
The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Unmet Need for Effective Amyotrophic Lateral Sclerosis Therapies
2. ALS; a Complex Disorder, with Distinct Pathological and Molecular Manifestations
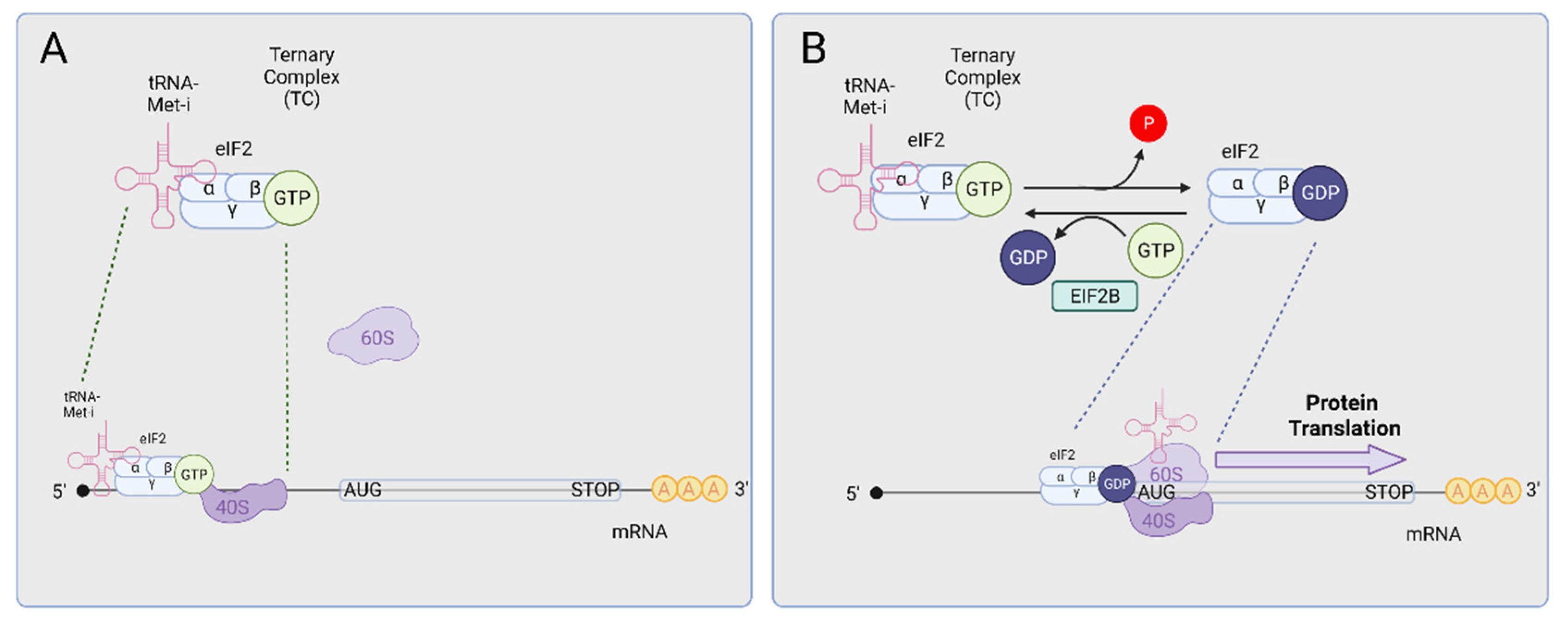
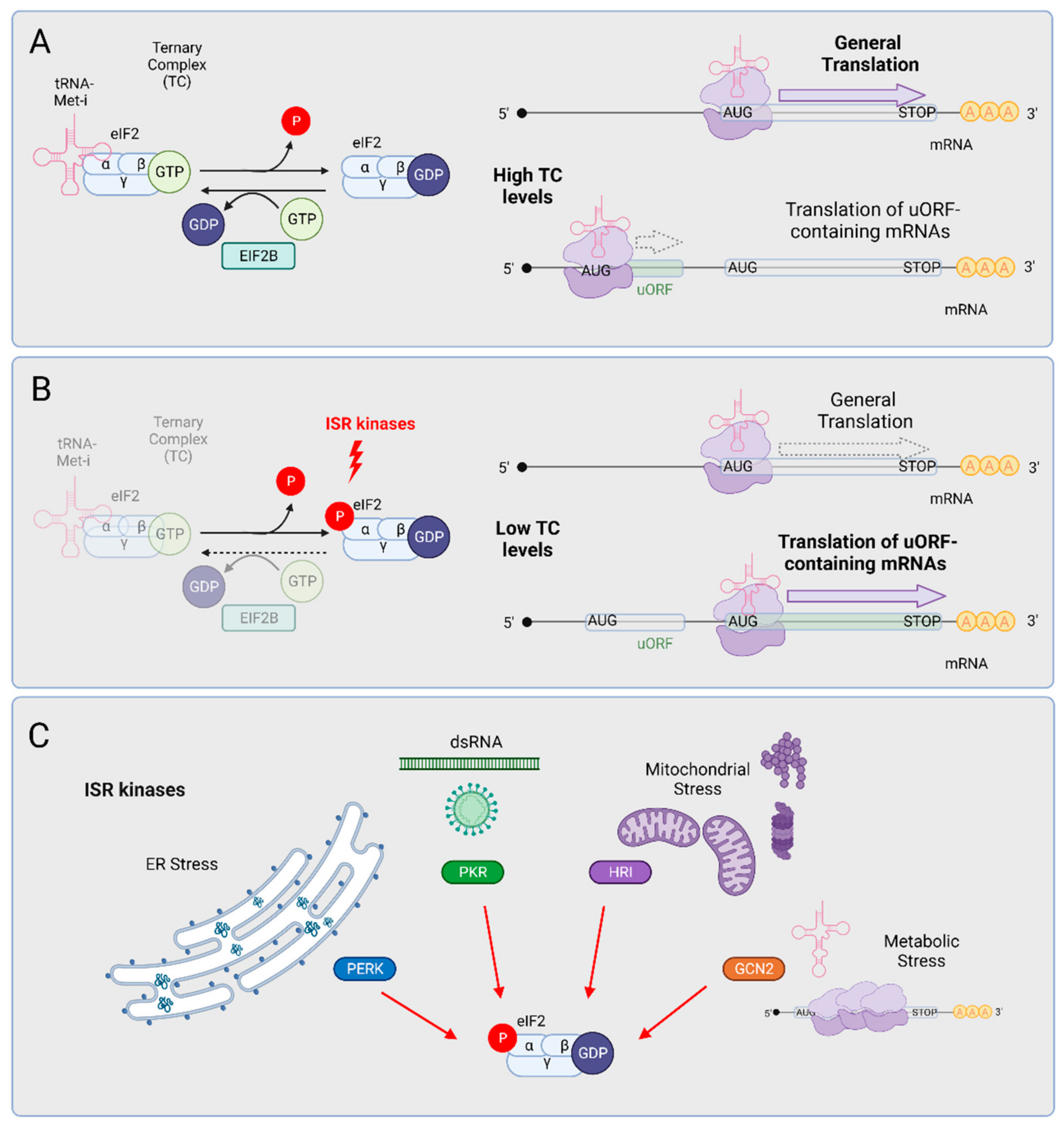
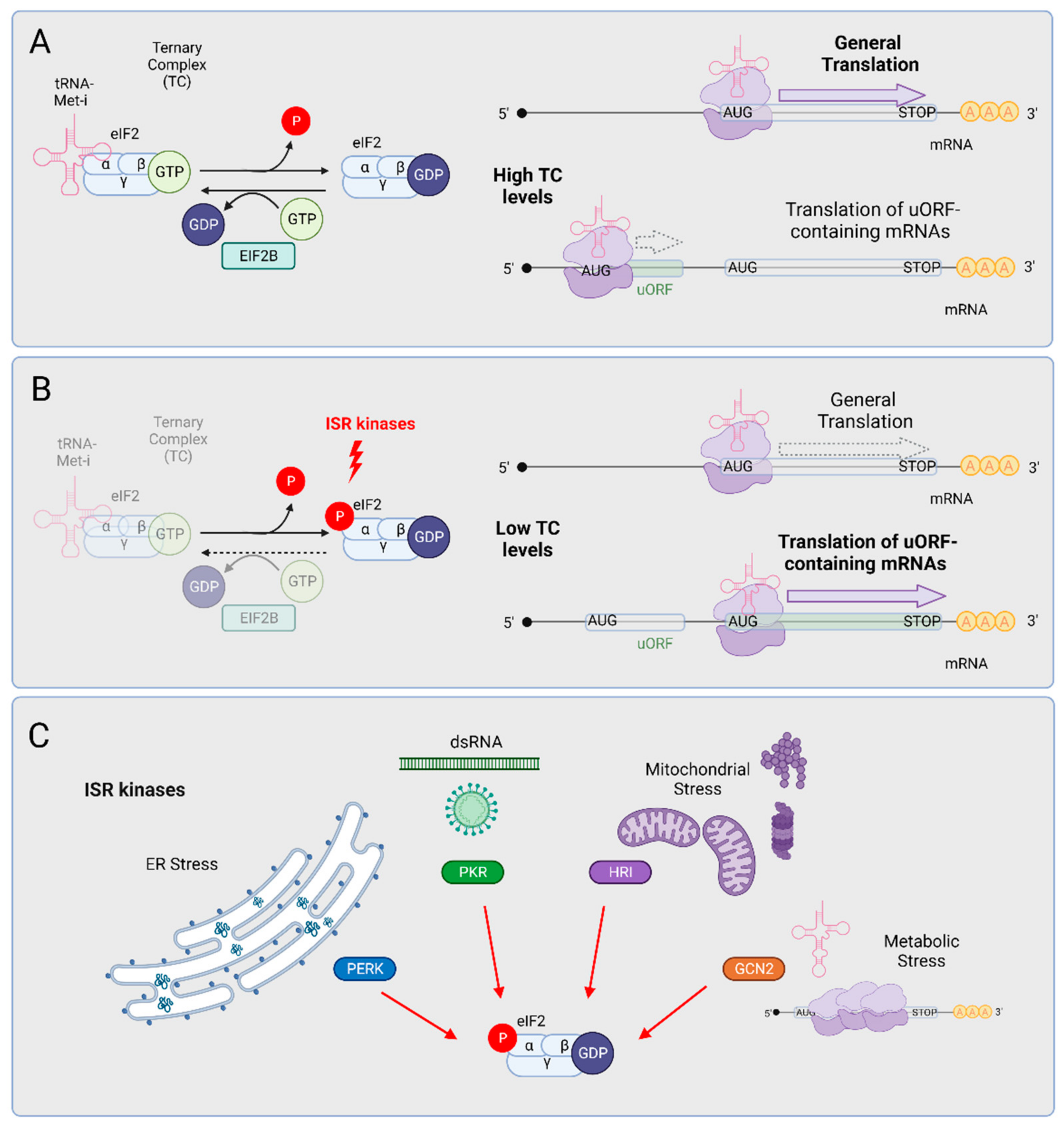
3. The Integrated Stress Response (ISR)
3.1. GCN2
3.2. HRI
3.3. PKR
