
Natural Cyclopeptides as Anticancer Agents in the Last 20 Years


Abstract
1. Introduction
2. Marine-Derived Anticancer Cyclopeptides
2.1. Anticancer Cyclopeptides Derived from Sponge
2.2. Anticancer Cyclopeptides Derived from Ascidians/Tunicates
2.3. Anticancer Cyclopeptides Derived from Mollusks
2.4. Anticancer Cyclopeptides Derived from Marine Algae

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Biological Source | Anticancer Activity | Reference |
|---|---|---|---|
| Anticancer Cyclopeptides Derived from Sponge | |||
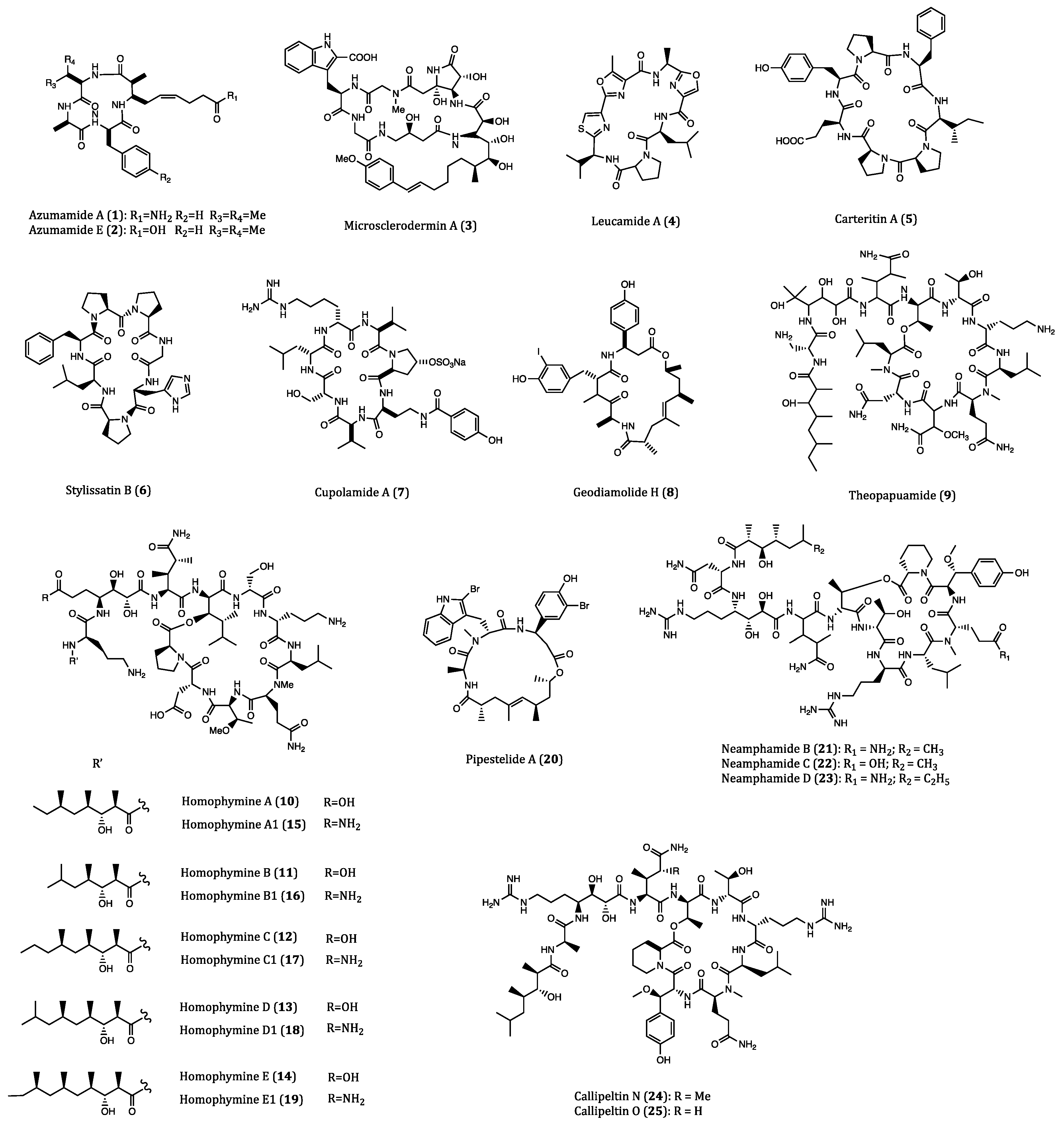
| Azumamide A (1) | Mycale izuensis | HDAC inhibitory activity against K562 cells (IC50 = 0.045 μM); Cytostatic effects on WiDr (IC50 = 5.8 μM) and K562 (IC50 = 4.5 μM) cells | [35,36,37,38,39] |
| Azumamide E (2) | Mycale izuensis | HDAC inhibitory activity against K562 cells (IC50 = 0.045 μM) | [35,36,37,38,39] |
| Microsclerodermin A (3) | Microscleroderma herdmani | Induction of apoptosis in AsPC-1 (IC50 = 2.3 µM), BxPC-3 (IC50 = 0.8 µM) and PANC-1 (IC50 = 4.0 µM) cells. | [40,41,42] |
| Leucamide A (4) | Leucetta microraphis | Inhibitory activities to HM02 (GI50 = 8.5 µM), HepG2 (GI50 = 9.7 µM) and Huh7 (GI50 = 8.3 µM) cells. | [43,44] |
| Carteritin A (5) | Sponge, Stylissa carteri | Cytotoxicity against HCT116 (IC50 = 1.3 µM), RAW264 (IC50 = 1.5 µM) and HeLa (IC50 = 0.7 µM) cells | [45] |
| Stylissatin B (6) | Stylissa massa | Inhibitory effects on HCT116, HepG2, BGC823, NCI-H1650, A2780 and MCF7 cells. (IC50 = 2.3 to 10.6 µM) | [46] |
| Cupolamide A (7) | Theonella cupola | Cytotoxicity against P388 (IC50 = 7.5 µM) cell. | [47] |
| Geodiamolide H (8) | Geodia corticostylifera | Anti-proliferative activitiy against T47D (EC50 = 38.36 nM) and MCF7 (EC50 = 89.96 nM) cells | [48,49,50] |
