
Increase of Circulating Monocyte–Platelet Conjugates in Rheumatoid Arthritis Responders to IL-6 Blockage

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
2.1. Circulating CD14+PLT+ in RA Patients
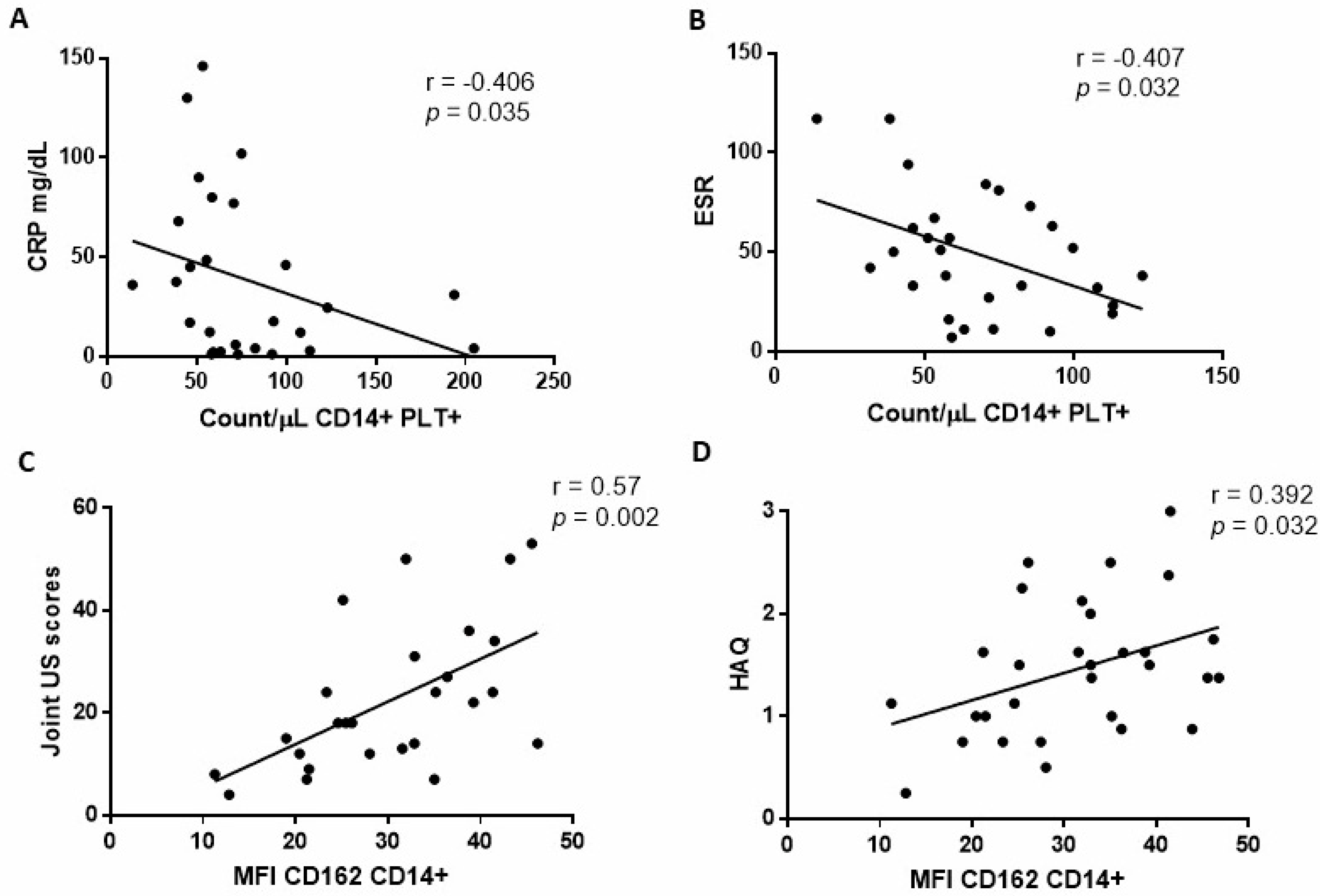
2.2. Relationship between CD14+PLT+ and mCD162 Expression on Monocytes with Clinical RA Parameters
2.3. Relationship between CD14+PLT+ and mCD162 Expression on Monocytes with Clinical Outcome in RA Patients after TCZ Treatment
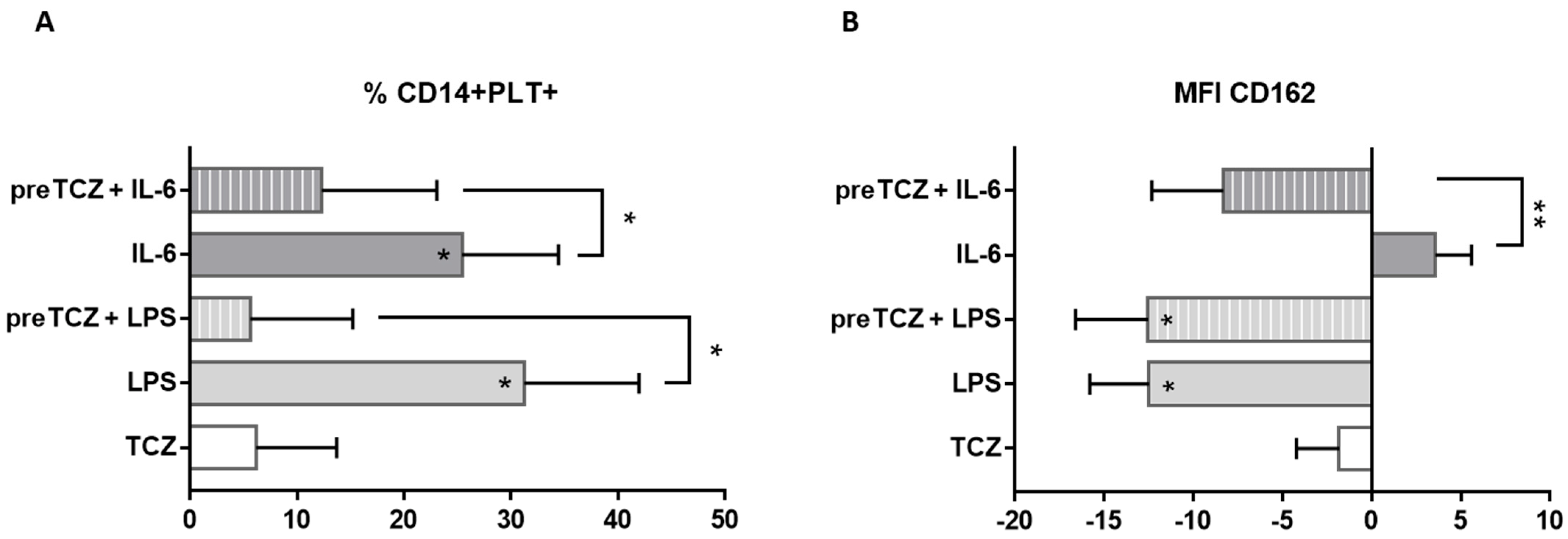
2.4. IL-6 and TCZ Effects over PLT Binding to Monocytes, mCD162 Expression on Monocytes and PLT Activation
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Peripheral Blood Staining and Flow Cytometry Analysis
4.3. Plasma Analysis of RF, ACPAs, and Cytokines
4.4. Peripheral Blood Cultures
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muravyev, Y.V. Extra-articular manifestations of rheumatoid arthritis. Nauchno-Prakt. Revmatol. 2018, 56, 356–362. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Mechanism of Disease the Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Lioté, F.; Boval-Boizard, B.; Weill, D.; Kuntz, D.; Wautier, J.L. Blood monocyte activation in rheumatoid arthritis: Increased monocyte adhesiveness, integrin expression, and cytokine release. Clin. Exp. Immunol. 1996, 106, 13–19. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Panizzi, P.; Figueiredo, J.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; Aikawa, E.; et al. Identification Monocytes Inflammatory of Splenic Reservoir and Their Deployment Sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Boutet, M.A.; Courties, G. Novel insights into macrophage diversity in rheumatoid arthritis synovium. Autoimmun. Rev. 2021, 20, 102758. [Google Scholar] [CrossRef]
- Nakashima, T.; Takayanagi, H. The dynamic interplay between osteoclasts and the immune system. Arch. Biochem. Biophys. 2008, 473, 166–171. [Google Scholar] [CrossRef]
- Semple, J.W.; Freedman, J. Platelets and innate immunity. Cell. Mol. Life Sci. 2010, 67, 499–511. [Google Scholar] [CrossRef]
- Smith, T.L.; Weyrich, A.S. Platelets as central mediators of systemic inflammatory responses. Thromb. Res. 2011, 127, 4. [Google Scholar] [CrossRef]
- Tunjungputri, R.N.; van der Ven, A.J. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2- and TLR4-mediated inflammatory responses. Thromb. Haemost. 2015, 113, 1035–1045. [Google Scholar]
