
Modeling Sialidosis with Neural Precursor Cells Derived from Patient-Derived Induced Pluripotent Stem Cells

Abstract
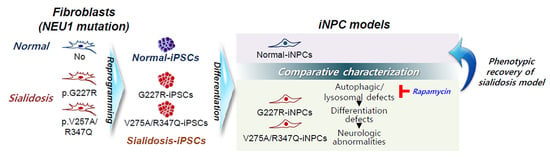
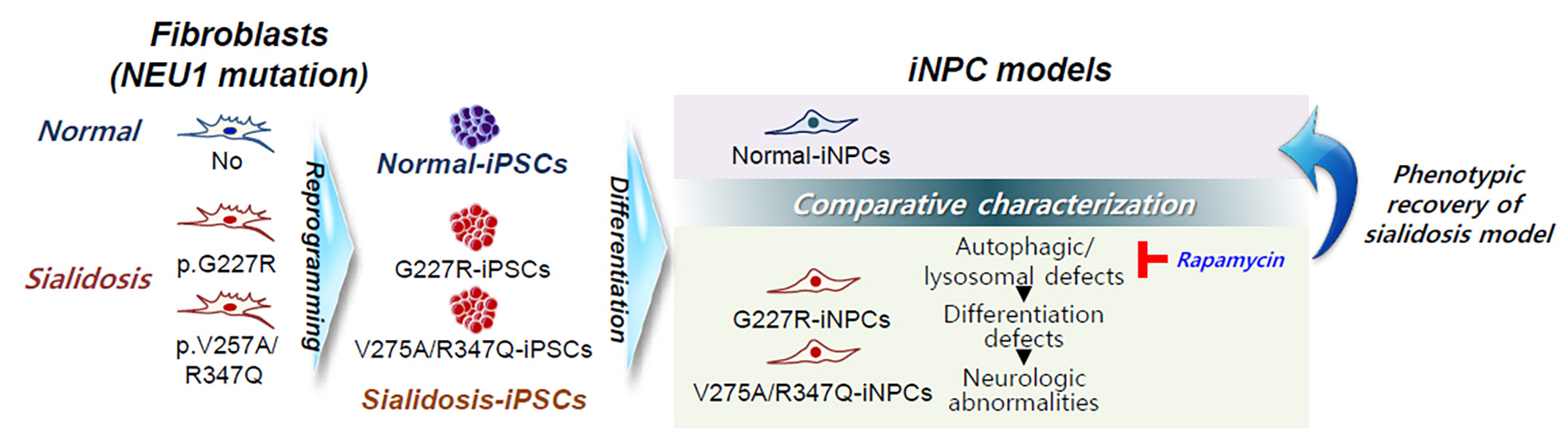{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
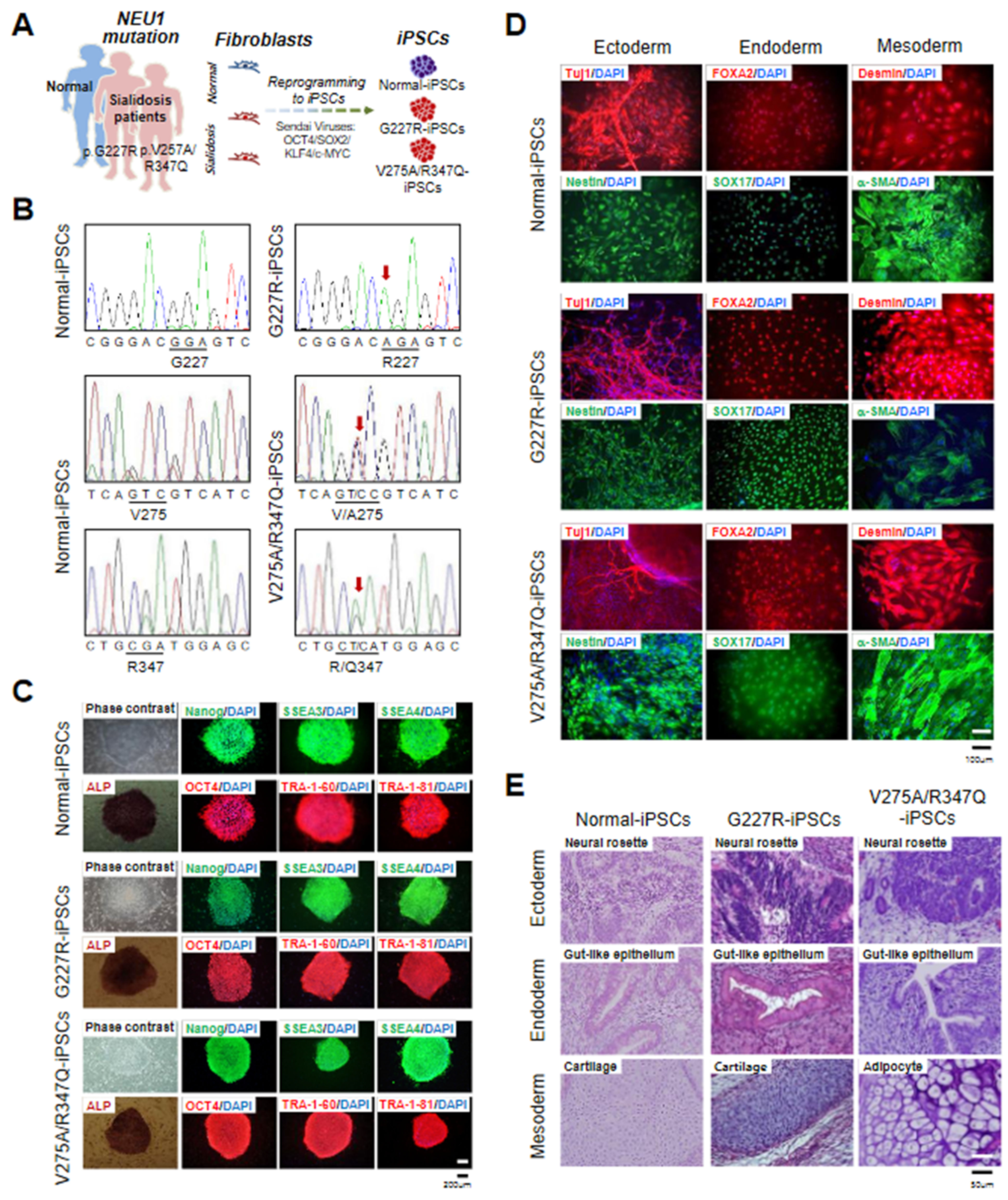
2.1. Generation and Characterization of Sialidosis-iPSCs
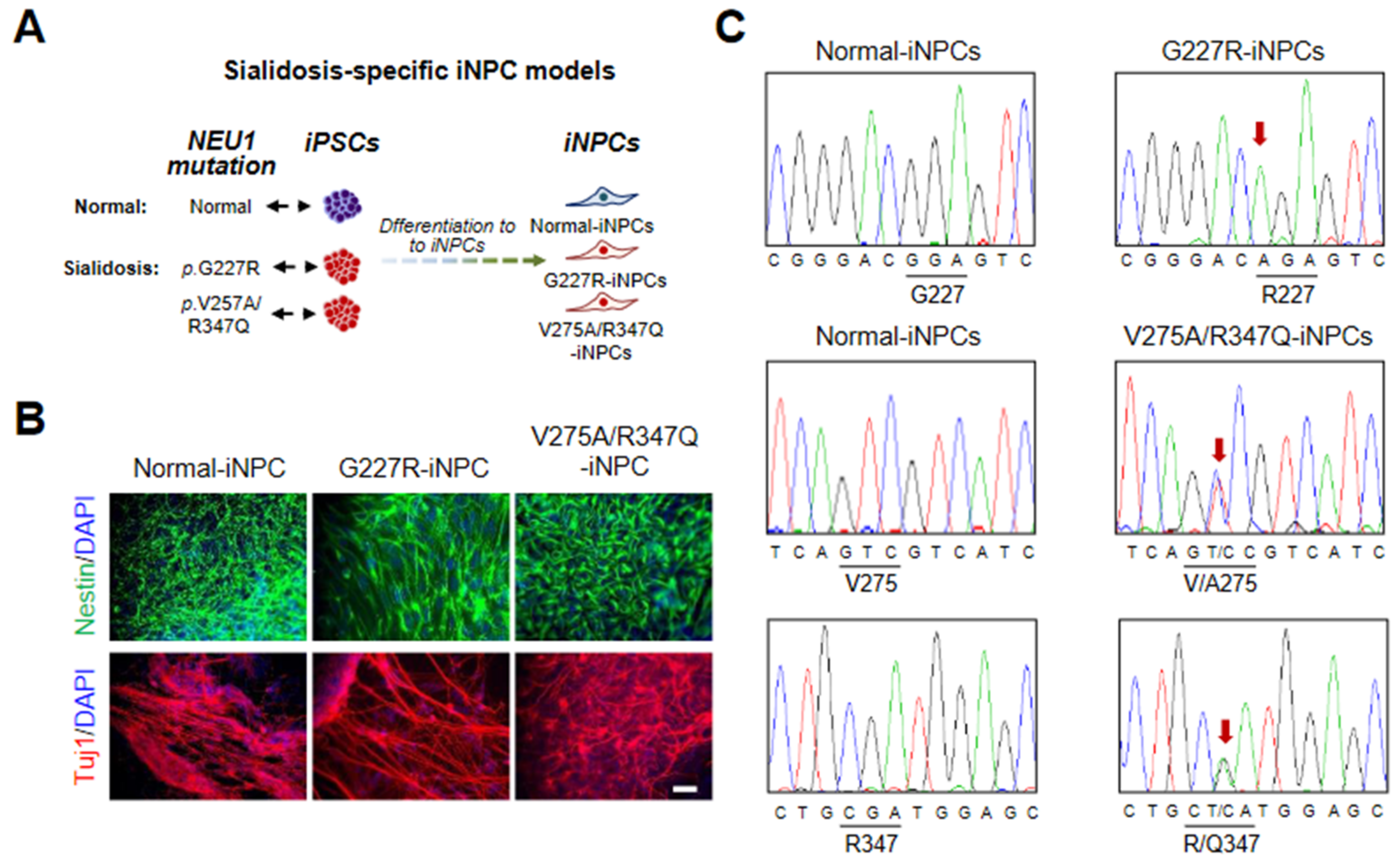
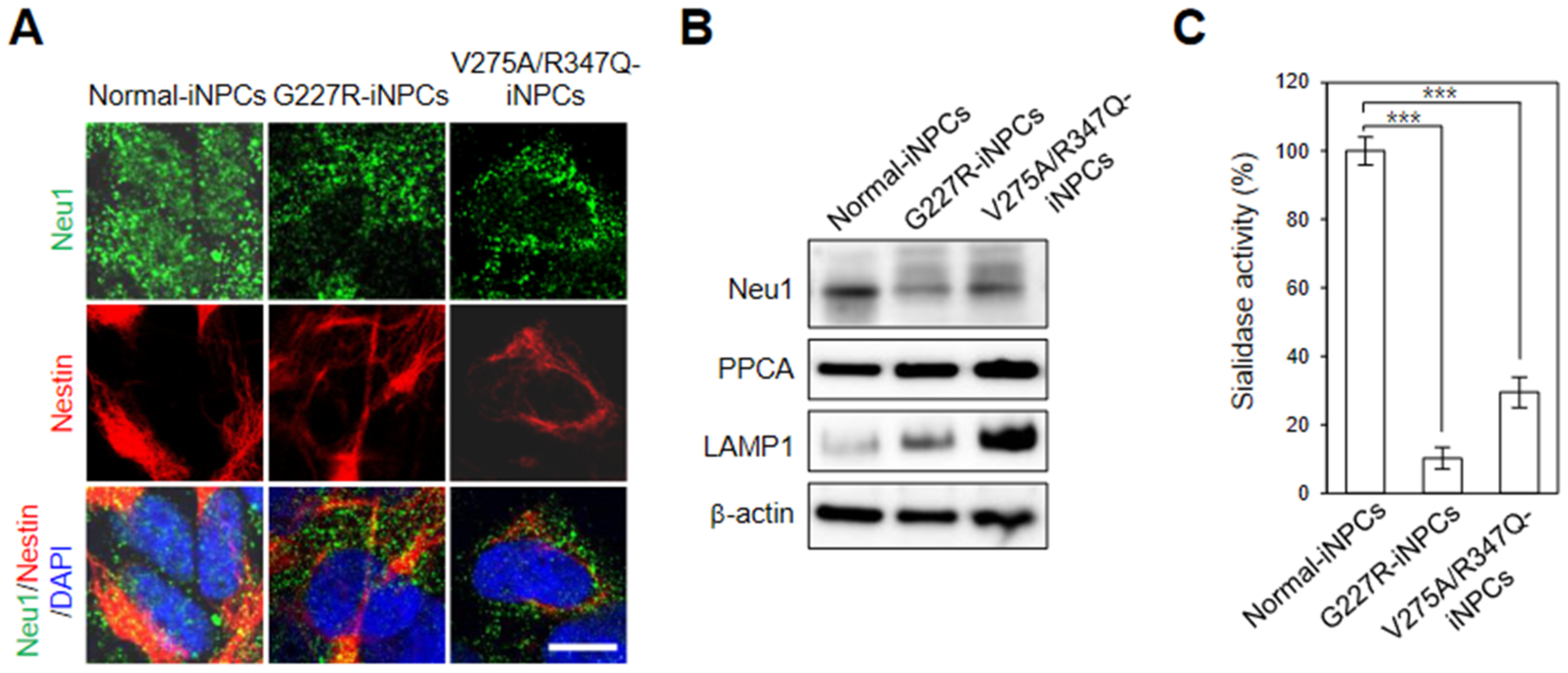
2.2. Generation and Characterization of Sialidosis-iNPCs
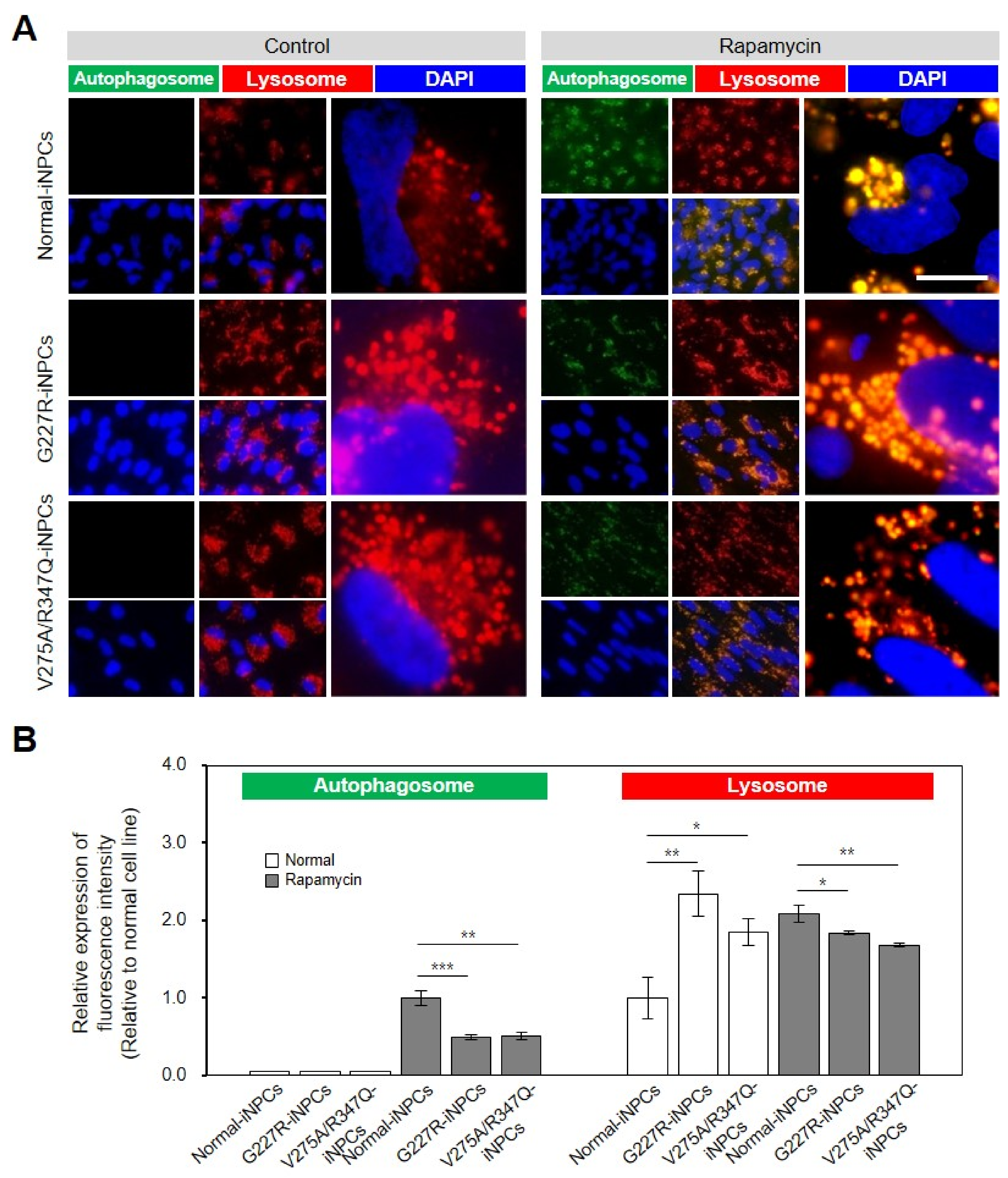
2.3. NEU1 Deficiency-Associated Autophagy–Lysosomal Dysfunction in Sialidosis-iNPCs
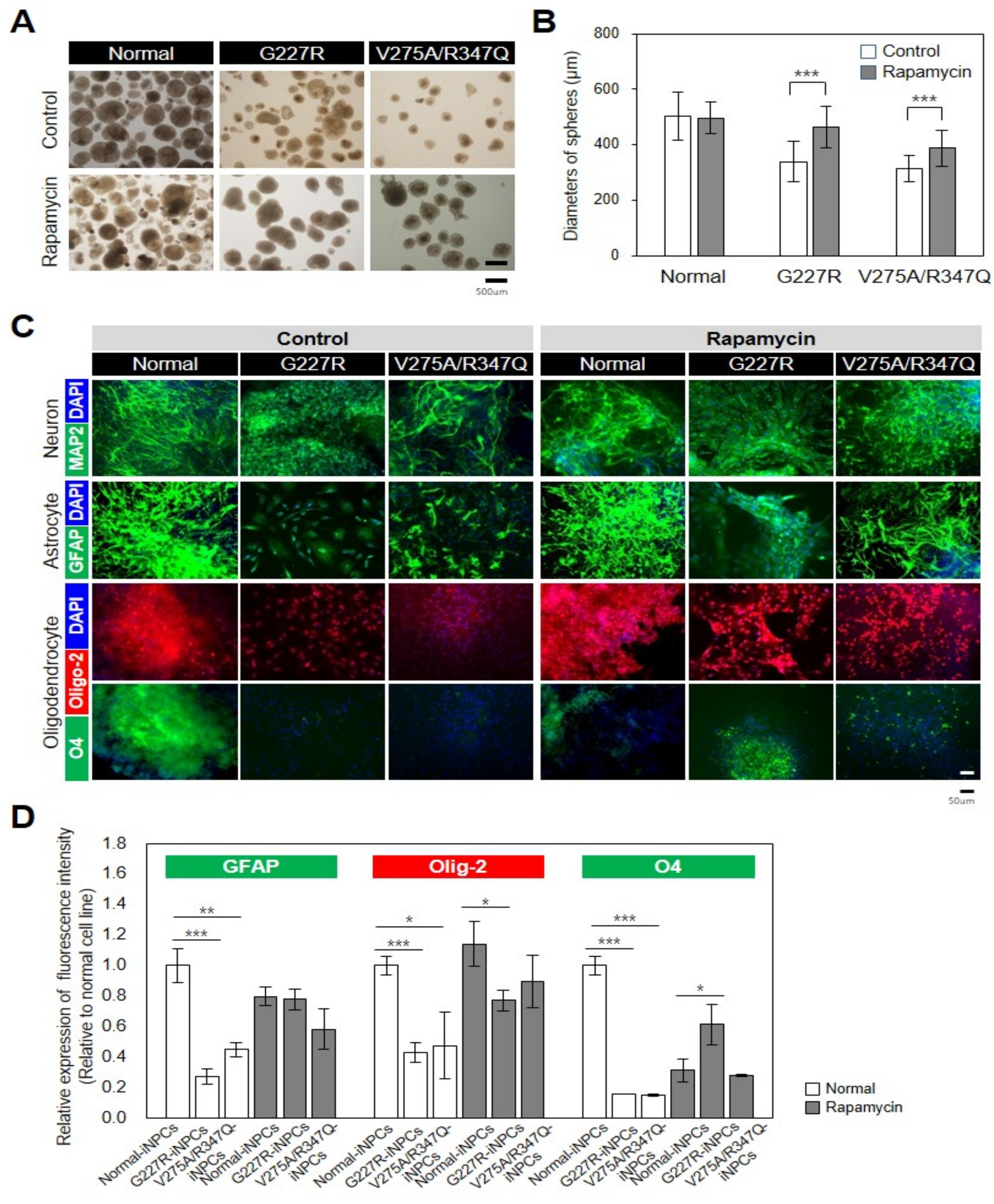
2.4. Rapamycin Partly Restores Impaired Function of Sialidosis-iNPCs
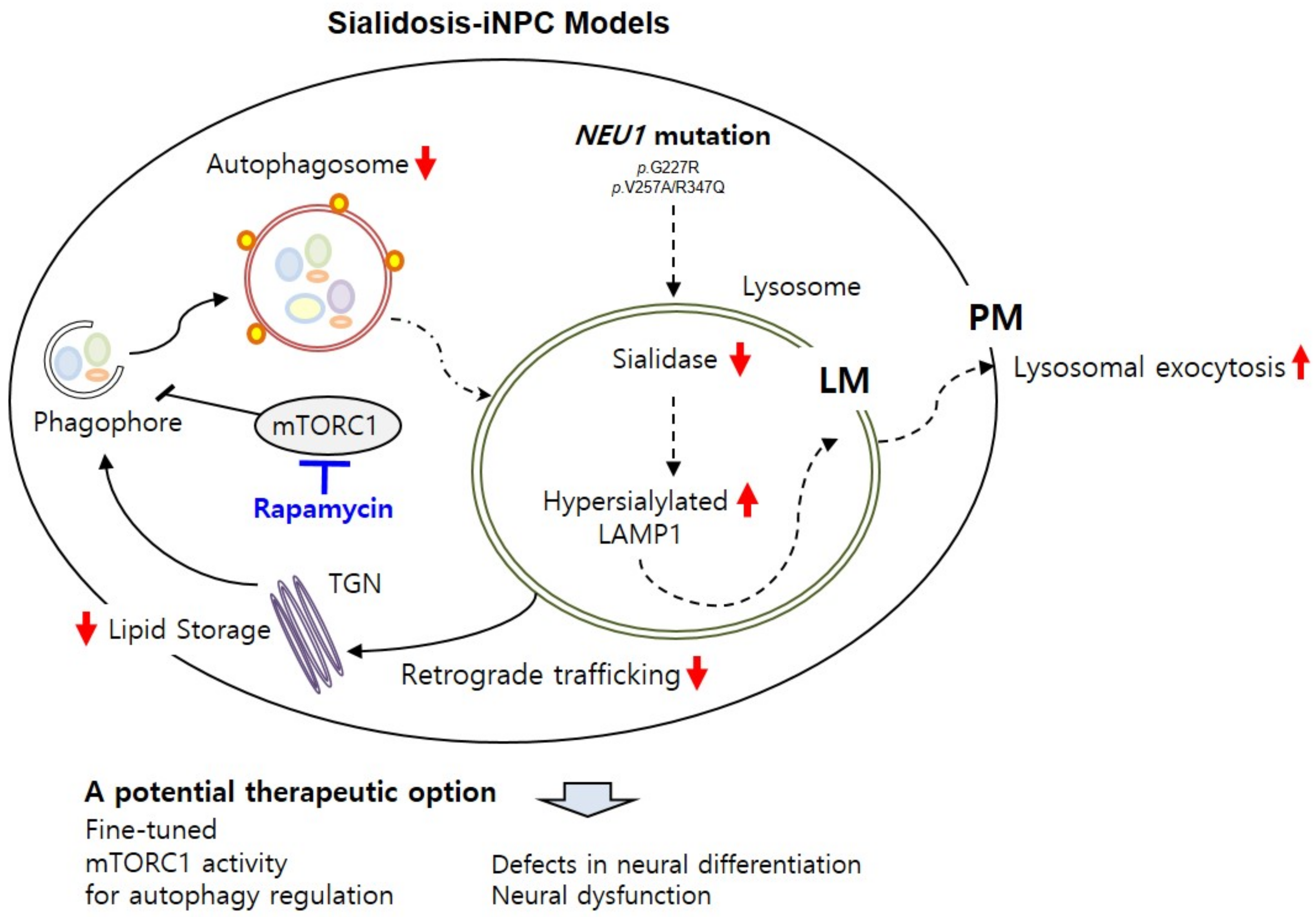
3. Discussion
4. Materials and Methods
4.1. Human Fibroblasts
4.2. cDNA Sequencing
4.3. Normal- and Sialidosis-iPSCs
4.4. Alkaline Phosphatase (ALP) Analysis
4.5. In Vitro EB Formation and Trilineage Differentiation
4.6. G-Banding Karyotyping and Short Tandem Repeat (STR) Profiling
4.7. Teratoma Analysis
4.8. Immunocytochemistry
4.9. Normal and Sialidosis-iNPCs
4.10. Sialidase Assay
4.11. Western Blot
4.12. Autophagy and Lysosome Staining
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sphranger, J.; Gehler, J.; Cantz, M. Mucolipidosis I—A sialidosis. Am. J. Med. Genet. 1977, 1, 21–29. [Google Scholar] [CrossRef]