| Theopapuamide (9) | Theonella swinhoei | Cytotoxicity against CEM-TART (EC50 = 0.5 µM) and HCT116 (EC50 = 0.9 µM) cells. | [51] |
| Homophymines A-E (10–14) & A1-E1 (15–19) | Homophymia sp. | Anti-proliferative against several cancer cell lines (IC50 = 2 to 100 nM), among which PC3 and OV3 are the most sensitive. | [52,53] |
| Pipestelide A (20) | Pipestela candelabra | Cytotoxicity against KB (IC50 = 0.1 µM) cell. | [54] |
| Neamphamides B-D (21–23) | Neamphius huxleyi | Cytotoxicity against A549, LNCaP and PC3 cells. (IC50 = 91 to 230 nM) | [55,56] |
| Callipeltins N (24) and O (25) | Asteropus sp. | Cytotoxicity against A2058, HT29, MCF7 and MRC-5 cells. (IC50 = 0.16 to 0.21 µM and 0.48 to 2.08 µM, respectively) | [57] |
| Pipecolidepsins A (26) and B (27) | Homophymia lamellosa | Cytotoxicity against A549 (GI50 = 0.6 and 0.04 µM, respectively), HT29 (GI50 = 1.12 and 0.01 µM, respectively) and MDA-MB-231 (GI50 = 0.7 and 0.02 µM, respectively) cells. | [58,59,60,61] |
| Halipeptin D (28) | Leiosella cf. arenifibrosa | Cytotoxicity against HCT116 cell line (IC50 = 7 nM) and BMS ODCP with an average IC50 value of 420 nM. | [62,63] |
| Reniochalistatin E (29) | Reniochalina stalagmitis | Cytotoxicity against RPMI-8226 (IC50 = 4.9 μM) and MGC-803 (IC50 = 9.7 μM) cells. | [64,65] |
| Callyaerin G (30) | Callyspngia aerizusa | Cytotoxicity against L5178Y (ED50 = 4.1 mM) and HeLa (ED50 = 41.8 mM) cells. | [66] |
| Callyaerins E (31) and H (32) | Callyspngia aerizusa | Cytotoxicity against L5178Y (ED50 = 0.39 and 0.48 μM, respectively). | [67] |
| Scleritodermin A (33) | Scleritoderma nodosum | Cytotoxicity against HCT116 (IC50 = 1.9 μM), HCT116/VM46 (IC50 = 5.6 μM), A2780 (IC50 = 0.94 μM) and SKBR3 (IC50 = 0.67 μM) cells. | [68,69,70] |