- Zamora, C.; Cantó, E. Functional consequences of platelet binding to T lymphocytes in inflammation. J. Leukoc. Biol. 2013, 94, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Nguyen, K.A. The inflammatory role of platelets via their TLRs and Siglec receptors. Front. Immunol. 2015, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Habets, K.L.L.; Trouw, L.A. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 209. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef]
- Cafaro, G.; Bartoloni, E. Platelets: A potential target for rheumatoid arthritis treatment? Expert Rev. Clin. Immunol. 2019, 15, 1–3. [Google Scholar] [CrossRef]
- Yun, S.H.; Sim, E.H. Platelet activation: The mechanisms and potential biomarkers. Biomed Res. Int. 2016, 2016, 10–15. [Google Scholar] [CrossRef]
- Pamuk, G.E.; Vural, Ö. Increased platelet activation markers in rheumatoid arthritis: Are they related with subclinical atherosclerosis? Platelets 2008, 19, 146–154. [Google Scholar] [CrossRef]
- Boilard, E.; Blanco, P. Platelets: Active players in the pathogenesis of arthritis and SLE. Nat. Rev. Rheumatol. 2012, 8, 534–542. [Google Scholar] [CrossRef]
- Bunescu, A.; Seideman, P. Enhanced Fc γ Receptor I, α M ß 2 Integrin Receptor Expression by Monocytes and Neutrophils in Rheumatoid Arthritis: Interaction with Platelets. J. Rheumatol. 2004, 31, 2345–2355. [Google Scholar]
- Stephen, J.; Emerson, B. The uncoupling of monocyte-platelet interactions from the induction of proinflammatory signaling in monocytes. J. Immunol. 2013, 191, 5677–5683. [Google Scholar] [CrossRef]
- da Costa Martins, P.A.; van Gils, J.M. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. J. Leukoc. Biol. 2006, 79, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.I.; Chen, Z. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, E.; Tenno, T. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Asch, A.S. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J. Clin. Investig. 1989, 84, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Von Brühl, M.L. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J. Thromb. Haemost. 2011, 9, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Rong, M.; Wang, C. Platelets induce a proinflammatory phenotype in monocytes via the CD147 pathway in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, 478. [Google Scholar] [CrossRef] [PubMed]
- van Gils, J.M.; Zwaginga, J.J. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 2008, 85, 195–204. [Google Scholar] [CrossRef]
- Larsen, E.; Celi, A. PADGEM protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 1989, 59, 305–312. [Google Scholar] [CrossRef]
- Mariscal, A.; Zamora, C. Phenotypic and Functional Consequences of PLT Binding to Monocytes and Its Association with Clinical Features in SLE. Int. J. Mol. Sci. 2021, 22, 4719. [Google Scholar] [CrossRef]
