
The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Atorvastatin Reduces Neuronal Death after Global Cerebral Ischemia
2.2. Atorvastatin Reduces Ischemia-Induced Oxidative Stress
2.3. Atorvastatin Reduces Microglia and Astrocyte Activation after Global Cerebral Ischemia
2.4. Atorvastatin Prevents Blood–Brain Barrier Disruption after Global Cerebral Ischemia
2.5. Atorvastatin Prevents Endothelial Cell Loss and Vasa Vasorum Proliferation after Global Cerebral Ischemia
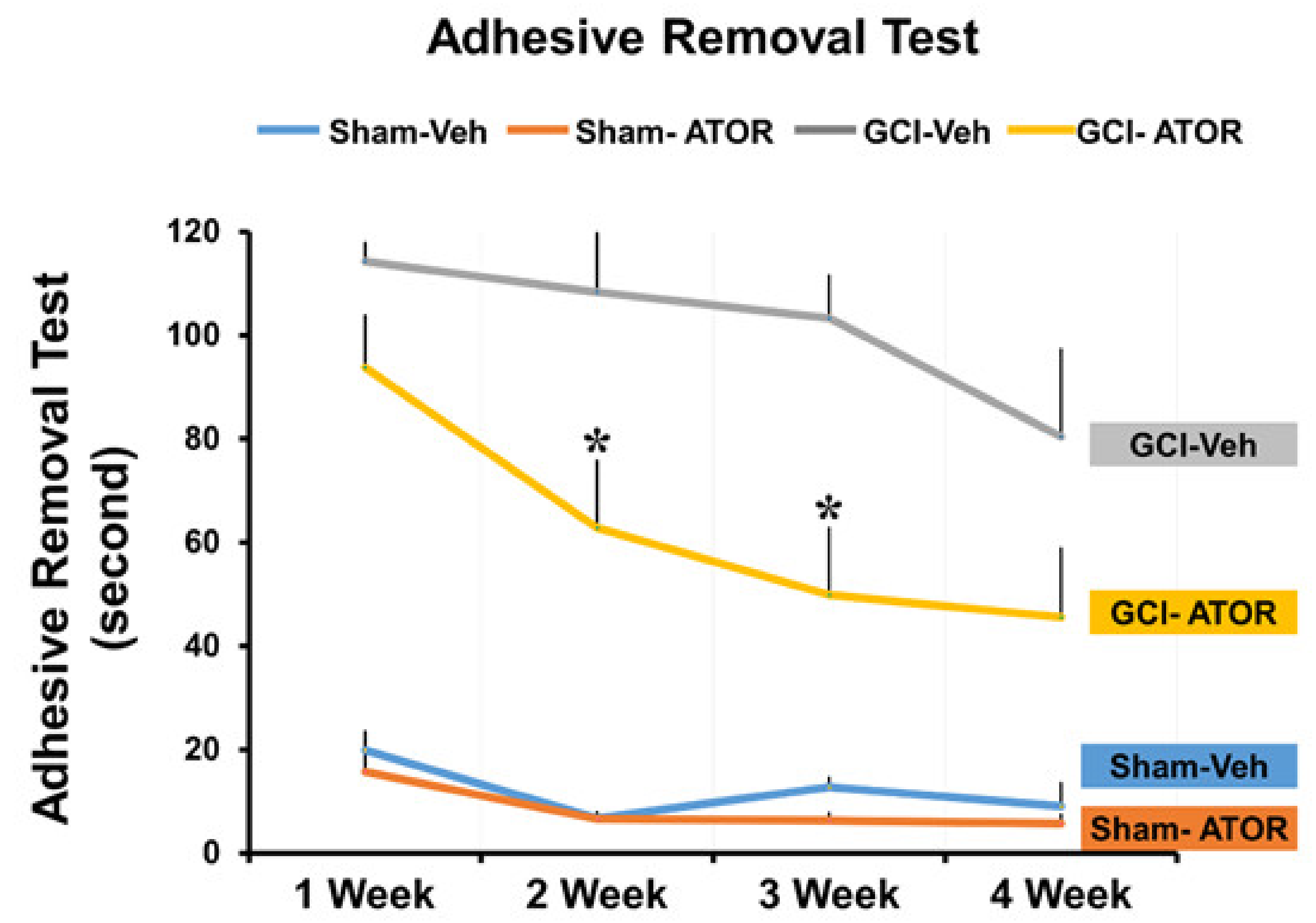
2.6. Atorvastatin Improves the Cognitive Function after Global Cerebral Ischemia
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Global Cerebral Ischemia Surgery
4.3. Statin Administration
4.4. Brain Sample Procedure
4.5. Analysis of Hippocampal Neuronal Death
4.6. Immunohistochemistry
4.7. Immunofluorescence Analysis
4.8. Behavioral Testing
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harukuni, I.; Bhardwaj, A. Mechanisms of Brain Injury after Global Cerebral Ischemia. Neurol. Clin. 2006, 24, 1–21. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Gopal, P.; Parker, J.R.; Downs, R.K.; Parker, J.C.; Dawn, B. Global cerebral ischemia due to circulatory arrest: Insights into cellular pathophysiology and diagnostic modalities. Mol. Cell. Biochem. 2016, 426, 111–127. [Google Scholar] [CrossRef]
- Laver, S.; Farrow, C.; Turner, D.; Nolan, J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensiv. Care Med. 2004, 30, 2126–2128. [Google Scholar] [CrossRef]
- Nolan, J.P.; Neumar, R.W.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation 2008, 79, 350–379. [Google Scholar] [PubMed]
- Meyer, F.B.; Sundt, T.M.; Yanagihara, T.; Anderson, R.E. Focal Cerebral Ischemia: Pathophysiologic Mechanisms and Rationale for Future Avenues of Treatment. Mayo Clin. Proc. 1987, 62, 35–55. [Google Scholar] [CrossRef]
- Kurisu, K.; Yenari, M.A. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology 2018, 134, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. N. Engl. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef]
- Nielsen, N.; Wetterslev, J.; Cronberg, T.; Erlinge, D.; Gasche, Y.; Hassager, C.; Horn, J.; Hovdenes, J.; Kjaergaard, J.; Kuiper, M.; et al. Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. N. Engl. J. Med. 2013, 369, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Colio, L.M.; Tuñón, J.; Martín-Ventura, J.L. Jes Anti-inflammatory and immunomodulatory effects of statins. Kidney Int. 2003, 63, 12–23. [Google Scholar] [CrossRef]
- Mekontso-Dessap, A.; Brun-Buisson, C. Statins: The next step in adjuvant therapy for sepsis? Intensiv. Care Med. 2006, 32, 11–14. [Google Scholar] [CrossRef]
- Li, Q.; Zhuang, Q.-K.; Yang, J.-N.; Zhang, Y.-Y. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: The potential molecular mechanisms. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1113–1126. [Google Scholar] [PubMed]