| Calyxamides A (34) and B (35) | Discodermia calyx | Cytotoxicity against P388 (IC50 = 3.9 and 0.9 μM, respectively) cell. | [71] |
| Keramamide E (36) | Theonella sp. | Cytotoxicity against L1210 (IC50 = 1.42 µM) and KB (IC50 = 1.38 µM) cells. | [72] |
| Keramamides K (37) and L (38) | Theonella sp. | Cytotoxicity against L1210 (IC50 = 0.77 and 0.5 µM, respectively) and KB (IC50 = 0.45 and 0.97 µM, respectively) cells. | [73] |
| Keramamides M (39) and N (40) | Theonella sp. | Cytotoxicity against L1210 (IC50 = 2.02 and 2.33 µM, respectively) and KB (IC50 = 5.05 and 6.23 µM, respectively) cells. | [74,75] |
| Axinellins A (41) and B (42) | Axinella carteri | Cytotoxicity against NSLC-N6 (IC50 = 16.7 and 29.8 µM, respectively) cell. | [76,77] |
| Stylopeptide 2 (43) | Stylotella sp. | Inhibition of 23% BT-549 cell growth and 44% Hs578T cell growth at one dose (10−5 M). | [78] |
| Stylissamide X (44) | Stylissa sp. | Anti-migration effects on HeLa cell. | [79,80] |
| Aciculitins A-C (45–47) | Aciculites orientalis | Cytotoxicity against HCT116 (IC50 = 0.37 µM) cell. | [81] |
| Nazumazoles A-C (48–50) | Theonella swinhoei | Cytotoxicity against P388 (IC50 = 0.83 µM) cell. | [82] |
| Theonellamide G (51) | Theonella swinhoei | Cytotoxicity against HCT116 (IC50 = 6.0 µM) cell. | [83] |
| Koshikamide B (52) | Theonella sp. | Cytotoxicity against HCT116 (IC50 = 3.62 µM) and P388 (IC50 = 0.22 µM) cells. | [84] |
| Anticancer Cyclopeptides Derived from Ascidians/Tunicates | |||
| Mollamide (53) | Ascidian, Didemnum molle | Cytotoxicity against P388 (IC50 = 1.24 µM), A549 (IC50 = 3.1 µM), HT29 (IC50 = 3.1 µM) and CV1 (IC50 = 3.1 µM) cells. | [87,88] |
| Mollamide B (54) | Tunicate, Didemnum molle | Growth inhibition of H460, MCF7 and SF-268 cells. | [89] |
| Trunkamide A (55) | Ascidian, Lissoclinum sp. | Cytotoxicity against A549, P388, HT29 and MEL28 cells. (IC50 = 0.6 to 1.19 µM) | [90,91] |
