
Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism


Abstract
:1. Introduction
2. Results
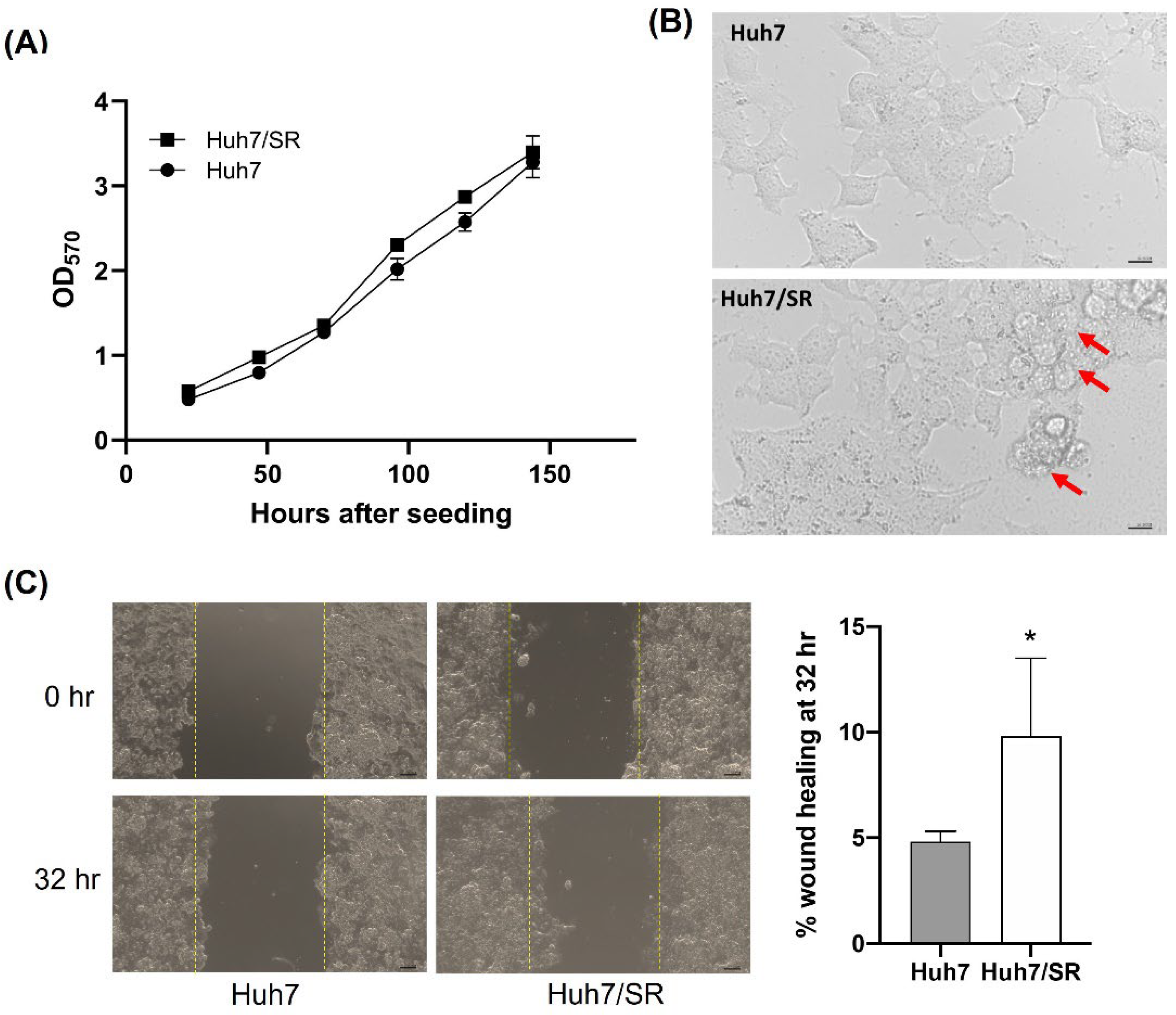
2.1. Huh7/SR Cells Show Higher Migratory Abilities than Parental Huh7 Cells
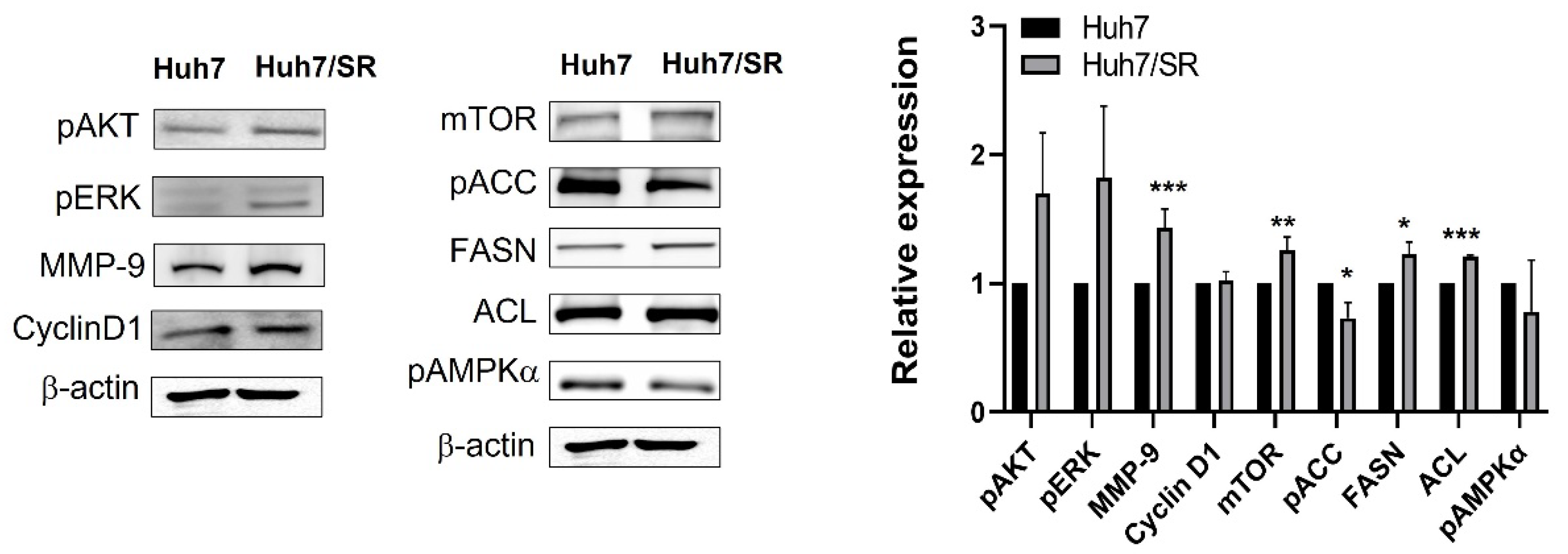
2.2. Protein Expression Profiles Are Different between Huh7 and Huh7/SR Cells
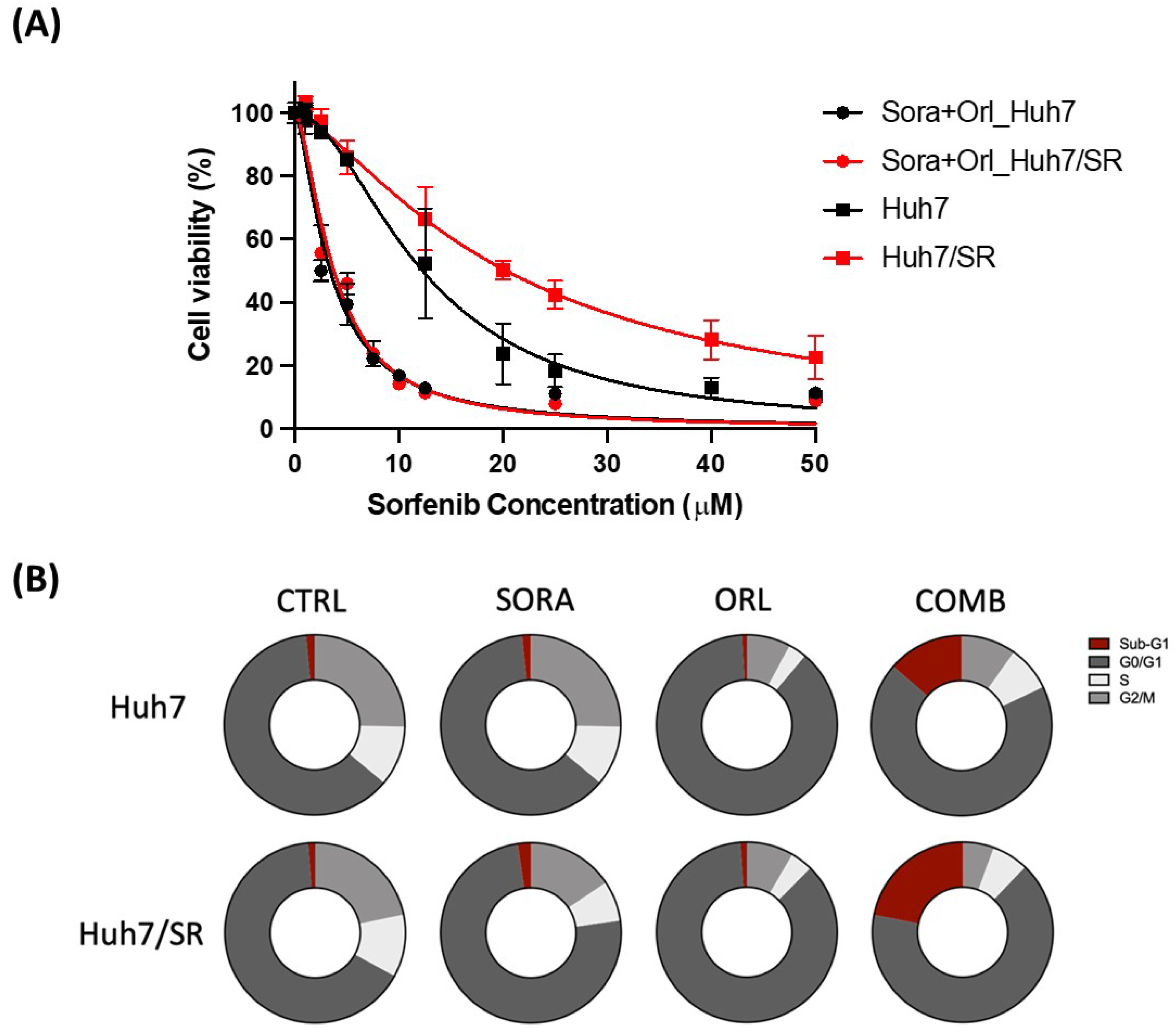
2.3. Orlistat Enhances Cytotoxicity of Sorafenib in Both Huh7 and Huh7-SR Cells
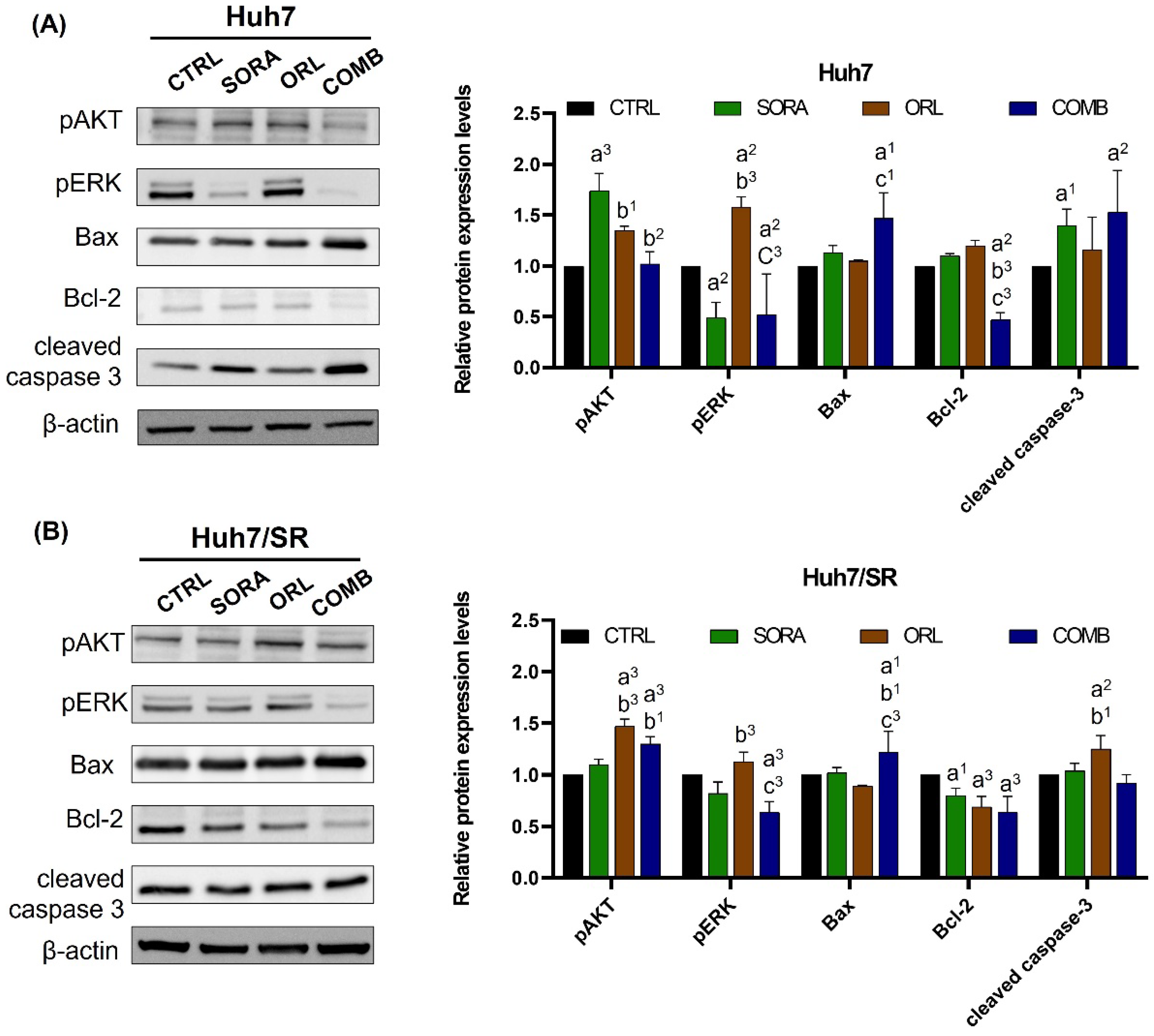
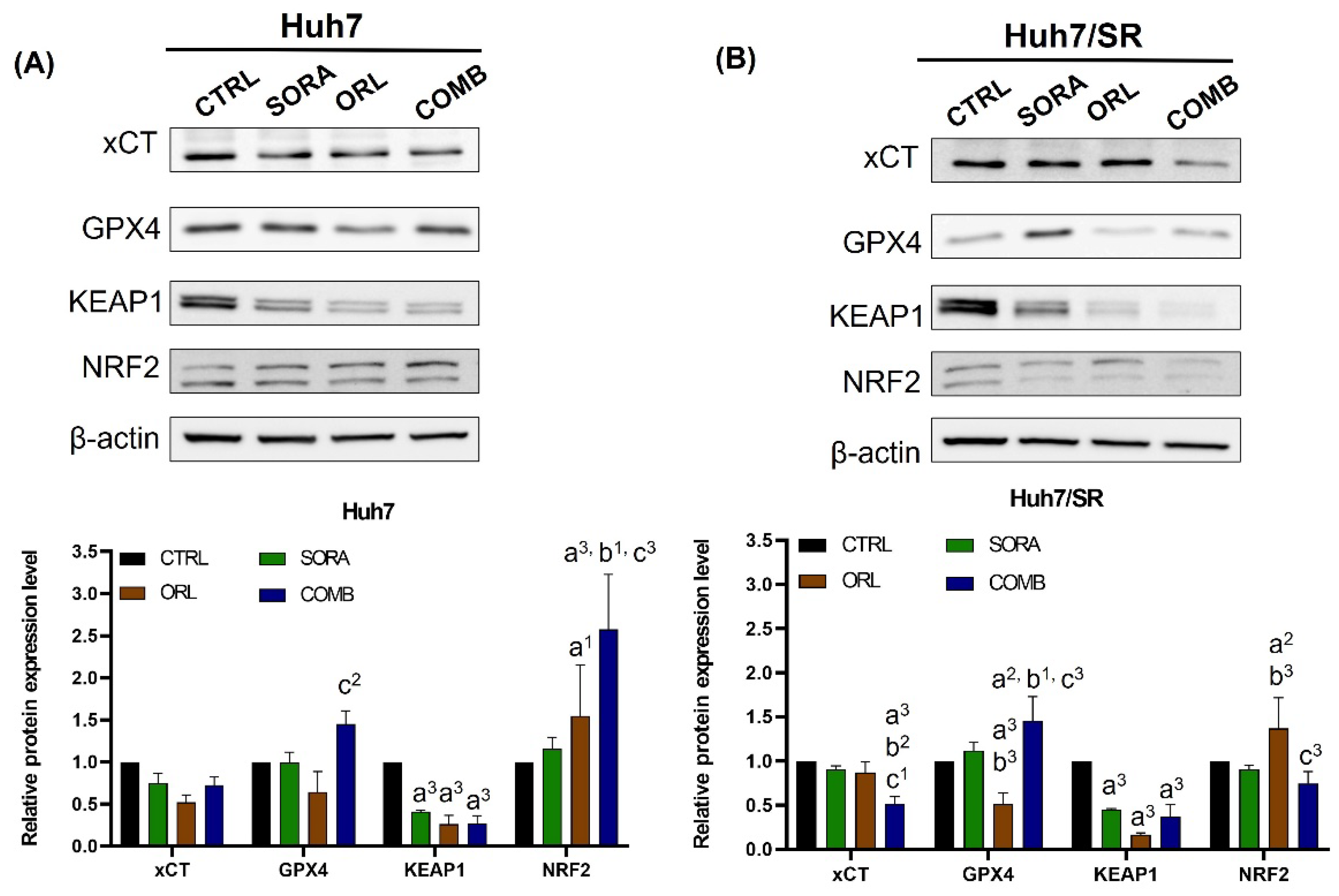
2.4. Combination Treatment Changes Protein Expressions Differently in Huh7 and Huh7/SR Cells
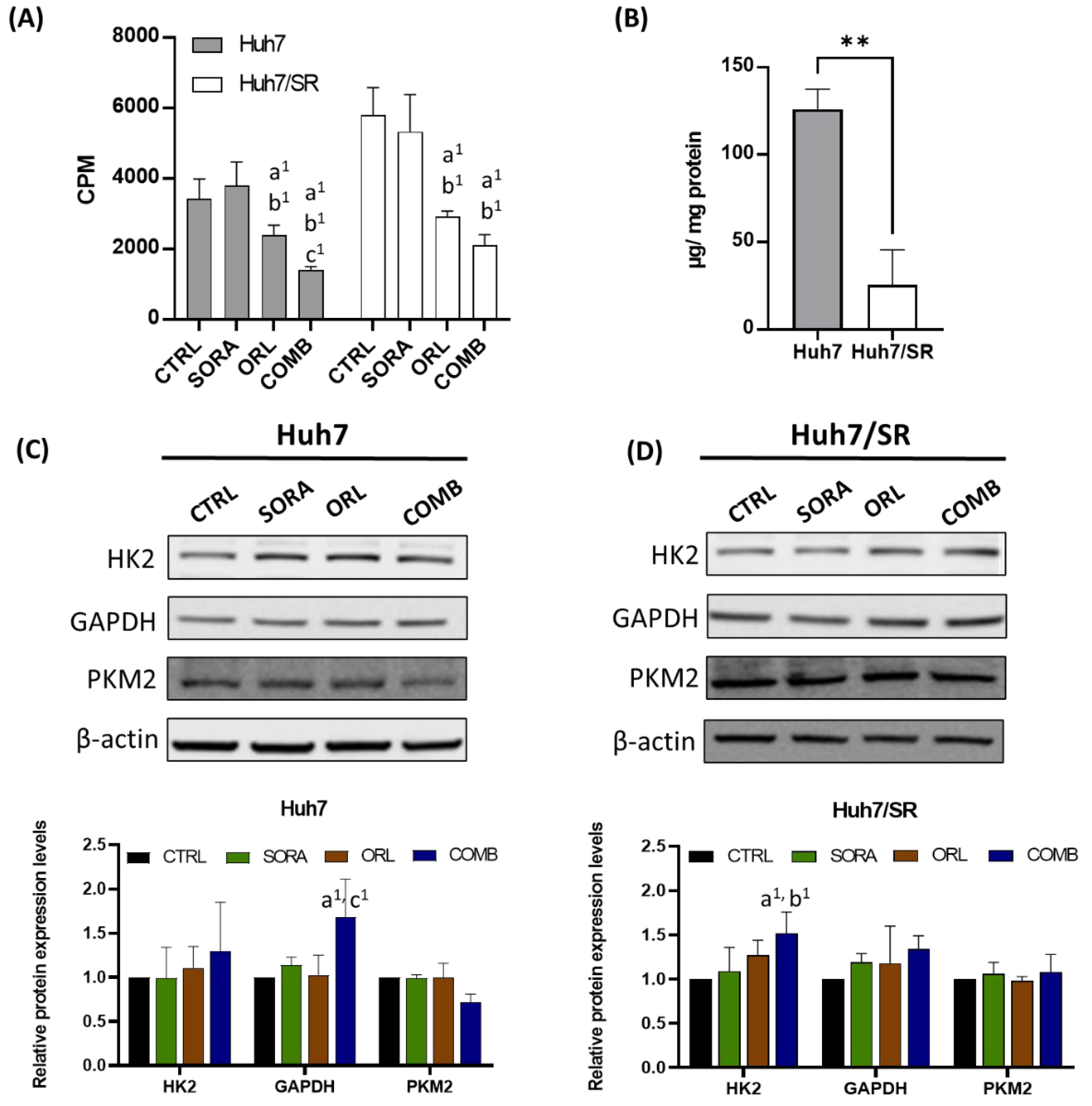
2.5. Combination Treatment Represses 18F-FDG Uptake in Huh7 and Huh7/SR Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Drugs
4.3. Cell Viability Assay
4.4. Wound Healing
4.5. Flow Cytometry
4.6. Western Blot
4.7. 18F-FDG Uptake
4.8. Intracellular Glucogen Measurement
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ozakyol, A. Global Epidemiology of Hepatocellular Carcinoma (HCC Epidemiology). J. Gastrointest. Cancer 2017, 48, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2020, 51, 102610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinnen, J.V.; Heemers, H.; de Sande, T.V.; Schrijver, E.D.; Brusselmans, K.; Heyns, W.; Verhoeven, G. Androgens, lipogenesis and prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 273–279. [Google Scholar] [CrossRef]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.; Loizzi, D.; Serviddio, G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Zhu, X.; Qin, X.; Fei, M.; Hou, W.; Greshock, J.; Bachman, K.E.; Wooster, R.; Kang, J.; Qin, C.Y. Combined Phosphatase and Tensin Homolog (PTEN) Loss and Fatty Acid Synthase (FAS) Overexpression Worsens the Prognosis of Chinese Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2012, 13, 9980–9991. [Google Scholar] [CrossRef]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef]