- Lowden, J.A.; O’Brien, J.S. Sialidosis: A review of human neuraminidase deficiency. Am. J. Hum. Genet. 1979, 31, 1–18. [Google Scholar] [PubMed]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Pattison, S.; Pankarican, M.; Rupar, C.A.; Graham, F.L.; Igdoura, S.A. Five novel mutations in the lysosomal sialidase gene (NEU1) in type II sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum. Mutat. 2004, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef]
- Bonten, E.J.; Campos, Y.; Zaitsev, V.; Nourse, A.; Waddell, B.; Lewis, W.; Taylor, G.; d’Azzo, A. Heterodimerization of the sialidase NEU1 with the chaperone protective protein/cathepsin A prevents its premature oligomerization. J. Biol. Chem. 2009, 284, 28430–28441. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Landry, K.; Elsliger, M.A.; Chang, Y.; Lefrancois, S.; Morales, C.R.; Pshezhetsky, A.V. Mutations in sialidosis impair sialidase binding to the lysosomal multienzyme complex. J. Biol. Chem. 2001, 276, 17286–17290. [Google Scholar] [CrossRef]
- Monti, E.; Bonten, E.; D’Azzo, A.; Bresciani, R.; Venerando, B.; Borsani, G.; Schauer, R.; Tettamanti, G. Sialidases in vertebrates: A family of enzymes tailored for several cell functions. Adv. Carbohydr. Chem. Biochem. 2010, 64, 403–479. [Google Scholar] [PubMed]
- Mohammad, A.N.; Bruno, K.A.; Hines, S.; Atwal, P.S. Type 1 sialidosis presenting with ataxia, seizures and myoclonus with no visual involvement. Mol. Genet. Metab. Rep. 2018, 15, 11–14. [Google Scholar] [CrossRef]