- Liang, H.; Duan, Z. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell. Mol. Immunol. 2015, 12, 435–443. [Google Scholar] [CrossRef]
- Marsik, C.; Mayr, F. Endotoxin down-modulates P-selectin glycoprotein ligand-1 (PSGL-1, CD162) on neutrophils in humans. J. Clin. Immunol. 2004, 24, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Zamora, C.; Canto, E. Inverse Association Between Circulating Monocyte-Platelet Complexes and Inflammation in Ulcerative Colitis Patients. Inflamm. Bowel Dis. 2018, 24, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Davenpeck, K.L.; Brummet, M.E. Activation of Human Leukocytes Reduces Surface P-Selectin Glycoprotein Ligand-1 (PSGL-1, CD162) and Adhesion to P-Selectin In Vitro. J. Immunol. 2000, 165, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Passacquale, G.; Vamadevan, P. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS ONE 2011, 6, e25595. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Dohle, F. Down-Regulation of Monocyte Apoptosis by Phagocytosis of Platelets: Involvement of a Caspase-9, Caspase-3, and Heat Shock Protein 70-Dependent Pathway. J. Immunol. 2002, 168, 6152–6158. [Google Scholar] [CrossRef]
- Ammon, C.; Kreutz, M. Platelets induce monocyte differentiation in serum-free coculture. J. Leukoc. Biol. 1998, 63, 469–476. [Google Scholar] [CrossRef]
- Gudbrandsdottir, S.; Hasselbalch, H.C. Activated platelets enhance IL-10 secretion and reduce TNF-α secretion by monocytes. J. Immunol. 2013, 191, 4059–4067. [Google Scholar] [CrossRef]
- Schwartzkopff, F.; Petersen, F. CXC chemokine ligand 4 (CXCL4) down-regulates CC chemokine receptor expression on human monocytes. Innate Immun. 2012, 18, 124–139. [Google Scholar] [CrossRef]
- Sarma, J.; Laan, C.A. Increased Platelet Binding to Circulating Monocytes in Acute Coronary Syndromes. Circulation 2002, 105, 2166–2171. [Google Scholar] [CrossRef]
- Harding, S.A.; Sommerfield, A.J. Increased CD40 ligand and platelet—monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis 2004, 176, 321–325. [Google Scholar] [CrossRef]
- Joseph, J.E.; Harrison, P. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Pamuk, G.E.; Turgut, B. Increased circulating platelet-leucocyte complexes in patients with primary Raynaud’s phenomenon and Raynaud’s phenomenon secondary to systemic sclerosis: A comparative study. Blood Coagul. Fibrinolysis 2007, 18, 297–302. [Google Scholar] [CrossRef]