- Saito, T.; Nito, C.; Ueda, M.; Inaba, T.; Kamiya, F.; Muraga, K.; Katsura, K.-I.; Katayama, Y. Continuous oral administration of atorvastatin ameliorates brain damage after transient focal ischemia in rats. Life Sci. 2014, 94, 106–114. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Anoopkumar-Dukie, S.; Arora, D.S.; Grant, G.D.; McDermott, C.M.; Perkins, A.V.; Davey, A.K. Molecular Mechanisms Underlying the Effects of Statins in the Central Nervous System. Int. J. Mol. Sci. 2014, 15, 607. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Z.G.; Li, Y.; Wang, Y.; Wang, L.; Jiang, H.; Bs, C.Z.; Lü, M.; Bs, M.K.; Feldkamp, C.S.; et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann. Neurol. 2003, 53, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Vizcaychipi, M.P.; Watts, H.R.; O’Dea, K.P.; Lloyd, D.G.; Penn, J.W.; Wan, Y.; Pac-Soo, C.; Takata, M.; Ma, D. The Therapeutic Potential of Atorvastatin in a Mouse Model of Postoperative Cognitive Decline. Ann. Surg. 2014, 259, 1235–1244. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Purushothaman, K.R.; Sirol, M.; Levy, A.P.; Fuster, V. Neovascularization in human atherosclerosis. Circulation 2006, 113, 2245–2252. [Google Scholar] [CrossRef]
- Kampschulte, M.; Buch, T.; Bohle, R.M.; Langheinrich, A.C. Vasa vasorum and atherosclerosis—Quid novi? Thromb. Haemost. 2007, 97, 873–879. [Google Scholar] [CrossRef]
- O’Brien, K.D.; McDonald, T.O.; Chait, A.; Allen, M.D.; Alpers, C.E. Neovascular Expression of E-Selectin, Intercellular Adhesion Molecule-1, and Vascular Cell Adhesion Molecule-1 in Human Atherosclerosis and Their Relation to Intimal Leukocyte Content. Circulation 1996, 93, 672–682. [Google Scholar] [CrossRef]
- Moulton, K.S. ArePlaque angiogenesis and atherosclerosis. Curr. Atheroscler. Rep. 2001, 3, 225–233. [Google Scholar] [CrossRef]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [Google Scholar]
- Herculano-Houzel, S.; Lent, R. Isotropic fractionator: A simple, rapid method for the quantification of total cell and neuron numbers in the brain. J. Neurosci. 2005, 25, 2518–2521. [Google Scholar] [CrossRef] [PubMed]
- Dent, M.A.; Segura-Anaya, E.; Alva-Medina, J.; Aranda-Anzaldo, A. NeuN/Fox-3 is an intrinsic component of the neuronal nuclear matrix. FEBS Lett. 2010, 584, 2767–2771. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhang, Y.-P.; Hou, Z.; Huang, C.; Zhu, H.; Zhang, C.-Q.; Yin, Q. Novel Insights into NeuN: From Neuronal Marker to Splicing Regulator. Mol. Neurobiol. 2015, 53, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, T.; Fang, B.; Wang, A.; Zhang, M.; Hussain, S.; Luo, L.; Zhang, R.; Zhang, Q.; Wu, J.; et al. NeuN-Specific Fluorescent Probe Revealing Neuronal Nuclei Protein and Nuclear Acids Association in Living Neurons under STED Nanoscopy. ACS Appl. Mater. Interfaces 2018, 10, 31959–31964. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gao, W.; Cheng, S.; Yin, D.; Li, F.; Wu, Y.; Sun, D.; Zhou, S.; Wang, D.; Zhang, Y.; et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J. Neuroinflammation 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Xu, M.; Zhou, J.; Ge, X.; Chen, G.; Guo, L.; Luo, L.; Li, K.; Zhu, Z.; Zhang, F. Atorvastatin Attenuates Ischemia/Reperfusion-Induced Hippocampal Neurons Injury Via Akt-nNOS-JNK Signaling Pathway. Cell Mol. Neurobiol. 2017, 37, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, X.; Zhai, L.; Sheng, X.; Zheng, W.; Chu, H.; Zhang, G. Atorvastatin Attenuates Cognitive Deficits and Neuroinflammation Induced by Abeta1-42 Involving Modulation of TLR4/TRAF6/NF-kappaB Pathway. J. Mol. Neurosci. 2018, 64, 363–373. [Google Scholar] [CrossRef]