- Seyrantepe, V.; Poupetova, H.; Froissart, R.; Zabot, M.T.; Maire, I.; Pshezhetsky, A.V. Molecular pathology of NEU1 gene in sialidosis. Hum. Mutat. 2003, 22, 343–352. [Google Scholar] [CrossRef]
- Zanoteli, E.; van de Vlekkert, D.; Bonten, E.J.; Hu, H.; Mann, L.; Gomero, E.M.; Harris, A.J.; Ghersi, G.; d’Azzo, A. Muscle degeneration in neuraminidase 1-deficient mice results from infiltration of the muscle fibers by expanded connective tissue. Biochim. Biophys. Acta 2010, 1802, 659–672. [Google Scholar] [CrossRef] [PubMed]
- De Geest, N.; Bonten, E.; Mann, L.; de Sousa-Hitzler, J.; Hahn, C.; d’Azzo, A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 2002, 11, 1455–1464. [Google Scholar] [CrossRef]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; d’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 2734. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Yogalingam, G.; Hu, H.; Gomero, E.; van de Vlekkert, D.; d’Azzo, A. Chaperone-mediated gene therapy with recombinant AAV-PPCA in a new mouse model of type I sialidosis. Biochim. Biophys. Acta 2013, 1832, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Wang, D.; Toy, J.N.; Mann, L.; Mignardot, A.; Yogalingam, G.; D’Azzo, A. Targeting macrophages with baculovirus-produced lysosomal enzymes: Implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. FASEB J. 2004, 18, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Mazzulli, J.R. Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol. Dis. 2019, 127, 147–162. [Google Scholar] [CrossRef]
- Huang, H.P.; Chuang, C.Y.; Kuo, H.C. Induced pluripotent stem cell technology for disease modeling and drug screening with emphasis on lysosomal storage diseases. Stem. Cell Res. Ther. 2012, 3, 34. [Google Scholar] [CrossRef]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. Alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef]
- Liu, S.P.; Hsu, Y.H.; Huang, C.Y.; Ho, M.C.; Cheng, Y.C.; Wen, C.H.; Lu, H.E.; Tsai, C.H.; Shyu, W.C.; Hsieh, P.C.H. Generation of novel induced pluripotent stem cell (iPSC) line from a 16-year-old sialidosis patient with NEU-1 gene mutation. Stem Cell Res. 2018, 28, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Davaadorj, O.; Akatsuka, H.; Yamaguchi, Y.; Okada, C.; Ito, M. Impaired Autophagy in Retinal Pigment Epithelial Cells Induced from iPS Cells obtained from a Patient with Sialidosis. Cell Dev. Biol. 2017, 6, 2. [Google Scholar] [CrossRef]
- Han, M.J.; Annunziata, I.; Weesner, J.; Campos, Y.; Salie, M.; O’Reilly, C.; d’Azzo, A. Generation of human induced pluripotent stem cells (hIPSCs) from sialidosis types I and II patients with pathogenic neuraminidase 1 mutations. Stem Cell Res. 2020, 46, 101836. [Google Scholar] [CrossRef] [PubMed]