| Prepatellamide A (56) | Ascidian, Lissoclinum patella | Cytotoxicity against P388 cells. (IC50~6.57 µM) | [92] |
| Vitilevuamide (57) | Ascidian, Didemnum cuculiferum | Cytotoxicity against HCT116 (IC50 = 6.24 nM), A549 (IC50 = 0.12 µM), SKMEL-5 (IC50 = 0.31 µM) and A498 (IC50 = 3.12 µM) cells. | [93,94] |
| Tamandarin A (58) | Ascidian, Didemnidae sp. | Cytotoxicity against BX-PC3 (IC50 = 1.69 nM), DU145 (IC50 = 1.29 nM), UM-SCC-10B (IC50 = 0.94 nM) cells. | [95,96,97] |
| Comoramides A-B (59–60) & Mayotamides A-B (61–62) | Ascidian, Didemnum molle | Cytotoxicity against A549, HT29 and MEL28 cells. (IC50 = 7.22 to 14.97 µM) | [98] |
| Patellin 6 (63) | Ascidian, Lissoclinum patella | Cytotoxicity against P388, A549, HT29 and CV1 cells. (IC50~2.08 µM) | [99] |
| Cycloforskamide (64) | Sea slug, Pleurobranchus forskalii | Cytotoxicity against P388 cell (IC50 = 5.8 µM). | [100] |
| Anticancer Cyclopeptides Derived from Mollusks | |||
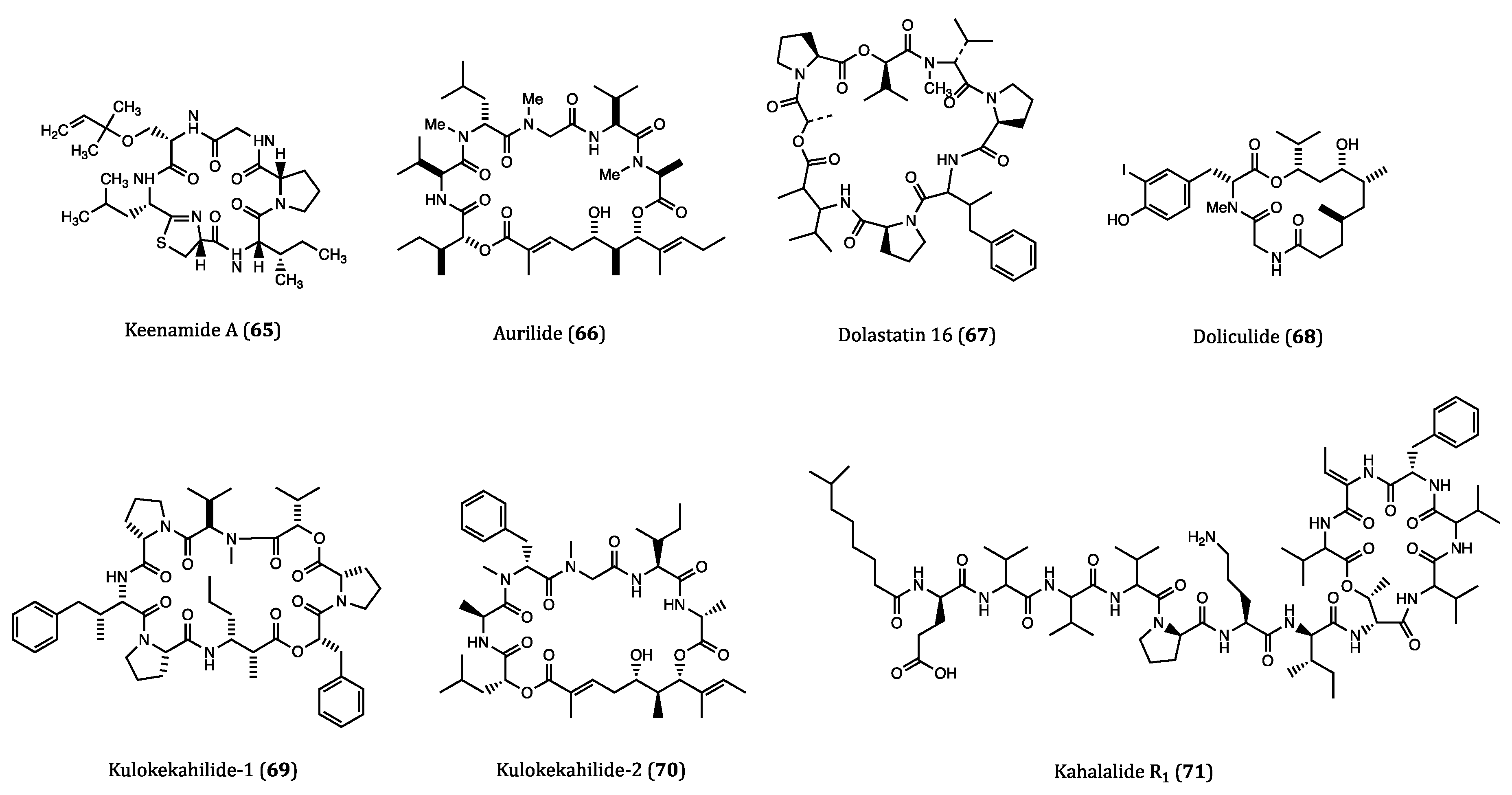
| Keenamide A (65) | Mollusk, Pleurobranchus forskali | Anti-proliferative activity against A549, P388, MEL20 and HT29 cells. (IC50 = 4.03 to 8.05 µM) | [103] |
| Aurilide (66) | Sea hare, Dolabella auricularia | Cytotoxicity against HeLa S3 (IC50 = 0.013 µM) cell. Inhibitory activity against ovarian, renal and prostate cancer cells in NCI 60 cell lines. | [104,105] |
| Dolastatin 16 (67) | Sea hare, Dolabella auricularia | Cytotoxicity against H460 (GI50 = 1.09 nM), KM20L2 (GI50 = 1.37 nM), SF-295 (GI50 = 5.92 nM) and SKMEL-5 (GI50 = 3.75 nM) cells. Anti-proliferative effects against 5 human leukemia cell lines | [106] |
| Doliculide (68) | Sea hare, Dolabella auricularia | Cytotoxicity against HeLa S3 (IC50 = 1.62 nM) cell. | [107,108,109,110] |