3.4. PERK, the ISR Arm of the UPR
4. ISR Activation Is a Molecular Hallmark of ALS
5. Is the ISR a Driver of Neurodegeneration?
6. The Logic for Therapeutic ISR Modulation in Different fALS Models
6.1. SOD1
6.2. C9ORF72
6.3. TDP43 and FUS
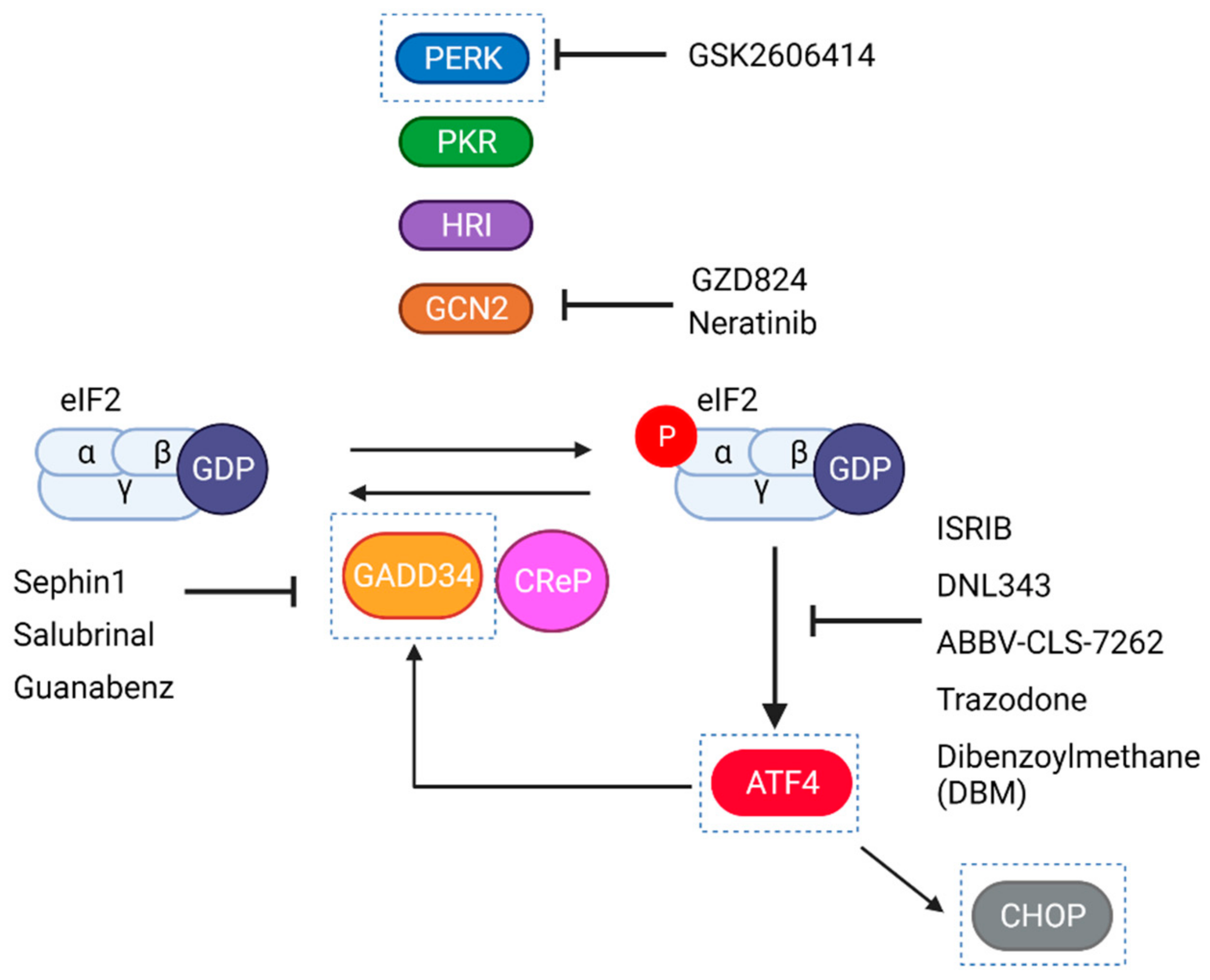
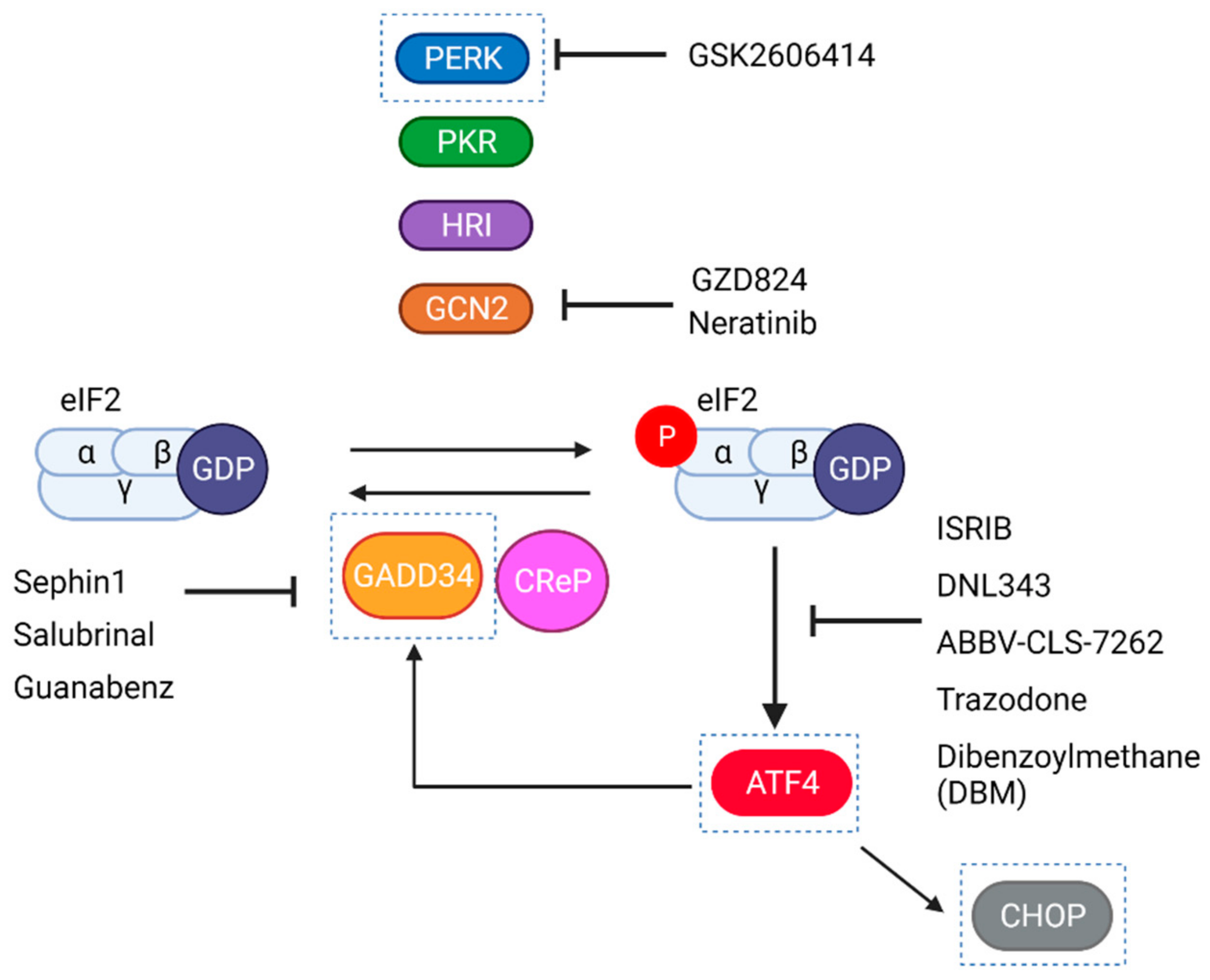
6.4. Clinical Trials with Pharmacological Modulators of the ISR
7. Conclusions and Open Questions
7.1. Should ISR Therapy of ALS Be Personalized?
7.2. Is ISR Modulation Therapeutically Effective after Disease Diagnosis?
7.3. A good Opportunity for ALS Gene Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladman, M.; Zinman, L. The economic impact of amyotrophic lateral sclerosis: A systematic review. Expert Rev. Pharmacoecon. Outcomes Res. 2015, 15, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Schönfelder, E.; Osmanovic, A.; Müschen, L.H.; Petri, S.; Schreiber-Katz, O. Costs of illness in amyotrophic lateral sclerosis (ALS): A cross-sectional survey in Germany. Orphanet J. Rare Dis. 2020, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [Google Scholar] [CrossRef]
- FDA Approves Drug to Treat ALS. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-als (accessed on 11 July 2022).