- Odaka, H.; Numakawa, T.; Soga, M.; Kido, J.; Matsumoto, S.; Kajihara, R.; Okumiya, T.; Tani, N.; Tanoue, Y.; Fukuda, T.; et al. An iPSC-based neural model of sialidosis uncovers glycolytic impairment-causing presynaptic dysfunction and deregulation of Ca(2+) dynamics. Neurobiol. Dis. 2021, 152, 105279. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.E.; Son, M.Y.; Son, Y.S.; Son, M.J.; Cho, Y.S. Biochemical and molecular characterization of novel mutations in GLB1 and NEU1 in patient cells with lysosomal storage disorders. Biochem. Biophys. Res. Commun. 2015, 457, 554–560. [Google Scholar] [CrossRef]
- Yogalingam, G.; Bonten, E.J.; van de Vlekkert, D.; Hu, H.; Moshiach, S.; Connell, S.A.; d’Azzo, A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 2008, 15, 74–86. [Google Scholar] [CrossRef]
- Wu, X.; Steigelman, K.A.; Bonten, E.; Hu, H.; He, W.; Ren, T.; Zuo, J.; d’Azzo, A. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim. Biophys. Acta 2010, 1802, 259–268. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Rubinsztein, D.C.; Ballabio, A. Lysosomal storage diseases as disorders of autophagy. Autophagy 2008, 4, 113–114. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Asghari, A.M.; Castillo-Quan, J.I. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen. Res. 2017, 12, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Arts, W.F.; Beck, M.; Covanis, A.; Donati, M.A.; Parini, R.; Zammarchi, E.; d’Azzo, A. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum. Mol. Genet. 2000, 9, 2715–2725. [Google Scholar] [CrossRef]