- Epstein, J.I.; Carmichael, M.; Partin, A.W. OA-519 (fatty acid synthase) as an independent predictor of pathologic stage in adenocarcinoma of the prostate. Urology 1995, 45, 81–86. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty Acid Synthase Over Expression is an Indicator of Tumor Aggressiveness and Poor Prognosis in Renal Cell Carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef]
- Notarnicola, M.; Tutino, V.; Calvani, M.; Lorusso, D.; Guerra, V.; Caruso, M.G. Serum levels of fatty acid synthase in colorectal cancer patients are associated with tumor stage. J. Gastrointest. Cancer 2012, 43, 508–511. [Google Scholar] [CrossRef]
- Pandey, P.R.; Okuda, H.; Watabe, M.; Pai, S.K.; Liu, W.; Kobayashi, A.; Xing, F.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res. Treat. 2011, 130, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellberg, E.A.; Anderson, S.M. FASNating targets of metformin in breast cancer stem-like cells. Horm. Cancer 2014, 5, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M.; et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Chao, W.-T.; Liao, C.-C.; Shih, J.-H.; Lai, Y.-S.; Hsu, Y.-H.; Liu, Y.-H. The Roles of Angiogenesis and Cancer Stem Cells in Sorafenib Drug Resistance in Hepatocellular Carcinoma. Onco Targets Ther. 2019, 12, 8217–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, H.W.; Leung, C.O.N.; Lau, E.Y.; Chung, K.P.S.; Mok, E.H.; Lei, M.M.L.; Leung, R.W.H.; Tong, M.; Keng, V.W.; Ma, C.; et al. EPHB2 Activates β-Catenin to Enhance Cancer Stem Cell Properties and Drive Sorafenib Resistance in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 3229–3240. [Google Scholar] [CrossRef]
- Bhat, M.; Sonenberg, N.; Gores, G.J. The mTOR pathway in hepatic malignancies. Hepatology 2013, 58, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Ren, Q.-N.; Zhang, H.; Sun, C.-Y.; Zhou, Y.-F.; Yang, X.-F.; Long, J.-W.; Li, X.-X.; Mai, S.-J.; Zhang, M.-Y.; Zhang, H.-Z.; et al. Phosphorylation of Androgen Receptor by mTORC1 Promotes Liver Steatosis and Tumorigenesis. Hepatology 2022, 75, 1123–1138. [Google Scholar] [CrossRef]
