Model. 2013, 45, 65–83. [Google Scholar] [CrossRef]
- Gupta, P.; Roy, N.; Garg, P. Docking-based 3D-QSAR study of HIV-1 integrase inhibitors. Eur. J. Med. Chem. 2009, 44, 4276–4287. [Google Scholar] [CrossRef]
- Chhatbar, D.M.; Chaube, U.J.; Vyas, V.K.; Bhatt, H.G. CoMFA, CoMSIA, Topomer CoMFA, HQSAR, molecular docking and molecular dynamics simulations study of triazine morpholino derivatives as mTOR inhibitors for the treatment of breast cancer. Comput. Biol. Chem. 2019, 80, 351–363. [Google Scholar] [CrossRef]
- Kang, T.; Gao, S.; Zhao, L.X.; Zhai, Y.; Ye, F.; Fu, Y. Design, synthesis and SAR of novel 1,3-disubstituted imidazolidine or hexahydropyrimidine derivatives as herbicide safeners. J. Agric. Food Chem. 2021, 69, 45–54. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, L.P.; Kai, Z.P.; Wu, F.H.; Liu, Z.Y.; Cai, M.F. Molecular modeling studies of atorvastatin analogues as HMGR inhibitors using 3D-QSAR, molecular docking and molecular dynamics simulations. Bioorg. Med. Chem. Lett. 2014, 24, 3869–3876. [Google Scholar] [CrossRef]
- Kristam, R.; Rao, S.N.; D’Cruz, A.S.; Mahadevan, V.; Viswanadhan, V.N. TRPV1 antagonism by piperazinyl-aryl compounds: A topomer-CoMFA study and its use in virtual screening for identification of novel antagonists. J. Mol. Graph. Model. 2017, 72, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Bhatt, H.; Patel, B. Structural insights on 2-phenylquinazolin-4-one derivatives as tankyrase inhibitors through CoMFA, CoMSIA, topomer CoMFA and HQSAR studies. J. Mol. Struct. 2022, 1249, 131636. [Google Scholar] [CrossRef]
- Tintori, C.; Magnani, M.; Schenone, S.; Botta, M. Docking, 3D-QSAR studies and in silico ADME prediction on c-Src tyrosine kinase inhibitors. Eur. J. Med. Chem. 2009, 44, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Zhou, H.Y.; Mo, X.W.; Zhang, L.; Li, J. In silico study of febuxostat analogs as inhibitors of xanthine oxidoreductase: A combined 3D-QSAR and molecular docking study. J. Mol. Struct. 2019, 1181, 428–435. [Google Scholar] [CrossRef]
- Tong, J.; Jiang, G.; Li, L.; Li, Y. Molecular virtual screening studies of herbicidal sulfonylurea analogues using molecular docking and topomer CoMFA research. J. Struct. Chem. 2019, 60, 210–218. [Google Scholar] [CrossRef]
- Heravi, Y.E.; Sereshti, H.; Saboury, A.A.; Ghasemi, J.; Amirmostofian, M.; Supuran, C.T. 3D QSAR studies, pharmacophore modeling, and virtual screening of diarylpyrazole-benzenesulfonamide derivatives as a template to obtain new inhibitors, using human carbonic anhydrase II as a model protein. J. Enzym. Inhib. Med. Chem. 2017, 32, 688–700. [Google Scholar] [CrossRef]
- Liu, Y.X.; Gao, S.; Ye, T.; Li, J.Z.; Ye, F.; Fu, Y. Combined 3D-quantitative structure–activity relationships and topomer technology-based molecular design of human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Future Med. Chem. 2020, 12, 795–811. [Google Scholar] [CrossRef]
- Tripathi, H.; Khan, F. Identification of potential inhibitors against nuclear dam1 complex subunit ask1 of Candida albicans using virtual screening and MD simulations. Comput. Biol. Chem. 2018, 72, 33–44. [Google Scholar] [CrossRef]