- Pamuk, G.E.; Pamuk, Ö.N. Elevated platelet-monocyte complexes ın patıents wıth psorıatıc arthrıtıs. Platelets 2009, 20, 493–497. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Weber, K.S.C. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Linke, B.; Schreiber, Y. Activated Platelets Induce an Anti-Inflammatory Response of Monocytes/Macrophages through Cross-Regulation of PGE2 and Cytokines. Mediat. Inflamm. 2017, 2017, 1463216. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.; Walsh, D. Outcome in rheumatoid arthritis patients with continued conventional therapy for moderate disease activity-the early RA network (ERAN). Rheumatology 2011, 50, 926–931. [Google Scholar] [CrossRef]
- Tono, T.; Aihara, S. Effects of anti-IL-6 receptor antibody on human monocytes. Mod. Rheumatol. 2015, 25, 79–84. [Google Scholar] [CrossRef]
- Lee, J.S.; Oh, J.S. Tocilizumab-induced Thrombocytopenia in Patients with Rheumatoid Arthritis. J. Rheum. Dis. 2019, 26, 186. [Google Scholar] [CrossRef]
- Gibiansky, L.; Frey, N. Linking interleukin-6 receptor blockade with tocilizumab and its hematological effects using a modeling approach. J. Pharmacokinet. Pharmacodyn. 2012, 39, 5–16. [Google Scholar] [CrossRef]
- Veale, D.J.; Maple, C. Soluble cell adhesion molecules—P-selectin and ICAM-1, and disease activity in patients receiving sulphasalazine for active rheumatoid arthritis. Scand. J. Rheumatol. 1998, 27, 296–299. [Google Scholar]
- Lee, S.J.; Yoon, B.R. Activated Platelets Convert CD14+CD16- Into CD14+CD16+ Monocytes With Enhanced FcγR-Mediated Phagocytosis and Skewed M2 Polarization. Front. Immunol. 2021, 11, 3363. [Google Scholar] [CrossRef] [PubMed]
- Torsteinsdóttir, I.; Arvidson, N.G. Monocyte activation in rheumatoid arthritis (RA): Increased integrin, Fcγ and complement receptor expression and the effect of glucocorticoids. Clin. Exp. Immunol. 1999, 115, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Higuchi, Y. IL-6 plays an essential role in neutrophilia under inflammation. Cytokine 2011, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Zamora, C.; Cantó, E. Binding of Platelets to Lymphocytes: A Potential Anti-Inflammatory Therapy in Rheumatoid Arthritis. J. Immunol. 2017, 198, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Paré, A. Platelets can enhance vascular permeability. Blood 2012, 120, 1334–1343. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Uemura, M. Clinical usefulness of the 1987 revised criteria for rheumatoid arthritis by American Rheumatism Association. Nihon Naika Gakkai Zasshi 1988, 77, 742–743. [Google Scholar] [CrossRef][Green Version]
- Sanmartí, R.; García-Rodríguez, S. Actualización 2014 del Documento de Consenso de la Sociedad Española de Reumatología sobre el uso de terapias biológicas en la artritis reumatoide. Reumatol. Clin. 2015, 11, 279–294. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Botar-Jid, C.; Bolboaca, S. Gray scale and power Doppler ultrasonography in evaluation of early rheumatoid arthritis. Med. Ultrason. 2010, 12, 300–305. [Google Scholar]