- Zhou, W.; Han, W.F.; Landree, L.E.; Thupari, J.N.; Pinn, M.L.; Bililign, T.; Kim, E.K.; Vadlamudi, A.; Medghalchi, S.M.; El Meskini, R.; et al. Fatty Acid Synthase Inhibition Activates AMP-Activated Protein Kinase in SKOV3 Human Ovarian Cancer Cells. Cancer Res. 2007, 67, 2964. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-H.; Tsai, S.-J.; Wang, Y.-J.; Pan, M.-H.; Kao, J.-Y.; Way, T.-D. EGCG inhibits protein synthesis, lipogenesis, and cell cycle progression through activation of AMPK in p53 positive and negative human hepatoma cells. Mol. Nutr. Food Res. 2009, 53, 1156–1165. [Google Scholar] [CrossRef]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Jiang, W.-W.; Luo, X.-F.; Dai, T.-Y.; Peng, L.; Song, S.; Li, L.-F.; Tao, L.; Shi, C.-Y.; et al. Moringa oleifera Leaf Petroleum Ether Extract Inhibits Lipogenesis by Activating the AMPK Signaling Pathway. Front. Pharmacol. 2018, 9, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bort, A.; Sánchez, B.G.; Mateos-Gómez, P.A.; Díaz-Laviada, I.; Rodríguez-Henche, N. Capsaicin Targets Lipogenesis in HepG2 Cells Through AMPK Activation, AKT Inhibition and PPARs Regulation. Int. J. Mol. Sci. 2019, 20, 1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Huang, B.; Sun, L.; Wang, X.; Zhou, B.; Tang, H.; Geng, W. MK8722 an AMPK activator, inhibiting carcinoma proliferation, invasion and migration in human pancreatic cancer cells. Biomed. Pharmacother. 2021, 144, 112325. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-I.; Zeng, Y.-S.; Lin, Y.-C.J.; Huang, J.-W.; Lin, C.-L.; Lee, M.-H.; Yang, F.-W.; Fang, T.-P.; Mar, A.-C.; Su, J.-C. A novel AMPK activator shows therapeutic potential in hepatocellular carcinoma by suppressing HIF1α-mediated aerobic glycolysis. Mol. Oncol. 2022, 16, 2274–2294. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic Significance of AMPK Activation and Therapeutic Effects of Metformin in Hepatocellular Carcinoma. Clin. Cancer Res. 2013, 19, 5372. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef]