- Rybova, J.; Ledvinova, J.; Sikora, J.; Kuchar, L.; Dobrovolny, R. Neural cells generated from human induced pluripotent stem cells as a model of CNS involvement in mucopolysaccharidosis type II. J. Inherit. Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Aguisanda, F.; Yeh, C.D.; Chen, C.Z.; Li, R.; Beers, J.; Zou, J.; Thorne, N.; Zheng, W. Neural stem cells for disease modeling of Wolman disease and evaluation of therapeutics. Orphanet J. Rare Dis. 2017, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for Tay-Sachs disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen 2014, 19, 1164–1173. [Google Scholar] [CrossRef]
- Trilck, M.; Hubner, R.; Seibler, P.; Klein, C.; Rolfs, A.; Frech, M.J. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 2013, 8, 144. [Google Scholar] [CrossRef]
- Son, M.Y.; Kwak, J.E.; Seol, B.; Lee, D.Y.; Jeon, H.; Cho, Y.S. A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 2015, 237, 98–110. [Google Scholar] [CrossRef]
- Matsushita, K.; Numakawa, T.; Odaka, H.; Kajihara, R.; Soga, M.; Ozasa, S.; Nakamura, K.; Mizuta, H.; Era, T. Presynaptic Dysfunction in Neurons Derived from Tay-Sachs iPSCs. Neuroscience 2019, 414, 128–140. [Google