- Laufs, U.; Gertz, K.; Huang, P.; Nickenig, G.; Böhm, M.; Dirnagl, U.; Endres, M. Atorvastatin Upregulates Type III Nitric Oxide Synthase in Thrombocytes, Decreases Platelet Activation, and Protects From Cerebral Ischemia in Normocholesterolemic Mice. Stroke 2000, 31, 2442–2449. [Google Scholar] [CrossRef]
- Hong, K.-S.; Lee, J.S. Statins in Acute Ischemic Stroke: A Systematic Review. J. Stroke 2015, 17, 282–301. [Google Scholar] [CrossRef]
- Amin-Hanjani, S.; Stagliano, N.E.; Yamada, M.; Huang, P.L.; Liao, J.K.; Moskowitz, M.A. Mevastatin, an HMG-CoA Reductase Inhibitor, Reduces Stroke Damage and Upregulates Endothelial Nitric Oxide Synthase in Mice. Stroke 2001, 32, 980–986. [Google Scholar] [CrossRef]
- Asahi, M.; Huang, Z.; Thomas, S.S.; Yoshimura, S.-I.; Sumii, T.; Mori, T.; Qiu, J.; Amin-Hanjani, S.; Huang, P.L.; Liao, J.K.; et al. Protective Effects of Statins Involving Both eNOS and tPA in Focal Cerebral Ischemia. Br. J. Pharmacol. 2005, 25, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Gertz, K.; Laufs, U.; Lindauer, U.; Nickenig, G.; Böhm, M.; Dirnagl, U.; Endres, M. Withdrawal of Statin Treatment Abrogates Stroke Protection in Mice. Stroke 2003, 34, 551–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ni Chroinin, D.; Callaly, E.L.; Duggan, J.; Merwick, A.; Hannon, N.; Sheehan, O.; Marnane, M.; Horgan, G.; Williams, E.B.; Harris, D.; et al. Association between acute statin therapy, survival, and improved functional outcome after ischemic stroke: The North Dublin Population Stroke Study. Stroke 2011, 42, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- European Stroke Organisation (ESO) Executive Committee; ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc. Dis. 2008, 25, 457–507. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Giovannini, M.G. An Overview on the Differential Interplay Among Neurons–Astrocytes–Microglia in CA1 and CA3 Hippocampus in Hypoxia/Ischemia. Front. Cell. Neurosci. 2020, 14, 585833. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; Yang, S.; Huang, J.; Xue, X.; Zheng, Y.; Shang, G.; Tao, J.; Chen, L. Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci. 2016, 151, 313–322. [Google Scholar] [CrossRef]
- Macin, S.M.; Perna, E.R.; Farías, E.F.; Franciosi, V.; Cialzeta, J.R.; Brizuela, M.; Medina, F.; Tajer, C.; Doval, H.; Badaracco, R. Atorvastatin has an important acute anti-inflammatory effect in patients with acute coronary syndrome: Results of a randomized, double-blind, placebo-controlled study. Am. Hear. J. 2005, 149, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.K.; Ridker, P.M. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat. Rev. Drug Discov. 2005, 4, 977–987. [Google Scholar] [CrossRef]
- Kaur, C.; Ling, E.A. Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc. Res. 2008, 5, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Van Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood–Brain Barrier Breakdown in Acute and Chronic Cerebrovascular Disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, R.; Kaya, M.; Elmas, I.; Arican, N.; Ahishali, B.; Uzun, H.; Bilgic, B.; Kucuk, M.; Kudat, H. Effects of atorvastatin on blood–brain barrier permeability during l-NAME hypertension followed by angiotensin-II in rats. Brain Res. 2005, 1042, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Ritman, E.L.; Lerman, A. The dynamic vasa vasorum. Cardiovasc. Res. 2007, 75, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Nishimiya, K.; Matsumoto, Y.; Takahashi, J.; Uzuka, H.; Wang, H.; Tsuburaya, R.; Hao, K.; Ohyama, K.; Odaka, Y.; Miyata, S.; et al. Enhanced Adventitial Vasa Vasorum Formation in Patients With Vasospastic Angina: Assessment With OFDI. J. Am. Coll. Cardiol. 2016, 67, 598–600. [Google Scholar] [CrossRef]