- Breiner, A.; Zinman, L.; Bourque, P.R. Edaravone for amyotrophic lateral sclerosis: Barriers to access and lifeboat ethics. CMAJ 2020, 192, E319–E320. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Tanabe, M.; Corporation, P.; Head, M.; Osaka, O. Withdrawal of Marketing Authorization Application of Edaravone for ALS in the European Union; Mitsubishi Tanabe Pharma. Europe Ltd.: London, UK, 2019; pp. 1–2. Available online: https://www.mt-pharma.co.jp/e/news/assets/pdf/e_MTPC190530.pdf (accessed on 11 July 2022).
- FDA Approves Oral Form for the Treatment of Adults with Amyotrophic Lateral Sclerosis (ALS). Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-oral-form-treatment-adults-amyotrophic-lateral-sclerosis-als (accessed on 11 July 2022).
- Charcot, J.-M.; Joffroy, A. Deux cas d’atrophie musculaire progressive avec lésions de la substance grise et des faiseaux antéro-latéraux de la moelle épinière. Arch. Physiol. Norm. Pathol. 1869, 2, 744–760. [Google Scholar]
- Siddique, T.; Figlewigz, D.A.; Pericak-Vance, M.A.; Haines, J.L.; Rouleau, G.; Jeffers, A.J.; Sapp, P.; Hung, W.-Y.; Bebout, J.; McKenna-Yasek, D.; et al. Linkage of a Gene Causing Familial Amyotrophic Lateral Sclerosis to Chromosome 21 and Evidence of Genetic-Locus Heterogeneity. N. Engl. J. Med. 1991, 324, 1381–1384. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Chen, L.X.; Xu, H.F.; Wang, P.S.; Yang, X.X.; Wu, Z.Y.; Li, H.F. SOD1 Mutation Spectrum and Natural History of ALS Patients in a 15-Year Cohort in Southeastern China. Front. Genet. 2021, 12, 1891. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Oh, K.W.; Nordin, A.; Panthi, S.; Kim, S.H.; Nordin, F.; Freischmidt, A.; Ludolph, A.C.; Ki, C.S.; Forsberg, K.; et al. De novo mutations in SOD1 are a cause of ALS. J. Neurol. Neurosurg. Psychiatry 2022, 93, 201–206. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet. Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, G.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Todd, T.W.; McEachin, Z.T.; Chew, J.; Burch, A.R.; Jansen-West, K.; Tong, J.; Yue, M.; Song, Y.; Castanedes-Casey, M.; Kurti, A.; et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration In Vivo. Cell Rep. 2020, 31, 107616. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Jovičič, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W.; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [Green Version]
- Arzberger, T.; Schludi, M.H.; Lehmer, C.; Schmid, B.; Edbauer, D. RNA versus protein toxicity in C9orf72 ALS/FTLD. Acta Neuropathol. 2018, 135, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frottin, F.; Pérez-Berlanga, M.; Hartl, F.U.; Hipp, M.S. Multiple pathways of toxicity induced by c9orf72 dipeptide repeat aggregates and g4c2 rna in a cellular model. eLife 2021, 10, e62718. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Shu, S.; Wang, R.R.; Liu, F.; Cui, B.; Guo, X.N.; Lu, C.X.; Li, X.G.; Liu, M.S.; Peng, B.; et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology 2016, 87, 1763–1769. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta-Mol. Basis Dis. 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- de Martínez-Silva, M.L.; Imhoff-Manuel, R.D.; Sharma, A.; Heckman, C.J.; Shneider, N.A.; Roselli, F.; Zytnicki, D.; Manuel, M. Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. eLife 2018, 7, e30955. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Kapur, M.; Monaghan, C.E.; Ackerman, S.L. Regulation of mRNA Translation in Neurons—A Matter of Life and Death. Neuron 2017, 96, 616–637. [Google Scholar] [CrossRef] [Green Version]
- Chesnokova, E.; Bal, N.; Kolosov, P. Kinases of eIF2a Switch Translation of mRNA Subset during Neuronal Plasticity. Int. J. Mol. Sci. 2017, 18, 2213. [Google Scholar] [CrossRef]
- English, A.M.; Green, K.M.; Moon, S.L. A (dis)integrated stress response: Genetic diseases of eIF2α regulators. Wiley Interdiscip. Rev. RNA 2022, 13, e1689. [Google Scholar] [CrossRef]
- Ishimura, R.; Nagy, G.; Dotu, I.; Chuang, J.H.; Ackerman, S.L. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. eLife 2016, 5, e14295. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, E.L.; Hines, T.J.; Bais, P.; Tadenev, A.L.D.; Schneider, R.; Jewett, D.; Pattavina, B.; Pratt, S.L.; Morelli, K.H.; Stum, M.G.; et al. The integrated stress response contributes to tRNA synthetase–associated peripheral neuropathy. Science 2021, 373, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Ohno, M. Deletion of the eIF2α Kinase GCN2 Fails to Rescue the Memory Decline Associated with Alzheimer’s Disease. PLoS ONE 2013, 8, e77335. [Google Scholar] [CrossRef] [Green Version]
- Han, A.P.; Yu, C.; Lu, L.; Fujiwara, Y.; Browne, C.; Chin, G.; Fleming, M.; Leboulch, P.; Orkin, S.H.; Chen, J.J. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001, 20, 6909–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-J.; Zhang, S. Translational control by heme-regulated elF2α kinase during erythropoiesis. Curr. Opin. Hematol. 2022, 29, 103–111. [Google Scholar] [CrossRef]