- You, A.; Cao, M.; Guo, Z.; Zuo, B.; Gao, J.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J. Hematol. Oncol. 2016, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Schulte, L.; Scheiner, B.; Voigtländer, T.; Koch, S.; Schweitzer, N.; Marhenke, S.; Ivanyi, P.; Manns, M.P.; Rodt, T.; Hinrichs, J.B.; et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019, 39, 714–726. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Mazière, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer. Res. 2014, 34, 6417–6422. [Google Scholar]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Kuang, S.; Cao, R.; Wang, J.; Peng, Q.; Sun, C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 2019, 33, 10089–10103. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-Y.; Chang, Y.-F.; Hwang, J.-J. Antitumor effect of orlistat, a fatty acid synthase inhibitor, is via activation of caspase-3 on human colorectal carcinoma-bearing animal. Biomed. Pharmacother. 2011, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-Y.; Lee, Y.-P.; Lin, W.-C.; Lin, Y.-H.; Hwang, J.-J. Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-κB Pathway. Sci. Rep. 2019, 9, 13284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Sheng, L.; Yuan, M.; Hu, J.; Meng, Y.; Wu, Y.; Chen, L.; Yu, H.; Li, S.; Zheng, G.; et al. Orlistat delays hepatocarcinogenesis in mice with hepatic co-activation of AKT and c-Met. Toxicol. Appl. Pharmacol. 2020, 392, 114918. [Google Scholar] [CrossRef]
- Tyan, Y.-S.; Lee, Y.-P.; Chuang, H.-Y.; Wang, W.-H.; Hwang, J.-J. Effects of orlistat combined with enzalutamide and castration through inhibition of fatty acid synthase in a PC3 tumor-bearing mouse model. Biosci. Rep. 2021, 41, BSR20204203. [Google Scholar] [CrossRef]
