
Emerging Roles of Exosomes in Huntington’s Disease


Abstract
1. Introduction
2. Exosome Biogenesis
3. Content of Exosomes
4. Huntington’s Disease
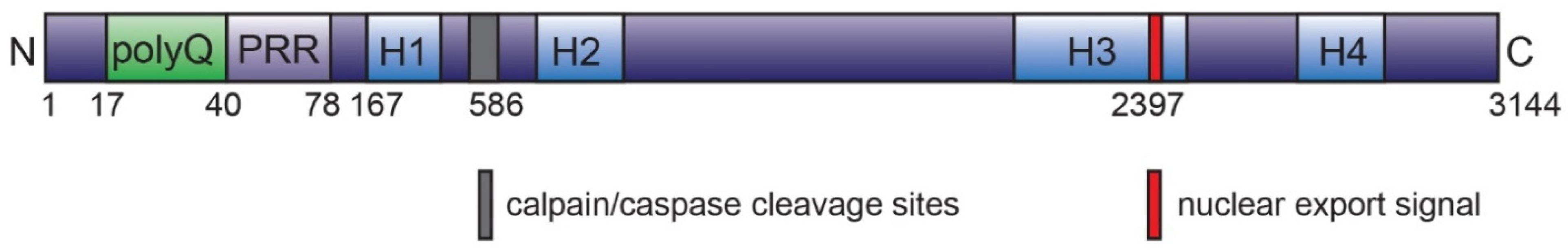
4.1. Huntingtin Protein
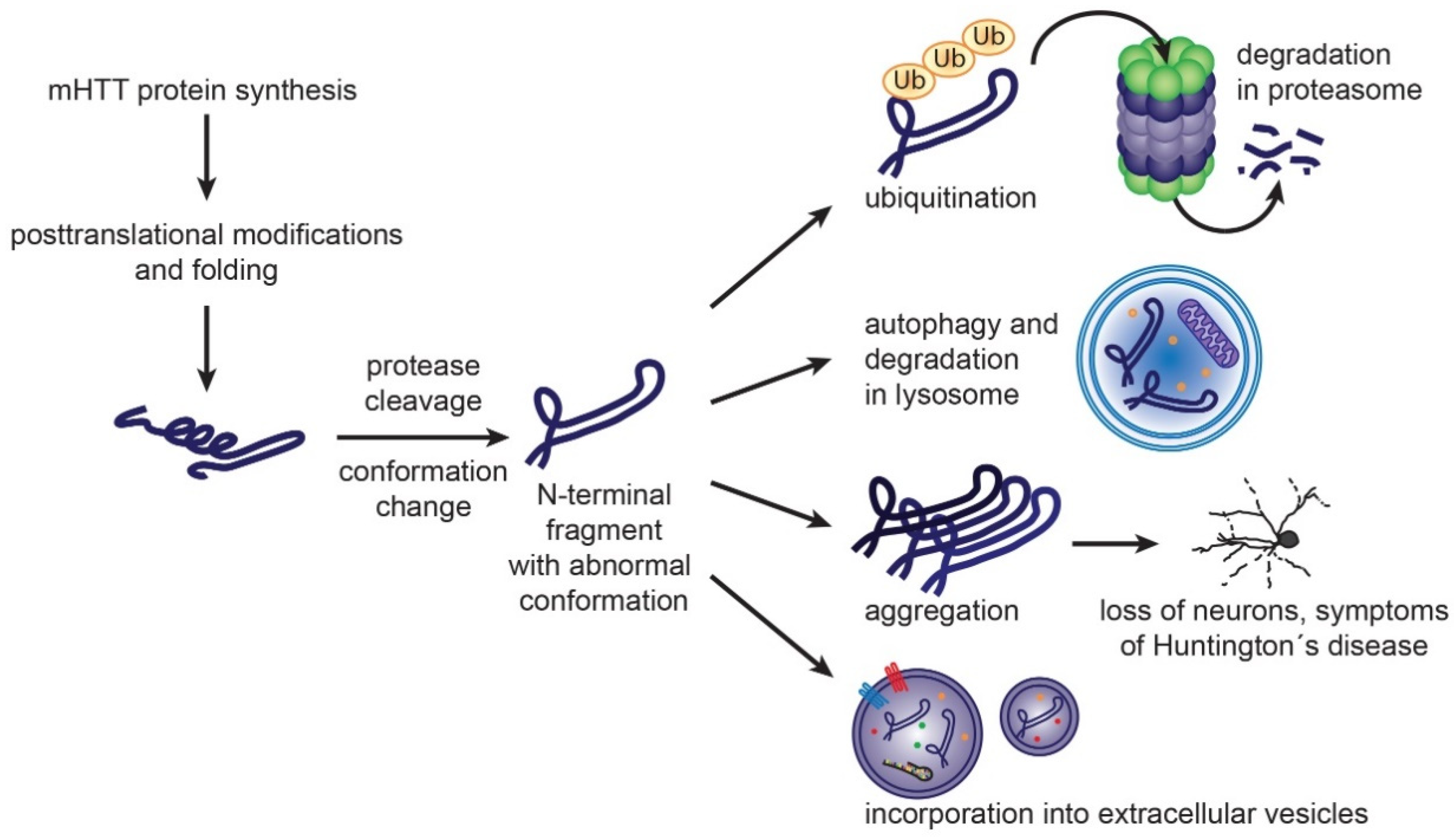
4.2. Pathogenesis of Huntington’s Disease
5. Exosomes in Huntington’s Disease
5.1. Exosomes in Misfolded Protein Spreading
5.2. Diagnostic Potential of Exosomes
5.3. Exosomes in Delivery of Therapeutics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Lakhal, S.; Wood, M.J.A. Exosome nanotechnology: An emerging paradigm shift in drug delivery: Exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. BioEssays 2011, 33, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Chopp, M. Exosomes in stroke pathogenesis and therapy. J. Clin. Investig. 2016, 126, 1190–1197. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of MRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Malm, T.; Loppi, S.; Kanninen, K.M. Exosomes in Alzheimer’s disease. Neurochem. Int. 2016, 97, 193–199. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Jan, A.; Rahman, S.; Khan, S.; Tasduq, S.; Choi, I. Biology, pathophysiological role, and clinical implications of exosomes: A critical appraisal. Cells 2019, 8, 99. [Google Scholar] [CrossRef]
- Yue, B.; Yang, H.; Wang, J.; Ru, W.; Wu, J.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Exosome biogenesis, secretion and function of exosomal MiRNAs in skeletal muscle myogenesis. Cell Prolif. 2020, 53, e12857. [Google Scholar] [CrossRef]
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel biomarkers for clinical diagnosis. Sci. World J. 2015, 2015, 657086. [Google Scholar] [CrossRef]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic Cph. Den. 2009, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant huntingtin inhibits AB-crystallin expression and impairs exosome secretion from astrocytes. J. Neurosci. 2017, 37, 9550–9563. [Google Scholar] [CrossRef]
- Properzi, F.; Ferroni, E.; Poleggi, A.; Vinci, R. The regulation of exosome function in the CNS: Implications for neurodegeneration. Swiss Med. Wkly. 2015, 145, w14204. [Google Scholar] [CrossRef]
- Zebrowska, A.; Skowronek, A.; Wojakowska, A.; Widlak, P.; Pietrowska, M. Metabolome of exosomes: Focus on vesicles released by cancer cells and present in human body fluids. Int. J. Mol. Sci. 2019, 20, 3461. [Google Scholar] [CrossRef]
- Van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.A.A.; van Solinge, W.W.; Wood, M.J.A.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 2012, 161, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Beach, A.; Zhang, H.-G.; Ratajczak, M.Z.; Kakar, S.S. Exosomes: An overview of biogenesis, composition and role in ovarian cancer. J. Ovarian Res. 2014, 7, 14. [Google Scholar] [CrossRef]
- Babst, M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Huang, Y.; Zhang, H.; Lu, H.; Zheng, J.C. Exosomal MiRNAs in central nervous system diseases: Biomarkers, pathological mediators, protective factors and therapeutic agents. Prog. Neurobiol. 2019, 183, 101694. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L. Circulating exosomal MiRNA as diagnostic biomarkers of neurodegenerative diseases. Front. Mol. Neurosci. 2020, 13, 53. [Google Scholar] [CrossRef]
- Manna, I.; De Benedittis, S.; Quattrone, A.; Maisano, D.; Iaccino, E.; Quattrone, A. Exosomal MiRNAs as potential diagnostic biomarkers in Alzheimer’s disease. Pharmaceuticals 2020, 13, 243. [Google Scholar] [CrossRef]