- Hou, J.Y.; Zou, Q.; Wang, Y.J.; Gao, Q.; Yao, W.H.; Yao, Q.Z.; Zhang, J. Screening for the selective inhibitors of MMP-9 from natural products based on pharmacophore modeling and molecular docking in combination with bioassay experiment, hybrid QM/MM calculation, and MD simulation. J. Biomol. Struct. Dyn. 2019, 37, 3135–3149. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.X.; Kang, T.; Sun, Y.N.; Li, J.Z.; Ye, F. Identifification of novel inhibitors of p-hydroxyphenylpyruvate dioxygenase using receptor-based virtual screening. J. Taiwan Inst. Chem. Eng. 2019, 103, 33–43. [Google Scholar] [CrossRef]
- Jia, L.; Gao, S.; Zhang, Y.Y.; Zhao, L.X.; Fu, Y.; Ye, F. Fragmenlt recombination design, synthesis, and safener activity of novel ester-substituted pyrazole derivatives. J. Agric. Food Chem. 2021, 69, 8366–8379. [Google Scholar] [CrossRef] [PubMed]
- Fatriansyah, J.F.; Rizqillah, R.K.; Yandi, M.Y.; Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud Univ. Sci. 2022, 34, 101707. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Y.X.; Yi, K.H.; Li, M.Q.; Li, J.Z.; Ye, F. Quantitative structure activity relationship studies and molecular dynamics simulations of 2-(aryloxyacetyl)cyclohexane-1,3-diones derivatives as 4-hydroxyphenylpyruvate dioxygenase inhibitors. Front Chem. 2019, 7, 556. [Google Scholar] [CrossRef]
- Brown, D.K.; Penkler, D.L.; Amamuddy, O.S.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Bishop, O.T. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Struct. Bioinform. 2017, 33, 2768–2771. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.J.; Kang, D.W.; Cherukupalli, S.; Zalloum, W.A.; Zhang, T.; Liu, X.Y.; Zhan, P. Discovery and optimizing polycyclic pyridone compounds as anti-HBV agents. Expert Opin. Ther. Pat. 2021, 30, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Edlin, C.D.; Morgans, G.; Winks, S.; Duffy, S.; Avery, V.M.; Wittlin, S.; Waterson, D.; Burrows, J.; Bryans, J. Identification and in-vitro ADME assessment of a series of novel anti-malarial agents suitable for hit-to-lead chemistry. ACS Med. Chem. Lett. 2012, 3, 570–573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mei, L.; Wu, F.; Hao, G.F.; Yang, G.F. Protocol for hit-to-lead optimization of compounds by auto in silico ligand directing evolution (AILDE) approach. STAR Protoc. 2021, 2, 100312. [Google Scholar] [CrossRef]
- Wu, F.X.; Zhuo, L.S.; Wang, F.; Huang, W.; Hao, G.F.; Yang, G.F. Auto in silico ligand directing evolution to facilitate the rapid and efficient discovery of drug lead. iScience 2020, 23, 101179. [Google Scholar] [CrossRef]
- Tong, J.B.; Qin, S.S.; Lei, S.; Wang, Y. Molecular modeling studies of HIV-1 non-nucleoside reverse transcriptase inhibitors using 3D-QSAR, virtual screening, and docking simulations. J. Serb. Chem. Soc. 2019, 84, 303–316. [Google Scholar] [CrossRef]
- Yu, S.L.; Zhou, Q.Q.; Zhang, X.L.; Jia, S.X.; Gan, Y.; Zhang, Y.H.; Shi, J.H.; Yuan, J.T. Hologram quantitative structure–activity relationship and topomer comparative molecular-field analysis to predict the affinities of azo dyes for cellulose fibers. Dyes Pigm. 2018, 153, 35–43. [Google Scholar] [CrossRef]