- Jin, B.-R.; Kim, H.-J.; Sim, S.-A.; Lee, M.; An, H.-J. Anti-Obesity Drug Orlistat Alleviates Western-Diet-Driven Colitis-Associated Colon Cancer via Inhibition of STAT3 and NF-κB-Mediated Signaling. Cells 2021, 10, 2060. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Iavarone, M.; Cabibbo, G.; Piscaglia, F.; Zavaglia, C.; Grieco, A.; Villa, E.; Cammà, C.; Colombo, M.; SOFIA Study Group. Field-practice study of sorafenib therapy for hepatocellular carcinoma: A prospective multicenter study in Italy. Hepatology 2011, 54, 2055–2063. [Google Scholar] [CrossRef]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Takasu, C.; Miyazaki, K.; Shimada, M. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 2021, 16, e0256755. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, X.; Liang, Q.; Yu, Y.; Shen, X.; Sun, G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2020, 126, 105820. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.-Z.; Chen, C.-T.; Li, N.-C.; Lin, L.-C.; Huang, B.-S.; Chang, Y.-H.; Chow, L.-P. Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 224. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Choi, Y.-K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.-G.; Jeon, H.-J.; Kim, J.-H.; Kim, J.-G.; et al. PPARδ Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol. Cancer Res. 2017, 15, 1230. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Zhou, L.; Zuo, H.; Le, W.; Hu, J.; Zhang, T.; Li, M.; Yuan, Y. Overriding sorafenib resistance via blocking lipid metabolism and Ras by sphingomyelin synthase 1 inhibition in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2021, 87, 217–228. [Google Scholar] [CrossRef]
- Souchek, J.J.; Davis, A.L.; Hill, T.K.; Holmes, M.B.; Qi, B.; Singh, P.K.; Kridel, S.J.; Mohs, A.M. Combination Treatment with Orlistat-Containing Nanoparticles and Taxanes Is Synergistic and Enhances Microtubule Stability in Taxane-Resistant Prostate Cancer Cells. Mol. Cancer Ther. 2017, 16, 1819–1830. [Google Scholar] [CrossRef] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.-L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers 2021, 13, 2284. [Google Scholar] [CrossRef]