- Helder, D.I.; Kaptein, A.A.; van Kempen, G.M.J.; van Houwelingen, J.C.; Roos, R.A.C. Impact of Huntington’s disease on quality of life. Mov. Disord. 2001, 16, 325–330. [Google Scholar] [CrossRef]
- Coulson, N.S.; Buchanan, H.; Aubeeluck, A. Social support in cyberspace: A content analysis of communication within a Huntington’s disease online support group. Patient Educ. Couns. 2007, 68, 173–178. [Google Scholar] [CrossRef]
- Dayalu, P.; Albin, R.L. Huntington disease. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef]
- Wynford-Thomas, R.; Robertson, N.P. The economic burden of chronic neurological disease. J. Neurol. 2017, 264, 2345–2347. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The prevalence of Huntington’s disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef]
- Ha, A.D.; Jankovic, J. Exploring the correlates of intermediate CAG repeats in huntington disease. Postgrad. Med. 2011, 123, 116–121. [Google Scholar] [CrossRef]
- Schneider, S.A.; Bird, T. Huntington’s disease, Huntington’s disease look-alikes, and benign hereditary chorea: What’s new? Mov. Disord. Clin. Pract. 2016, 3, 342–354. [Google Scholar] [CrossRef]
- Capiluppi, E.; Romano, L.; Rebora, P.; Nanetti, L.; Castaldo, A.; Gellera, C.; Mariotti, C.; Macerollo, A.; Cislaghi, M.G. Late-onset Huntington’s disease with 40–42 CAG expansion. Neurol. Sci. 2020, 41, 869–876. [Google Scholar] [CrossRef]
- Testa, C.M.; Jankovic, J. Huntington disease: A quarter century of progress since the gene discovery. J. Neurol. Sci. 2019, 396, 52–68. [Google Scholar] [CrossRef]
- Craufurd, D.; MacLeod, R.; Frontali, M.; Quarrell, O.; Bijlsma, E.K.; Davis, M.; Hjermind, L.E.; Lahiri, N.; Mandich, P.; Martinez, A.; et al. Diagnostic genetic testing for Huntington’s disease. Pract. Neurol. 2015, 15, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.S.; La Spada, A.R. Therapy development in Huntington disease: From current strategies to emerging opportunities. Am. J. Med. Genet. A 2018, 176, 842–861. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Feigin, A. Huntington’s disease: New frontiers in therapeutics. Curr. Neurol. Neurosci. Rep. 2021, 21, 10. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef]
- Shannon, K.M. Recent advances in the treatment of Huntington’s disease: Targeting DNA and RNA. CNS Drugs 2020, 34, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Bashir, H. Emerging therapies in Huntington’s disease. Expert Rev. Neurother. 2019, 19, 983–995. [Google Scholar] [CrossRef]
- Przybyl, L.; Wozna-Wysocka, M.; Kozlowska, E.; Fiszer, A. What, when and how to measure—Peripheral biomarkers in therapy of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 1561. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 2014, 289, 6709–6726. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.R.; Chaibva, M.; Legleiter, J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomol. Concepts 2015, 6, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Datson, N.A.; González-Barriga, A.; Kourkouta, E.; Weij, R.; van de Giessen, J.; Mulders, S.; Kontkanen, O.; Heikkinen, T.; Lehtimäki, K.; van Deutekom, J.C.T. The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 2017, 12, e0171127. [Google Scholar] [CrossRef]