| Kulokekahilide-1 (69) | Mollusk, Philinopsis speciosa | Cytotoxicity against P388 (IC50 = 2.2 µM) cell. | [111] |
| Kulokekahilide-2 (70) | Mollusk, Philinopsis speciosa | Cytotoxicity against P388 (IC50 = 4.2 nM), SK-OV-3 (IC50 = 7.5 nM), MDA-MB-435 (IC50 = 14.6 nM) and A-10 (IC50 = 59.1 nM) cells. | [112,113,114] |
| Kahalalide R1 (71) | Sea slug, Elysia grandifolia | Cytotoxicity against MCF7 (IC50 = 0.14 µM), L1578Y (IC50 = 4.26 nM) cells. | [117] |
| Anticancer Cyclopeptides Derived from Marine Algae | |||
| Galaxamide (72) | Algae, Galaxaura filamentosa | Inhibitory activity against GRC-1 (IC50 = 7.18 μM) and HepG2 (IC50 = 7.81 μM) cells. | [119,120] |
3. Terrestrial Plant-Derived Anticancer Cyclopeptides
| Name | Biological Source | Anticancer Activity | Reference |
|---|---|---|---|
| Cherimolacyclopeptides C-F (73–76) | Seeds of Annona cherimola | Cytotoxicity against KB cell. (IC50 = 0.017 to 0.97 µM) | [123,124,125] |
| Integerrimide C (77) | Latex of Jatropha integerrima | Cytotoxicity against KB (IC50 = 1.7 µM) cell. | [126] |
| LOB3 (78) | Flaxseed oil | Cytotoxicty against A375, SKBR3 and MCF7 cells. | [128,129] |
| Cambodine A (79) | Root bark of Ziziphus cambodiana | Cytotoxicity against BC-1 (IC50 = 11.1 µM) cell. | [130] |
| Longicalycinin A (80) | Dianthus superbus | Growth inhibitory activity against HepG2 (IC50 = 22.13 μM) cell. | [131,132,133] |
| Dianthin E (81) | Dianthus superbus | Cytotoxicity against HepG2 (IC50 = 3.51 μM) cell. | [134] |
| [1-8-NαC]-Zanriorb A1 (82) | Leaves of Zanthoxylum riedelianum | Cytotoxicity against Jurkat leukemia T cell (IC50 = 218 nM). | [136] |
| Justicianenes B-D (83–85) | Justicia procumbens L. | Justicianene D displayed cytotoxicity against MCF-7 cells (IC50 = 90 μM). | [137] |