- Heine, S.; Kleih, M.; Giménez, N.; Böpple, K.; Ott, G.; Colomer, D.; Aulitzky, W.E.; van der Kuip, H.; Silkenstedt, E. Cyclin D1-CDK4 activity drives sensitivity to bortezomib in mantle cell lymphoma by blocking autophagy-mediated proteolysis of NOXA. J. Hematol. Oncol. 2018, 11, 112. [Google Scholar] [CrossRef]
- Fardid, R.; Ghorbani, Z.; Haddadi, G.; Behzad-Behbahani, A.; Arabsolghar, R.; Kazemi, E.; Okhovat, M.A.; Hosseinimehr, S.J. Effects of Hesperidin as a Radio-protector on Apoptosis in Rat Peripheral Blood Lymphocytes after Gamma Radiation. J. Biomed. Phys. Eng. 2016, 6, 217–228. [Google Scholar]
- Moghadam, S.B.; Masoudi, R.; Monsefi, M. Salvia officinalis Induces Apoptosis in Mammary Carcinoma Cells through Alteration of Bax to Bcl-2 Ratio. Iran. J. Sci. Technol. Trans. A Sci. 2018, 42, 297–303. [Google Scholar] [CrossRef]
- Balusamy, S.R.; Perumalsamy, H.; Veerappan, K.; Huq, M.A.; Rajeshkumar, S.; Lakshmi, T.; Kim, Y.J. Citral Induced Apoptosis through Modulation of Key Genes Involved in Fatty Acid Biosynthesis in Human Prostate Cancer Cells: In Silico and In Vitro Study. Biomed. Res. Int. 2020, 2020, 6040727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumarola, C.; Caffarra, C.; La Monica, S.; Galetti, M.; Alfieri, R.R.; Cavazzoni, A.; Galvani, E.; Generali, D.; Petronini, P.G.; Bonelli, M.A. Effects of sorafenib on energy metabolism in breast cancer cells: Role of AMPK–mTORC1 signaling. Breast Cancer Res. Treat. 2013, 141, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Sun, L.; Wang, Z.; Zhang, Y.; He, Z.; Xu, C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. OncoTargets Ther. 2019, 12, 3339–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.H.; Choi, Y.-J.; Park, J.