- Alterman, J.F.; Hall, L.M.; Coles, A.H.; Hassler, M.R.; Didiot, M.-C.; Chase, K.; Abraham, J.; Sottosanti, E.; Johnson, E.; Sapp, E.; et al. Hydrophobically modified SiRNAs silence huntingtin MRNA in primary neurons and mouse brain. Mol. Ther. Nucleic Acids 2015, 4, e266. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated delivery of hydrophobically modified SiRNA for huntingtin MRNA silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Haraszti, R.A.; Echeverria, D.; Miller, R.; Didiot, M.-C.; Nikan, M.; Roux, L.; Aronin, N.; Khvorova, A. Hydrophobicity of lipid-conjugated SiRNAs predicts productive loading to small extracellular vesicles. Mol. Ther. 2018, 26, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yu, M.; Zhang, L.; Chen, X.; Pei, Z. I02 Systemic injection of exosomal sirna significantly reduced huntingtin expression in transgenic mice of Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, A88–A89. [Google Scholar] [CrossRef]
- Lee, S.-T.; Im, W.; Ban, J.-J.; Lee, M.; Jung, K.-H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-based delivery of MiR-124 in a Huntington’s disease model. J. Mov. Disord. 2017, 10, 45–52. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, characterization, and lead selection of therapeutic MiRNAs targeting huntingtin for development of gene therapy for Huntington’s disease. Mol. Ther. Nucleic Acids 2016, 5, e297. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-MiHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; van Deventer, S.J.; et al. AAV5-MiHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef]
- Pfister, E.; Dinardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial MiRNAs reduce human mutant huntingtin throughout the striatum in a transgenic sheep model of Huntington’s disease. Hum. Gene Ther. 2017, 29, 663–673. [Google Scholar] [CrossRef]
- Agustín-Pavón, C.; Mielcarek, M.; Garriga-Canut, M.; Isalan, M. Deimmunization for gene therapy: Host matching of synthetic zinc finger constructs enables long-term mutant huntingtin repression in mice. Mol. Neurodegener. 2016, 11, 64. [Google Scholar] [CrossRef]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.-M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef]
- Roche Provides Update on Tominersen Programme in Manifest Huntington’s Disease. Available online: https://www.roche.com/media/releases/med-cor-2021-03-22b.htm (accessed on 29 March 2021).
- Saudou, F.; Humbert, S. The biology of huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Zheng, Z.; Diamond, M.I. Huntington disease and the huntingtin protein. Prog. Mol. Biol. Transl. Sci. 2012, 107, 189–214. [Google Scholar] [CrossRef]
- Warby, S.C.; Doty, C.N.; Graham, R.K.; Carroll, J.B.; Yang, Y.-Z.; Singaraja, R.R.; Overall, C.M.; Hayden, M.R. Activated Caspase-6 and Caspase-6-Cleaved Fragments of Huntingtin Specifically Colocalize in the Nucleus. Hum. Mol. Genet. 2008, 17, 2390–2404. [Google Scholar] [CrossRef] [PubMed]
- Tebbenkamp, A.T.N.; Crosby, K.W.; Siemienski, Z.B.; Brown, H.H.; Golde, T.E.; Borchelt, D.R. Analysis of proteolytic processes and enzymatic activities in the generation of huntingtin N-terminal fragments in an HEK293 cell model. PLoS ONE 2012, 7, e50750. [Google Scholar] [CrossRef] [PubMed]