- Alvarez-Castelao, B.; Tom Dieck, S.; Fusco, C.M.; Donlin-Asp, P.; Perez, J.D.; Schuman, E.M. The switch-like expression of heme-regulated kinase 1 mediates neuronal proteostasis following proteasome inhibition. eLife 2020, 9, e52714. [Google Scholar] [CrossRef]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.J.; Arnoult, D.; et al. The eIF2α kinase HRI triggers the autophagic clearance of cytosolic protein aggregates. J. Biol. Chem. 2020, 296, 100050. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Carneiro, L.A.M.; Downey, J.; Tsalikis, J.; Outlioua, A.; Prescott, D.; Da Costa, L.S.; Hovingh, E.S.; Farahvash, A.; Gaudet, R.G.; et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 2019, 365, eaaw4144. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Canonne, M.; Arnould, T.; Renard, P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 2015, 21, 58–68. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol. Cell 2019, 74, 877–890.e6. [Google Scholar] [CrossRef]
- Gomez, E.; Powell, M.L.; Bevington, A.; Herbert, T.P. A decrease in cellular energy status stimulates PERK-dependent eIF2alpha phosphorylation and regulates protein synthesis in pancreatic beta-cells. Biochem. J. 2008, 410, 485–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: The 2’-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Youssef, O.A.; Safran, S.A.; Nakamura, T.; Nix, D.A.; Hotamisligil, G.S.; Bass, B.L. Potential role for snoRNAs in PKR activation during metabolic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 5023–5028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.A.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef] [Green Version]
- Romano, P.R.; Garcia-Barrio, M.T.; Zhang, X.; Wang, Q.; Taylor, D.R.; Zhang, F.; Herring, C.; Mathews, M.B.; Qin, J.; Hinnebusch, A.G. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol. Cell. Biol. 1998, 18, 2282–2297. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Jiang, T.; Mu, P.; Chen, X.; Wen, X.; Hu, Z.; Tang, S.; Wen, J.; Deng, Y. Cell fate determined by the activation balance between PKR and SPHK1. Cell Death Differ. 2021, 28, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yang, M.; May, W.S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 1999, 274, 15427–15432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, A.; Haque, S.J.; Mogensen, T.; Silverman, R.H.; Williams, B.R. RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J. Immunol. 2001, 166, 6170–6180. [Google Scholar] [CrossRef] [Green Version]
- Kaempfer, R.; Namer, L.S.; Osman, F.; Ilan, L. Control of mRNA splicing by noncoding intragenic RNA elements that evoke a cellular stress response. Int. J. Biochem. Cell Biol. 2018, 105, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; van Beers, E.J. Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther. Adv. Hematol. 2021, 12, 20406207211066070. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress Sensor PERK Coordinates ER-Plasma Membrane Contact Site Formation through Interaction with Filamin-A and F-Actin Remodeling. Mol. Cell 2017, 65, 885–899.e6. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef]
- Walker, A.K.; Farg, M.A.; Bye, C.R.; McLean, C.A.; Horne, M.K.; Atkin, J.D. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 2010, 133, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Oyanagi, K.; Yamazaki, M.; Takahashi, H.; Watabe, K.; Wada, M.; Komori, T.; Morita, T.; Mizutani, T. Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from, and cisternal distention of the rough endoplasmic reticulum. Neuropathol. Appl. Neurobiol. 2008, 34, 650–658. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [Green Version]
- Perciballi, E.; Bovio, F.; Rosati, J.; Arrigoni, F.; D’Anzi, A.; Lattante, S.; Gelati, M.; De Marchi, F.; Lombardi, I.; Ruotolo, G.; et al. Characterization of the p.L145F and p.S135N Mutations in SOD1: Impact on the Metabolism of Fibroblasts Derived from Amyotrophic Lateral Sclerosis Patients. Antioxidants 2022, 11, 815. [Google Scholar] [CrossRef]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Guber, R.D.; Schindler, A.B.; Budron, M.S.; Chen, K.L.; Li, Y.; Fischbeck, K.H.; Grunseich, C. Nucleocytoplasmic transport defect in a North American patient with ALS8. Ann. Clin. Transl. Neurol. 2018, 5, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, J.; De Santis, R.; de Turris, V.; Morlando, M.; Laneve, P.; Calvo, A.; Caliendo, V.; Chiò, A.; Rosa, A.; Bozzoni, I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model. Mech. 2015, 8, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J.J.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H.J.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.S.T.; Chun, C.; Hesson, J.; Mathieu, J.; Valdmanis, P.N.; Mack, D.L.; Choi, B.-O.; Kim, D.-H.; Bothwell, M. Human Induced Pluripotent Stem Cell-Derived TDP-43 Mutant Neurons Exhibit Consistent Functional Phenotypes Across Multiple Gene Edited Lines Despite Transcriptomic and Splicing Discrepancies. Front. cell Dev. Biol. 2021, 9, 728707. [Google Scholar] [CrossRef]