Scholar] [CrossRef]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient iPSC-derived neural stem cells exhibit phenotypes in concordance with the clinical severity of mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626. [Google Scholar] [CrossRef] [PubMed]
- Kobolak, J.; Molnar, K.; Varga, E.; Bock, I.; Jezso, B.; Teglasi, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef]
- Bayo-Puxan, N.; Terrasso, A.P.; Creyssels, S.; Simao, D.; Begon-Pescia, C.; Lavigne, M.; Salinas, S.; Bernex, F.; Bosch, A.; Kalatzis, V.; et al. Lysosomal and network alterations in human mucopolysaccharidosis type VII iPSC-derived neurons. Sci. Rep. 2018, 8, 16644. [Google Scholar] [CrossRef] [PubMed]
- Schene, I.F.; Kalinina Ayuso, V.; de Sain-van der Velden, M.; van Gassen, K.L.; Cuppen, I.; van Hasselt, P.M.; Visser, G. Pitfalls in Diagnosing Neuraminidase Deficiency: Psychosomatics and Normal Sialic Acid Excretion. JIMD Rep. 2016, 25, 9–13. [Google Scholar] [PubMed]
- Sekijima, Y.; Nakamura, K.; Kishida, D.; Narita, A.; Adachi, K.; Ohno, K.; Nanba, E.; Ikeda, S. Clinical and serial MRI findings of a sialidosis type I patient with a novel missense mutation in the NEU1 gene. Intern. Med. 2013, 52, 119–124. [Google Scholar] [CrossRef]