-W. SIRT1 and AMPK Mediate Hypoxia-Induced Resistance of Non–Small Cell Lung Cancers to Cisplatin and Doxorubicin. Cancer Res. 2014, 74, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Ogura, T.; Kishimoto, A.; Minegishi, Y.; Nakajima, N.; Miyazaki, M.; Esumi, H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene 2002, 21, 6082–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, G.K.; Gifford, A.J.; Chen, Q.; Shen, Y.; Gabrielli, S.; Gill, A.J.; Stevenson, W.S.; Best, O.G. Fatty acid synthase and adenosine monophosphate-activated protein kinase regulate cell survival and drug sensitivity in diffuse large B-cell lymphoma. Leuk. Lymphoma 2020, 61, 1810–1822. [Google Scholar] [CrossRef]
- Zang, M.; Zuccollo, A.; Hou, X.; Nagata, D.; Walsh, K.; Herscovitz, H.; Brecher, P.; Ruderman, N.B.; Cohen, R.A. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004, 279, 47898–47905. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. eLife 2018, 7, e32656. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, J.; Shao, F.; Zhou, Y.; Yu, J.; Wu, H.; Du, J.; Ren, X. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem. Biophys. Res. Commun. 2021, 534, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, K.; Lu, Y.; Zhang, D.; Cheng, Y.; Li, L.; Huang, W.; He, G.; Liao, H.; Cai, L.; et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia 2021, 23, 1227–1239. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, L.; Wang, M.; Yang, K.; Jin, Z.; Zhao, C.; Shi, G. FNDC5 Causes Resistance to Sorafenib by Activating the PI3K/Akt/Nrf2 Pathway in Hepatocellular Carcinoma Cells. Front. Oncol. 2022, 12, 852095. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.-J.; et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, J.; Yan, M.; Wu, J.; Lian, S.; Sun, K.; Li, B.; Ma, J.; Xia, J.; Lian, C. Orlistat induces ferroptosis-like cell death of lung cancer cells. Front. Med. 2021, 15, 922–932. [Google Scholar] [CrossRef]
- Sorriento, D.; Rusciano, M.R.; Visco, V.; Fiordelisi, A.; Cerasuolo, F.A.; Poggio, P.; Ciccarelli, M.; Iaccarino, G. The Metabolic Role of GRK2 in Insulin Resistance and Associated Conditions. Cells 2021, 10, 167. [Google Scholar] [CrossRef]