- El-Daher, M.-T.; Hangen, E.; Bruyère, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin proteolysis releases non-PolyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015, 34, 2255–2271. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of huntingtin in huntington disease. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2011, 17, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.O.; Schmidt, M.E.; Nguyen, Y.T.; Lazic, N.; Hayden, M.R. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Cao, Q.; Zhang, Y.; Su, X.-D. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Schaffert, L.-N.; Carter, W.G. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: A systematic review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Huntington disease. In Neurobiology of Brain Disorders; Elsevier: Amsterdam, The Netherlands, 2015; pp. 303–320. ISBN 978-0-12-398270-4. [Google Scholar]
- Harding, R.J.; Tong, Y.-F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Thakur, A.K.; Jayaraman, M.; Mishra, R.; Thakur, M.; Chellgren, V.M.; Byeon, I.-J.L.; Anjum, D.H.; Kodali, R.; Creamer, T.P.; Conway, J.F.; et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009, 16, 380–389. [Google Scholar] [CrossRef]
- Li, S.; Li, X.-J. Multiple pathways contribute to the pathogenesis of huntington disease found. Mol. Neurodegener. 2006, 1, 19. [Google Scholar] [CrossRef][Green Version]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of huntingtin in health and disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.; Zhu, L.; Hua, W.; Zhang, Y.; Zhang, H.; Zhang, L.; Li, Z.; Xing, P.; Zhang, Y.; et al. Glia-derived exosomes: Promising therapeutic targets. Life Sci. 2019, 239, 116951. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, P. A novel cell-cell communication mechanism in the nervous system: Exosomes. J. Neurosci. Res. 2018, 96, 45–52. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular vesicle-mediated cell−cell communication in the nervous system: Focus on neurological diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.-S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S.B. Disruption of axonal transport by loss of huntingtin or expression of pathogenic PolyQ proteins in drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Rossetti, G.; Magistrato, A. Molecular mechanism of Huntington’s disease—A computational perspective. In Huntington’s Disease—Core Concepts and Current Advances; Ersoy Tunal, N., Ed.; InTech: London, UK, 2012; ISBN 978-953-307-953-0. [Google Scholar]
- Wyttenbach, A.; Carmichael, J.; Swartz, J.; Furlong, R.A.; Narain, Y.; Rankin, J.; Rubinsztein, D.C. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2898–2903. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Bali, J.; Barr, M.M.; Court, F.A.; Krämer-Albers, E.-M.; Picou, F.; Raposo, G.; van der Vos, K.E.; van Niel, G.; Wang, J.; et al. Emerging roles of extracellular vesicles in the nervous system. J. Neurosci. 2014, 34, 15482–15489. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Igarashi, Y. Physiological and pathological roles of exosomes in the nervous system. Biomol. Concepts 2016, 7, 53–68. [Google Scholar] [CrossRef]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013, 352, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. EBJ 2008, 37, 323–332. [Google Scholar] [CrossRef]
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism: Microglial MVs increase sphingolipid metabolism in neurons. EMBO J. 2012, 31, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cesca, F.; Loers, G.; Schweizer, M.; Buck, F.; Benfenati, F.; Schachner, M.; Kleene, R. Synapsin I Is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J. Neurosci. 2011, 31, 7275–7290. [Google Scholar] [CrossRef]