- Tang, H.J.; Zhao, D.S. Studies of febuxostat analogues as xanthine oxidase inhibitors through 3D-QSAR, Topomer CoMFA and molecular modeling. J. Iran. Chem. Soc. 2019, 16, 2659–2671. [Google Scholar] [CrossRef]
- He, B.; Dong, J.; Lin, H.Y.; Wang, M.Y.; Li, X.K.; Zheng, B.F.; Chen, Q.; Hao, G.F.; Yang, W.C.; Yang, G.F. Pyrazole-isoindoline-1,3-dione hybrid: A promising scaffold for 4-hydroxyphenylpyruvate dioxygenase inhibitors. J. Agric. Food Chem. 2019, 67, 10844–10852. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, M.; Zhao, L.X.; Zhang, S.Q.; Liu, Y.X.; Guo, Y.Y.; Zhang, D.; Gao, S.; Ye, F. Design, synthesis, herbicidal activity and CoMFA of aryl-formyl piperidinone HPPD inhibitors. Pestic. Biochem. Physiol. 2021, 174, 104811. [Google Scholar] [CrossRef]
- Emanuela, G.; John, G.M.; David, T.M. Theoretical hydrogen bonding parameters for drug design. J. Mol. Graph. Model. 2001, 19, 349–362. [Google Scholar]
- Zhu, T.; Jiao, Y.; Chen, Y.D.; Wang, X.; Li, H.F.; Zhang, L.Y.; Lu, T. Pharmacophore identification of Raf-1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2346–2350. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutting Method | q2 | r2 | N | F | SEE | Intercept |
|---|---|---|---|---|---|---|
| “split in two R-groups” | 0.703 | 0.957 | 6 | 95.338 | 0.046 | 4.98 |
| Comp. | Structure | -CDOCKER Energy | Comp. | Structure | -CDOCKER Energy |
|---|---|---|---|---|---|
| ZD-9 |  | 53.62 | Native ligand |  | 51.69 |
| Y1 |  | 62.76 | Y12 |  | 65.51 |
| Y2 |  | 51.79 | Y13 |  | 61.75 |
| Y3 |  | 51.66 | Y14 |  | 69.09 |
| Y4 |  | 63.96 | Y15 |  | 59.92 |
| Y5 |  | 54.27 | Y16 |  | 59.81 |
| Y6 |  | 59.48 | Y17 |  | 58.60 |
| Y7 |  | 65.57 | Y18 |  | 54.28 |
| Y8 |  | 53.29 | Y19 |  | 59.89 |
| Y9 |  | 52.83 | Y20 |  | 55.52 |
| Y10 |  | 58.18 | Y21 |  | 54.60 |
| Y11 |  | 65.12 | Y22 |  | 60.43 |
| Comp. | Solubility Level | Hepatotoxic | CYP2D6_ Applicability |
|---|---|---|---|
| Y1 | 3 | −2.52 | 13.33 |
| Y2 | 3 | −0.71 | 18.64 |
| Y3 | 3 | −0.99 | 15.80 |
| Y4 | 3 | −1.57 | 14.34 |
| Y5 | 3 | −0.46 | 13.20 |
| Y6 | 3 | −0.89 | 12.68 |
| Y7 | 3 | −2.85 | 15.91 |
| Y8 | 3 | −2.03 | 14.99 |
| Y9 | 3 | −0.43 | 17.18 |
| Y10 | 3 | −4.56 | 11.17 |
| Y11 | 4 | −3.20 | 12.81 |
| Y12 | 3 | −1.68 | 11.07 |
| Y13 | 3 | −2.06 | 13.78 |
| Y14 | 3 | −3.28 | 13.22 |
| Y15 | 3 | 0.84 | 15.14 |
| Y16 | 4 | −0.73 | 13.56 |
| Y17 | 3 | −2.25 | 14.67 |
| Y18 | 3 | −3.45 | 13.58 |
| Y19 | 4 | −1.24 | 15.39 |
| Y20 | 4 | −0.09 | 19.39 |
| Y21 | 4 | −1.01 | 18.93 |
| Y22 | 3 | −3.68 | 13.27 |
| Comp. | Degradability | Mutagenicity | Carcinogenicity |
|---|---|---|---|
| ZD-9 | √ | × | × |
| Y1 | × | × | × |
| Y2 | √ | × | × |
| Y3 | × | √ | × |
| Y4 | × | × | √ |
| Y5 | × | √ | × |
| Y6 | × | × | × |
| Y7 | √ | × | × |
| Y8 | × | √ | × |
| Y9 | √ | × | × |
| Y10 | √ | × | × |
| Y11 | × | × | × |
| Y12 | √ | × | × |
| Y13 | × | × | × |
| Y14 | √ | × | × |
| Y15 | √ | × | × |
| Y16 | × | × | × |
| Y17 | × | √ | × |
| Y18 | √ | × | × |
| Y19 | √ | × | × |
| Y20 | √ | × | × |
| Y21 | × | × | × |
| Y22 | × | × | × |
| Comp. | S1 | S2 | S3 | S4 | S5 |
|---|---|---|---|---|---|
| Structure |  |  |  |  |  |
| Hydrogen Number | 5871 | 5897 | 5897 | 5898 | 5903 |