- Bonifacino, T.; Zerbo, R.A.; Balbi, M.; Torazza, C.; Frumento, G.; Fedele, E.; Bonanno, G.; Milanese, M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 12236. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [Green Version]
- Prell, T.; Lautenschläger, J.; Witte, O.W.; Carri, M.T.; Grosskreutz, J. The unfolded protein response in models of human mutant G93A amyotrophic lateral sclerosis. Eur. J. Neurosci. 2012, 35, 652–660. [Google Scholar] [CrossRef]
- Sun, S.; Sun, Y.; Ling, S.C.; Ferraiuolo, L.; McAlonis-Downes, M.; Zou, Y.; Drenner, K.; Wang, Y.; Ditsworth, D.; Tokunaga, S.; et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. USA 2015, 112, E6993–E7002. [Google Scholar] [CrossRef] [Green Version]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Vehviläinen, P.; Koistinaho, J.; Gundars, G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Khoramian Tusi, S.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Raphael, A.R.; Ladow, E.S.; Mcgurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Medinas, D.B.; Rozas, P.; Martínez Traub, F.; Woehlbier, U.; Brown, R.H.; Bosco, D.A.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209–8214. [Google Scholar] [CrossRef] [Green Version]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, s41467s017. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [Green Version]
- Shelkovnikova, T.A.; An, H.; Skelt, L.; Tregoning, J.S.; Humphreys, I.R.; Buchman, V.L. Antiviral Immune Response as a Trigger of FUS Proteinopathy in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 29, 4496–4508.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayalakshmi, K.; Alladi, P.A.; Ghosh, S.; Prasanna, V.K.; Sagar, B.C.; Nalini, A.; Sathyaprabha, T.N.; Raju, T.R. Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2011, 41, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2013, 2, e00498. [Google Scholar] [CrossRef]
- Denali Therapeutics Announces Initiation of Phase 1b Study of EIF2B Activator DNL343 in ALS. Available online: https://www.globenewswire.com/news-release/2021/09/09/2294411/0/en/Denali-Therapeutics-Announces-Initiation-of-Phase-1b-Study-of-EIF2B-Activator-DNL343-in-ALS.html (accessed on 1 July 2022).
- A Phase 1 Study to Investigate the Safety and Pharmacokinetics of ABBV-CLS-7262 in Patients with Amyotrophic Lateral Sclerosis. Available online: https://clinicaltrials.gov/ct2/show/NCT04948645 (accessed on 1 July 2022).
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [Green Version]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef]
- Del Signore, S.J.; Amante, D.J.; Kim, J.; Stack, E.C.; Goodrich, S.; Cormier, K.; Smith, K.; Cudkowicz, M.E.; Ferrante, R.J. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler. 2009, 10, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Jansen-West, K.; Xu, Y.F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Min, J.H.; Hong, Y.H.; Sung, J.J.; Kim, S.M.; Lee, J.B.; Lee, K.W. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: A randomized cross-over trial. J. Korean Med. Sci. 2012, 27, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 2019, 127, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef]
- Seijffers, R.; Zhang, J.; Matthews, J.C.; Chen, A.; Tamrazian, E.; Babaniyi, O.; Selig, M.; Hynynen, M.; Woolf, C.J.; Brown, R.H. ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc. Natl. Acad. Sci. USA 2014, 111, 1622–1627. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Hernandez, M.; Tadokoro, T.; Navarro, M.R.; Platoshyn, O.; Kobayashi, Y.; Marsala, S.; Miyanohara, A.; Juhas, S.; Juhasova, J.; Skalnikova, H.; et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med. 2020, 26, 118–130. [Google Scholar] [CrossRef]
- Matus, S.; Nassif, M.; Glimcher, L.H.; Hetz, C. XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 2009, 5, 1226–1228. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Popko, B.; Roos, R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011, 20, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Popko, B.; Roos, R.P. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 2014, 23, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.Q.; Ren, M.; Jiang, H.Z.; Wang, J.; Zhang, J.; Yin, X.; Wang, S.Y.; Qi, Y.; Wang, X.D.; Feng, H.L. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 2014, 277, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ghadge, G.D.; Sonobe, Y.; Camarena, A.; Drigotas, C.; Rigo, F.; Ling, K.K.; Roos, R.P. Knockdown of GADD34 in neonatal mutant SOD1 mice ameliorates ALS. Neurobiol. Dis. 2020, 136, 104702. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.G.; Ping, Q.; Moreno, A.J.; Kidd, J.D.; Thompson, K.; Jiang, B.; Lincecum, J.M.; Wang, M.Z.; De Zutter, G.S.; Tassinari, V.R.; et al. Guanabenz Treatment Accelerates Disease in a Mutant SOD1 Mouse Model of ALS. PLoS ONE 2015, 10, e0135570. [Google Scholar] [CrossRef] [Green Version]