- Kanninen, K.M.; Bister, N.; Koistinaho, J.; Malm, T. Exosomes as new diagnostic tools in CNS diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 403–410. [Google Scholar] [CrossRef]
- Gharbi, T.; Zhang, Z.; Yang, G.-Y. The function of astrocyte mediated extracellular vesicles in central nervous system diseases. Front. Cell Dev. Biol. 2020, 8, 568889. [Google Scholar] [CrossRef] [PubMed]
- Mrowczynski, O.D.; Zacharia, B.E.; Connor, J.R. Exosomes and their implications in central nervous system tumor biology. Prog. Neurobiol. 2019, 172, 71–83. [Google Scholar] [CrossRef]
- Street, J.M.; Barran, P.E.; Mackay, C.L.; Weidt, S.; Balmforth, C.; Walsh, T.S.; Chalmers, R.T.A.; Webb, D.J.; Dear, J.W. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J. Transl. Med. 2012, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Banigan, M.G.; Kao, P.F.; Kozubek, J.A.; Winslow, A.R.; Medina, J.; Costa, J.; Schmitt, A.; Schneider, A.; Cabral, H.; Cagsal-Getkin, O.; et al. Differential expression of exosomal MicroRNAs in prefrontal cortices of schizophrenia and bipolar disorder patients. PLoS ONE 2013, 8, e48814. [Google Scholar] [CrossRef]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of pre-clinical Alzheimer’s disease by a profile of pathogenic proteins in neurally-derived blood exosomes: A case-control study. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed]
- Gassama, Y.; Favereaux, A. Emerging roles of extracellular vesicles in the central nervous system: Physiology, pathology, and therapeutic perspectives. Front. Cell. Neurosci. 2021, 15, 626043. [Google Scholar] [CrossRef] [PubMed]
- Blandford, S.N.; Galloway, D.A.; Moore, C.S. The roles of extracellular vesicle MicroRNAs in the central nervous system. Glia 2018, 66, 2267–2278. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell communication by extracellular vesicles: Focus on microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflam. 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.-H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Abounit, S.; Marzo, L.; Danckaert, A.; Chamoun, Z.; Roux, P.; Zurzolo, C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 2013, 126, 3678–3685. [Google Scholar] [CrossRef]
- Herrera, F.; Tenreiro, S.; Miller-Fleming, L.; Outeiro, T.F. Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. 2011, 3, RRN1210. [Google Scholar] [CrossRef]
- Yang, W.; Dunlap, J.R.; Andrews, R.B.; Wetzel, R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 2002, 11, 2905–2917. [Google Scholar] [CrossRef]
- Pearce, M.M.P.; Spartz, E.J.; Hong, W.; Luo, L.; Kopito, R.R. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the drosophila brain. Nat. Commun. 2015, 6, 6768. [Google Scholar] [CrossRef]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Cicchetti, F.; Lacroix, S.; Cisbani, G.; Vallières, N.; Saint-Pierre, M.; St-Amour, I.; Tolouei, R.; Skepper, J.N.; Hauser, R.A.; Mantovani, D.; et al. Mutant huntingtin is present in neuronal grafts in huntington disease patients: Transfer of mutant huntingtin to normal tissue. Ann. Neurol. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef]