- Futó, G.; Somogyi, A. Visualization of DAS28, SDAI, and CDAI: The magic carpets of rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 623–629. [Google Scholar] [CrossRef]
- Van Gestel, A.M.; Prevoo, M.L.L. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Arthritis Rheum. 1996, 39, 34–40. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RA (n = 35) | HD (n = 15) | p | |
|---|---|---|---|
| Sex, % (n), women | 80.6 (28) | 80 (12) | 0.96 |
| Age in years, mean ± sd | 53.82 ± 10.72 | 52.48 ± 8.1 | 0.72 |
| Years of evolution, median (IQR) | 12.5 (7–17) | ||
| Corticoids, % (n) | 67.7 (21) | ||
| Monotherapy, % (n) | 29 (9) | ||
| MTX, % (n) | 42 (13) | ||
| Previous biological therapies, % (n) | 64.5 (20) | ||
| HAQ, mean ± sd | 1.4 ± 0.67 | ||
| DAS28, mean ± sd | 5.7 ± 1.15 | ||
| ESR (mm/h), mean ± sd | 51.4 ± 32 | ||
| CRP (mg/L), median (IQR) | 24.6 (4–68) | ||
| ACPA+, % (n) | 61.3 (19) | ||
| ACPA (UI/mL), median (IQR) | 140.7 (54–318) | ||
| RF+, % (n) | 71 (22) | ||
| RF (UI/mL), median (IQR) | 161 (32.5–333.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariscal, A.; Zamora, C.; Díaz-Torné, C.; Ortiz, M.À.; Agustín, J.J.d.; Reina, D.; Estrada, P.; Moya, P.; Corominas, H.; Vidal, S. Increase of Circulating Monocyte–Platelet Conjugates in Rheumatoid Arthritis Responders to IL-6 Blockage. Int. J. Mol. Sci. 2022, 23, 5748. https://doi.org/10.3390/ijms23105748
Mariscal A, Zamora C, Díaz-Torné C, Ortiz MÀ, Agustín JJd, Reina D, Estrada P, Moya P, Corominas H, Vidal S. Increase of Circulating Monocyte–Platelet Conjugates in Rheumatoid Arthritis Responders to IL-6 Blockage. International Journal of Molecular Sciences. 2022; 23(10):5748. https://doi.org/10.3390/ijms23105748
Chicago/Turabian StyleMariscal, Anaís, Carlos Zamora, César Díaz-Torné, Mᵃ Àngels Ortiz, Juan José de Agustín, Delia Reina, Paula Estrada, Patricia Moya, Héctor Corominas, and Sílvia Vidal. 2022. "Increase of Circulating Monocyte–Platelet Conjugates in Rheumatoid Arthritis Responders to IL-6 Blockage" International Journal of Molecular Sciences 23, no. 10: 5748. https://doi.org/10.3390/ijms23105748
APA StyleMariscal, A., Zamora, C., Díaz-Torné, C., Ortiz, M. À., Agustín, J. J. d., Reina, D., Estrada, P., Moya, P., Corominas, H., & Vidal, S. (2022). Increase of Circulating Monocyte–Platelet Conjugates in Rheumatoid Arthritis Responders to IL-6 Blockage. International Journal of Molecular Sciences, 23(10), 5748. https://doi.org/10.3390/ijms23105748