- Dai, S.; Peng, Y.; Zhu, Y.; Xu, D.; Zhu, F.; Xu, W.; Chen, Q.; Zhu, X.; Liu, T.; Hou, C.; et al. Glycolysis promotes the progression of pancreatic cancer and reduces cancer cell sensitivity to gemcitabine. Biomed. Pharmacother. 2020, 121, 109521. [Google Scholar] [CrossRef]
- Sun, X.; Peng, Y.; Zhao, J.; Xie, Z.; Lei, X.; Tang, G. Discovery and development of tumor glycolysis rate-limiting enzyme inhibitors. Bioorg. Chem. 2021, 112, 104891. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-G1 | G0/G1 | S | G2/M | ||
|---|---|---|---|---|---|
| Huh7 | CTRL | 1.50 ± 0.12 | 61.24 ± 1.76 | 10.58 ± 0.96 | 24.81 ± 1.30 |
| SORA | 2.80 ± 0.89 | 67.01 ± 3.24 | 8.06 ± 1.72 | 21.64 ± 2.26 | |
| ORL | 0.84 ± 0.07 | 86.94 ± 1.70 | 3.22 ± 1.55 | 7.74 ± 1.41 | |
| COMB | 13.30 ± 6.81 | 66.86 ± 6.19 | 7.92 ± 2.64 | 9.61 ± 2.59 | |
| Huh7/SR | CTRL | 1.26 ± 0.28 | 65.87 ± 5.21 | 11.22 ± 1.45 | 21.76 ± 5.38 |
| SORA | 2.33 ± 1.90 | 73.98 ± 4.34 | 7.17 ± 1.20 | 15.39 ± 2.51 | |
| ORL | 1.16 ± 0.33 | 85.40 ± 6.14 | 3.96 ± 3.38 | 8.29 ± 1.78 | |
| COMB | 21.43 ± 7.51 | 64.51 ± 4.83 | 6.39 ± 0.62 | 5.46 ± 2.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shueng, P.-W.; Chan, H.-W.; Lin, W.-C.; Kuo, D.-Y.; Chuang, H.-Y. Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism. Int. J. Mol. Sci. 2022, 23, 6501. https://doi.org/10.3390/ijms23126501
Shueng P-W, Chan H-W, Lin W-C, Kuo D-Y, Chuang H-Y. Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism. International Journal of Molecular Sciences. 2022; 23(12):6501. https://doi.org/10.3390/ijms23126501
Chicago/Turabian StyleShueng, Pei-Wei, Hui-Wen Chan, Wei-Chan Lin, Deng-Yu Kuo, and Hui-Yen Chuang. 2022. "Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism" International Journal of Molecular Sciences 23, no. 12: 6501. https://doi.org/10.3390/ijms23126501
APA StyleShueng, P.-W., Chan, H.-W., Lin, W.-C., Kuo, D.-Y., & Chuang, H.-Y. (2022). Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism. International Journal of Molecular Sciences, 23(12), 6501. https://doi.org/10.3390/ijms23126501