- Briggs, D.I.; Defensor, E.; Memar Ardestani, P.; Yi, B.; Halpain, M.; Seabrook, G.; Shamloo, M. Role of Endoplasmic Reticulum Stress in Learning and Memory Impairment and Alzheimer’s Disease-Like Neuropathology in the PS19 and APP(Swe) Mouse Models of Tauopathy and Amyloidosis. eNeuro 2017, 4, 28721361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef] [Green Version]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Sonobe, Y.; Ghadge, G.; Masaki, K.; Sendoel, A.; Fuchs, E.; Roos, R.P. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol. Dis. 2018, 116, 155–165. [Google Scholar] [CrossRef]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non- AUG translation in C9orf72- ALS / FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef]
- Crowley, M.J.; Diamantidis, C.J.; McDuffie, J.R.; Cameron, B.; Stanifer, J.; Mock, C.K.; Kosinski, A.; Wang, X.; Tang, S.; Williams, J.W.J. Metformin Use in Patients with Historical Contraindications or Precautions; Department of Veterans Affairs: Washington, DC, USA, 2016. [Google Scholar]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Takeda, T.; Shibata, N.; Kobayashi, M. Alterations in subcellular localization of TDP-43 immunoreactivity in the anterior horns in sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 478, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Velde, C. Vande Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Litscher, G.; Watanabe, N.; Wei, W.; Hashimoto, T.; Iwatsubo, T.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked cytoplasmic FUS assemblies are compositionally different from physiological stress granules and sequester hnRNPA3, a novel modifier of FUS toxicity. Neurobiol. Dis. 2022, 162, 105585. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A.; et al. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P., 3rd; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Velde, C. Vande TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Vande Velde, C. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla Bella, E.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Filosto, M.; Giannini, F.; et al. The unfolded protein response in amyotrophic later sclerosis: Results of a phase 2 trial. Brain 2021, 144, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell. Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [Green Version]
- A Combined SAD and MAD Study to Investigate the Safety, Tolerability and Pharmacokinetic Profile of IFB-088. Available online: https://clinicaltrials.gov/ct2/show/NCT03610334 (accessed on 1 July 2022).
- A Study to Evaluate the Bioavailability and Safety of DNL343 in Healthy Volunteers. Available online: https://clinicaltrials.gov/ct2/show/NCT04581772?term=DNL343&draw=2&rank=3 (accessed on 1 July 2022).
- A Phase 1 Open-Label, Randomized, Crossover Study to Evaluate the Bioavailability, Effect of Food, Palatability, and Safety of Various DNL343 Oral Formulations in Healthy Participants. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04581772 (accessed on 1 July 2022).
- A Study to Determine the Safety, Pharmacokinetics, and Pharmacodynamics of DNL343 in Participants with Amyotrophic Lateral Sclerosis. Available online: https://clinicaltrials.gov/ct2/show/NCT05006352?term=DNL343&draw=2&rank=2 (accessed on 1 July 2022).
- Denali Therapeutics Announces Positive Clinical Results and Regulatory Progress for Development Programs in Amyotrophic Lateral Sclerosis (ALS). Available online: https://www.globenewswire.com/news-release/2021/10/06/2309876/0/en/Denali-Therapeutics-Announces-Positive-Clinical-Results-and-Regulatory-Progress-for-Development-Programs-in-Amyotrophic-Lateral-Sclerosis-ALS.html (accessed on 1 July 2022).
- Safety and Therapeutic Potential of the FDA-approved Drug Metformin for C9orf72 ALS/FTD. Available online: https://clinicaltrials.gov/ct2/show/NCT04220021 (accessed on 1 July 2022).
- Delaye, J.B.; Lanznaster, D.; Veyrat-Durebex, C.; Fontaine, A.; Bacle, G.; Lefevre, A.; Hergesheimer, R.; Lecron, J.C.; Vourc’h, P.; Andres, C.R.; et al. Behavioral, Hormonal, Inflammatory, and Metabolic Effects Associated with FGF21-Pathway Activation in an ALS Mouse Model. Neurother. J. Am. Soc. Exp. Neurother. 2020, 18, 297–308. [Google Scholar] [CrossRef]
- Adams, C.M.; Ebert, S.M.; Dyle, M.C. Role of ATF4 in skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 164–168. [Google Scholar] [CrossRef]
- Liu, D.; Zhu, M.; Zhang, Y.; Diao, Y. Crossing the blood-brain barrier with AAV vectors. Metab. Brain Dis. 2021, 36, 45–52. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marlin, E.; Viu-Idocin, C.; Arrasate, M.; Aragón, T. The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 7823. https://doi.org/10.3390/ijms23147823
Marlin E, Viu-Idocin C, Arrasate M, Aragón T. The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2022; 23(14):7823. https://doi.org/10.3390/ijms23147823
Chicago/Turabian StyleMarlin, Elías, Cristina Viu-Idocin, Montserrat Arrasate, and Tomás Aragón. 2022. "The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 23, no. 14: 7823. https://doi.org/10.3390/ijms23147823
APA StyleMarlin, E., Viu-Idocin, C., Arrasate, M., & Aragón, T. (2022). The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 23(14), 7823. https://doi.org/10.3390/ijms23147823