| ΔG (kcal/mol) | −1.23 | −0.98 | −1.87 | −1.53 | −1.23 |
| ΔH (kcal/mol) | −1.61 | −1.27 | −2.24 | −1.72 | −1.25 |
| −TΔS (kcal/mol) | 0.38 | 0.29 | 0.37 | 0.19 | 0.03 |
| Comp. | Log p a | pKa a | MW a | HBA a | HBD a | RB a | SA a | Electronegativity b |
|---|---|---|---|---|---|---|---|---|
| Y12 | 2.11 | 5.7 | 355.34 | 6 | 2 | 7 | 342.81 |  |
| S1 | 2.66 | 5.6 | 389.79 | 6 | 2 | 7 | 361.58 |  |
| S2 | 2.57 | 5.6 | 389.39 | 6 | 2 | 7 | 362.88 |  |
| S3 | 1.49 | 5.6 | 370.36 | 7 | 3 | 7 | 358.47 |  |
| S4 | 2.86 | 5.6 | 434.24 | 6 | 2 | 7 | 370.40 |  |
| S5 | 2.86 | 5.6 | 434.24 | 6 | 2 | 7 | 368.19 |  |
 | ||||
|---|---|---|---|---|
| Comp. | Ar | IC50 (μM) | pIC50 | |
| Obsd. | Obsd. | Pred. | ||
| ZD-1 |  | 1.247 | 5.904 | 5.840 |
| ZD-2 * |  | 1.276 | 5.894 | 5.877 |
| ZD-3 |  | 1.365 | 5.865 | 5.860 |
| ZD-4 |  | 0.998 | 6.001 | 5.997 |
| ZD-5 |  | 1.324 | 5.878 | 5.879 |
| ZD-6 |  | 5.000 | 5.301 | 5.402 |
| ZD-7 |  | 3.475 | 5.459 | 5.470 |
| ZD-8 * |  | 0.283 | 6.548 | 6.528 |
| ZD-9 |  | 0.261 | 6.583 | 6.579 |
| ZD-10 |  | 1.503 | 5.823 | 5.781 |
| ZD-11 |  | 1.346 | 5.871 | 5.903 |
| ZD-12 * |  | 0.352 | 6.454 | 6.471 |
| ZD-13 |  | 1.457 | 5.845 | 5.839 |
| ZD-14 |  | 1.203 | 5.938 | 5.890 |
| ZD-15 |  | 0.268 | 6.572 | 6.588 |
| ZD-16 * |  | 2.075 | 5.683 | 5.750 |
| ZD-17 |  | 0.561 | 6.251 | 6.205 |
| ZD-18 |  | 4.446 | 5.352 | 5.341 |
| ZD-19 |  | 3.327 | 5.478 | 5.480 |
| ZD-20 |  | 3.206 | 5.494 | 5.541 |
| ZD-21 * |  | 2.506 | 5.601 | 5.576 |
| ZD-22 |  | 3.076 | 5.512 | 5.533 |
| ZD-23 |  | 1.321 | 5.879 | 5.880 |
| ZD-24 |  | 1.202 | 5.920 | 5.930 |
| ZD-25 |  | 1.138 | 5.944 | 5.960 |
| ZD-26 |  | 0.998 | 6.001 | 6.082 |
| ZD-27 |  | 0.425 | 6.372 | 6.333 |
| ZD-28 |  | 1.782 | 5.749 | 5.891 |
| ZD-29 * |  | 1.589 | 5.799 | 5.888 |
| ZD-30 |  | 1.347 | 5.868 | 5.911 |
| ZD-31 |  | 2.238 | 5.676 | 5.590 |
| ZD-32 |  | 1.419 | 5.848 | 5.838 |
| ZD-33 * |  | 7.656 | 5.116 | 5.140 |
| ZD-34 |  | 4.256 | 5.371 | 5.380 |
| ZD-35 |  | 4.009 | 5.397 | 5.361 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, J.; Gao, S.; Wang, J.-Y.; Ye, T.; Yue, M.-L.; Fu, Y.; Ye, F. Computer-Aided and AILDE Approaches to Design Novel 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. Int. J. Mol. Sci. 2022, 23, 7822. https://doi.org/10.3390/ijms23147822
Shi J, Gao S, Wang J-Y, Ye T, Yue M-L, Fu Y, Ye F. Computer-Aided and AILDE Approaches to Design Novel 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. International Journal of Molecular Sciences. 2022; 23(14):7822. https://doi.org/10.3390/ijms23147822
Chicago/Turabian StyleShi, Juan, Shuang Gao, Jia-Yu Wang, Tong Ye, Ming-Li Yue, Ying Fu, and Fei Ye. 2022. "Computer-Aided and AILDE Approaches to Design Novel 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors" International Journal of Molecular Sciences 23, no. 14: 7822. https://doi.org/10.3390/ijms23147822
APA StyleShi, J., Gao, S., Wang, J.-Y., Ye, T., Yue, M.-L., Fu, Y., & Ye, F. (2022). Computer-Aided and AILDE Approaches to Design Novel 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. International Journal of Molecular Sciences, 23(14), 7822. https://doi.org/10.3390/ijms23147822