- Park, K.-H.; Sun, T.; Diez-Delhoyo, F.; Liu, Z.; Yang, S.-W.; Lennon, R.J.; Herrmann, J.; Gulati, R.; Rodriguez-Porcel, M.; Lerman, L.O.; et al. Association between coronary microvascular function and the vasa vasorum in patients with early coronary artery disease. Atherosclerosis 2016, 253, 144–149. [Google Scholar] [CrossRef]
- Holt, A.W.; Tulis, D.A. Experimental Rat and Mouse Carotid Artery Surgery: Injury and Remodeling Studies. ISRN Minim. Invasive Surg. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Li, X.-D.; Hong, M.-N.; Chen, J.; Lu, Y.-Y.; Ye, M.-Q.; Ma, Y.; Zhu, D.-L.; Gao, P.-J. Adventitial fibroblast-derived vascular endothelial growth factor promotes vasa vasorum-associated neointima formation and macrophage recruitment. Cardiovasc. Res. 2019, 116, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.-L.; Auer, R.N. The density and distribution of ischemic brain injury in the rat following 2?10 min of forebrain ischemia. Acta Neuropathol. 1984, 64, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Auer, R.N.; Olsson, Y.; Siesjo, B.K. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: A quantitative study. Diabetes 1984, 33, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Kim, J.H.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Sohn, M.; Suh, S.W. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetyl-l-cysteine (NAC). Amino Acids 2016, 49, 367–378. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Swanson, R.A. Poly(ADP-Ribose) Polymerase-1 Promotes Microglial Activation, Proliferation, and Matrix Metalloproteinase-9-Mediated Neuron Death. J. Immunol. 2005, 174, 2288–2296. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc Triggers Microglial Activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kho, A.R.; Hong, D.K.; Kang, B.S.; Park, W.-J.; Choi, K.C.; Park, K.-H.; Suh, S.W. The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death. Int. J. Mol. Sci. 2021, 22, 4385. https://doi.org/10.3390/ijms22094385
Kho AR, Hong DK, Kang BS, Park W-J, Choi KC, Park K-H, Suh SW. The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death. International Journal of Molecular Sciences. 2021; 22(9):4385. https://doi.org/10.3390/ijms22094385
Chicago/Turabian StyleKho, A Ra, Dae Ki Hong, Beom Seok Kang, Woo-Jung Park, Kyung Chan Choi, Kyoung-Ha Park, and Sang Won Suh. 2021. "The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death" International Journal of Molecular Sciences 22, no. 9: 4385. https://doi.org/10.3390/ijms22094385
APA StyleKho, A. R., Hong, D. K., Kang, B. S., Park, W.-J., Choi, K. C., Park, K.-H., & Suh, S. W. (2021). The Effects of Atorvastatin on Global Cerebral Ischemia-Induced Neuronal Death. International Journal of Molecular Sciences, 22(9), 4385. https://doi.org/10.3390/ijms22094385