- Van der Spoel, A.; Bonten, E.; d’Azzo, A. Transport of human lysosomal neuraminidase to mature lysosomes requires protective protein/cathepsin A. EMBO J. 1998, 17, 1588–1597. [Google Scholar] [CrossRef]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorder. Proc. Natl. Acad. Sci. USA 2012, 109, E2334–E2342. [Google Scholar] [CrossRef]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 2020, 143, 104963. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Choy, F.Y.M. A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene Editing Technology for Mutation Correction in Induced Pluripotent Stem Cells. Diseases 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seol, B.; Kim, Y.-D.; Cho, Y.S. Modeling Sialidosis with Neural Precursor Cells Derived from Patient-Derived Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2021, 22, 4386. https://doi.org/10.3390/ijms22094386
Seol B, Kim Y-D, Cho YS. Modeling Sialidosis with Neural Precursor Cells Derived from Patient-Derived Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2021; 22(9):4386. https://doi.org/10.3390/ijms22094386
Chicago/Turabian StyleSeol, Binna, Young-Dae Kim, and Yee Sook Cho. 2021. "Modeling Sialidosis with Neural Precursor Cells Derived from Patient-Derived Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 22, no. 9: 4386. https://doi.org/10.3390/ijms22094386
APA StyleSeol, B., Kim, Y.-D., & Cho, Y. S. (2021). Modeling Sialidosis with Neural Precursor Cells Derived from Patient-Derived Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 22(9), 4386. https://doi.org/10.3390/ijms22094386