- Zhang, X.; Abels, E.R.; Redzic, J.S.; Margulis, J.; Finkbeiner, S.; Breakefield, X.O. Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington’s disease: Background and evaluation in cell culture. Cell. Mol. Neurobiol. 2016, 36, 459–470. [Google Scholar] [CrossRef]
- Lee, M.; Liu, T.; Im, W.; Kim, M. Exosomes from adipose-derived stem cells ameliorate phenotype of Huntington’s disease in vitro model. Eur. J. Neurosci. 2016, 44, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Koutras, C.; Donnelier, J.; Alshehri, M.; Fotouhi, M.; Girard, M.; Casha, S.; McPherson, P.S.; Robbins, S.M.; Braun, J.E.A. Neurons export extracellular vesicles enriched in cysteine string protein and misfolded protein cargo. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Boukouris, S.; Mathivanan, S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom. Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef]
- Manterola, L.; Guruceaga, E.; Pérez-Larraya, J.G.; González-Huarriz, M.; Jauregui, P.; Tejada, S.; Diez-Valle, R.; Segura, V.; Samprón, N.; Barrena, C.; et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro-oncology 2014, 16, 520–527. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Sergeant, N.; Buée, L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer’s disease. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, P.R.; Zheng, Y.; Fischer, R.; Heidasch, R.; Gardiner, C.; Evetts, S.; Hu, M.; Wade-Martins, R.; Turner, M.R.; Morris, J.; et al. Identification of distinct circulating exosomes in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on extracellular vesicles: Exosomes and their role in protein trafficking and biomarker potential in Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-Y.; Lu, J.-M.; Zhao, Z.-Q.; Li, M.-C.; Lu, T.; An, X.-S.; Xue, L.-J. MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci. Lett. 2017, 644, 94–99. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Ikezu, T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol. Dis. 2019, 130, 104512. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Foster, B.P.; Balassa, T.; Benen, T.D.; Dominovic, M.; Elmadjian, G.K.; Florova, V.; Fransolet, M.D.; Kestlerova, A.; Kmiecik, G.; Kostadinova, I.A.; et al. Extracellular vesicles in blood, milk and body fluids of the female and male urogenital tract and with special regard to reproduction. Crit. Rev. Clin. Lab. Sci. 2016, 53, 379–395. [Google Scholar] [CrossRef]
- Espinosa-Parrilla, Y.; Gonzalez-Billault, C.; Fuentes, E.; Palomo, I.; Alarcón, M. Decoding the role of platelets and related MicroRNAs in aging and neurodegenerative disorders. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Weiss, A.; Chouinard, S.; Barker, R.A.; Boilard, E.; Cicchetti, F. Platelet-derived extracellular vesicles in Huntington’s disease. J. Neurol. 2018, 265, 2704–2712. [Google Scholar] [CrossRef]
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Rowley, J.W.; Cloutier, N.; Tremblay, M.-È.; Vincent, A.T.; Gould, P.V.; Chouinard, S.; et al. Platelet abnormalities in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2019, 90, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Baez, R.; Hernández-Ortega, K.; Martínez-Martínez, E. Insights into the proteomic profiling of extracellular vesicles for the identification of early biomarkers of neurodegeneration. Front. Neurol. 2020, 11, 580030. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kojima, K.; Mobley, J.A.; West, A.B. Proteomic analysis of urinary extracellular vesicles reveal biomarkers for neurologic disease. EBioMedicine 2019, 45, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal MiRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Palaniswamy, R.; Sevugan, K.; Sampathkumar Srisharnitha, A. Molecular signatures in exosomes as diagnostic markers for neurodegenerative disorders. Ann. Alzheimers Dement. Care 2020, 4, 12–17. [Google Scholar] [CrossRef]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific signatures of serum MiRNAs as potential biomarkers to discriminate clinically similar neurodegenerative and vascular-related diseases. Cell. Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-F.; Qu, M.-W.; Li, G.-C.; Zhang, F.-B.; Rui, H.-C. Circulating exosomal MiRNAs as diagnostic biomarkers in Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5278–5283. [Google Scholar] [CrossRef]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H. MicroRNAs in CSF as prodromal biomarkers for huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272. [Google Scholar] [CrossRef]
- Díez-Planelles, C.; Sánchez-Lozano, P.; Crespo, M.C.; Gil-Zamorano, J.; Ribacoba, R.; González, N.; Suárez, E.; Martínez-Descals, A.; Martínez-Camblor, P.; Álvarez, V.; et al. Circulating MicroRNAs in Huntington’s disease: Emerging mediators in metabolic impairment. Pharmacol. Res. 2016, 108, 102–110. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A MicroRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional MicroRNA MiR-9/MiR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef]
- Das, E.; Jana, N.R.; Bhattacharyya, N.P. MicroRNA-124 targets CCNA2 and regulates cell cycle in STHdhQ111/HdhQ111 cells. Biochem. Biophys. Res. Commun. 2013, 437, 217–224. [Google Scholar] [CrossRef]
- Cao, X.; Pfaff, S.L.; Gage, F.H. A functional study of MiR-124 in the developing neural tube. Genes Dev. 2007, 21, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Stewart, T.; Banks, W.A.; Zhang, J. The transport mechanism of extracellular vesicles at the blood-brain barrier. Curr. Pharm. Des. 2017, 23, 6206–6214. [Google Scholar] [CrossRef]
- Kumar, A.; Zhou, L.; Zhi, K.; Raji, B.; Pernell, S.; Tadrous, E.; Kodidela, S.; Nookala, A.; Kochat, H.; Kumar, S. Challenges in biomaterial-based drug delivery approach for the treatment of neurodegenerative diseases: Opportunities for extracellular vesicles. Int. J. Mol. Sci. 2020, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.A. Are extracellular vesicles new hope in clinical drug delivery for neurological disorders? Neurochem. Int. 2021, 144, 104955. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hou, K.; Ji, T.; Wang, X.; Liu, Y.; Zheng, Y.; Xu, J.; Hou, Y.; Chi, G. The role of exosomal MicroRNAs in central nervous system diseases. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Pereira, P.; Queiroz, J.A.; Figueiras, A.; Sousa, F. Current progress on MicroRNAs-based therapeutics in neurodegenerative diseases. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, Z.-M.; Guo, X.-M.; Su, D.-F.; Liu, X. An updated role of MicroRNA-124 in central nervous system disorders: A review. Front. Cell. Neurosci. 2015, 9, 193. [Google Scholar] [CrossRef]
- Ridolfi, B.; Abdel-Haq, H. Neurodegenerative disorders treatment: The MicroRNA role. Curr. Gene Ther. 2017, 17, 327–363. [Google Scholar] [CrossRef]
- Liu, T.; Im, W.; Mook-Jung, I.; Kim, M. MicroRNA-124 slows down the progression of Huntington’s disease by promoting neurogenesis in the striatum. Neural Regen. Res. 2015, 10, 786–791. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Haraszti, R.A.; Aronin, N.; Khvorova, A. Loading of extracellular vesicles with hydrophobically modified SiRNAs. Methods Mol. Biol. Clifton NJ 2018, 1740, 199–214. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Mechanism | Delivery Method | Ref. |
|---|---|---|---|
| RNA-based therapies | |||
| Antisense oligonucleotides | Inhibit HTT mRNA and thereby reduce the concentration of mHTT, such as Tominersen (previously IONIS-HTTRx) * | Intrathecal | [51] |
| Targeting the CAG repeats in HTT mRNA to reduce the mHTT level using ASO with the CUG-7 sequence | Intrathecal | [52] | |
| RNA interference (RNAi) | Targeting HTT mRNA using hydrophobically-modified siRNAs (hsiRNA) | Direct uptake in cell culture | [53] |
| Targeting HTT mRNA to reduce mHTT expression level using hsiRNAHTT | Exosomes | [54] | |
| Silence HTT mRNA using cholesterol-conjugated hsiRNAs | Exosomes | [55] | |
| Targeting human huntingtin exon 1 (HuHtt) transcript | Exosomes | [56] | |
| Targeting RE1-Silencing Transcription Factor (REST) using miR-124 | Exosomes | [57] | |
| Induce HTT gene silencing by targeting the heterozygous single nucleotide polymorphism (SNP) rs362331 in exon 50 or rs362307 in exon 67 linked to mHTT using artificial miRNAs (miHTTs) | Adeno-associated virus sero type 5 (AAV5) vector (Bilateral injection) | [58] | |
| Targeting HTT mRNA which then reduces and prevents the neuronal dysfunction using an engineered miRNA | AAV5 (Intracranial) | [59] | |
| Targeting SNP (single nucleotide polymorphism) using artificial miRNA (miR-451) to silence the mHTT allele which then results in suppression of mHTT aggregation and prevents neuronal dysfunction | AAV5 (Intracerebral) | [60] | |
| Targeting exon 48 of the human HTT mRNA using artificial miRNA (miR-155) | Adeno-associated virus serotype 9 (AAV9) (Striatal and cortical injection) | [61] | |
| DNA-based therapies | |||
| Zinc Finger Proteins (ZFPs) | Targeting mHTT gene expression using synthetic transcription repressor by binding to the expanded CAG repeats in mHTT gene | Adeno-associated virus (AAV) (Intraventricular injection) | [62] [63] |
| CRISPER-Cas9 | Genome editing by targeting SNPs to induce mHTT allele specific inactivation resulting in permanent inactivation of the HD mutation | Transfection to fibroblasts | [64] |
| Genome editing by the deletion of polyQ domain of the mHTT gene | AAV (Intracranial) | [65] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ananbeh, H.; Vodicka, P.; Kupcova Skalnikova, H. Emerging Roles of Exosomes in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 4085. https://doi.org/10.3390/ijms22084085
Ananbeh H, Vodicka P, Kupcova Skalnikova H. Emerging Roles of Exosomes in Huntington’s Disease. International Journal of Molecular Sciences. 2021; 22(8):4085. https://doi.org/10.3390/ijms22084085
Chicago/Turabian StyleAnanbeh, Hanadi, Petr Vodicka, and Helena Kupcova Skalnikova. 2021. "Emerging Roles of Exosomes in Huntington’s Disease" International Journal of Molecular Sciences 22, no. 8: 4085. https://doi.org/10.3390/ijms22084085
APA StyleAnanbeh, H., Vodicka, P., & Kupcova Skalnikova, H. (2021). Emerging Roles of Exosomes in Huntington’s Disease. International Journal of Molecular Sciences, 22(8), 4085. https://doi.org/10.3390/ijms22084085