| GCP-1 (86) | Ginseng | Cytotoxicity against SGC-7901 cells (IC50 = 37.8 μM). | [138] |
| Rubipodanin A (87) | Roots and rhizomes of Rubia podantha | Cytotoxicity against HeLa, A549 and SGC-7901 cells. (IC50 = 3.80 to 7.22 μM) | [143] |
| Rubipodanin B (88) | Rubia podantha | Cytotoxicity against MDA-MB-231 (IC50 = 1.47 μM), SW620 (IC50 = 0.69 μM) and HepG2 (IC50 = 3.37 μM) cells. | [144] |
| RA-XII (89) | Rubia yunnanensis | Inhibitory effects on 4T1 ((IC50 = 606 nM and 96 nM for 24 and 48 h, respectively), SW260 and HT29 cells. | [145,146,147] |
| RA-XXV (90) | Roots of Rubia cordifolia L. | Cytotoxicity against HL-60 ((IC50 = 62 nM) and HCT116 cells ((IC50 = 28 nM). | [149] |
| RA-XXVI (91) | Roots of Rubia cordifolia L. | Cytotoxicity against HL-60 ((IC50 = 66 nM) and HCT116 cells ((IC50 = 51 nM). | [149] |
| HB7 (92) | Hedyotis biflora | Cytotoxicity against BxPC3 (IC50 = 0.68 μM), Capan2 (IC50 = 0.45 μM), MOH-1 (IC50 = 0.33 μM) and PANC1 (IC50 = 0.36 μM) cells. | [150] |
| Cliotides T1-T4 (93–96), cliotide T7 (97), cliotide T10 (98), cliotide T12 (99) | Clitoria ternatea | Cliotides T1-T4: cytotoxicity against HeLa (IC50 = 0.6 to 8.0 μM) cell. Cliotides T2, T4, T7, T10 and T12: cytotoxicity against A549 (IC50 = 0.21 to 7.59 μM) and A549/paclitaxel (IC50 = 0.45 to 7.92 μM) cells. | [151,152] |
| psyle E (100) | Psychotria leptothyrsa var. longicarpa | Cytotoxicity against U937-GTB IC50 = 0.76 μM) cell. | [153] |
| Vigno 5 (101) | Viola ignobilis | Pro-apoptotic activity on HeLa cell. | [154,155] |
| Cycloviolacin O2 (CyO2) (102) | Viola odorata L. | Cytotoxicity against U937 GTB (IC50 = 0.75 μM), CCRF-CEM, NCI-H69 and HT29 cells. | [156,157,158,159,160] |
| Viphi A-G (103–109) | Viola philippica | Cytotoxicity against MM96L, HeLa, BGC-823, HFF-1 cells, but viphi D and E showed no activity against BGC-823 cell. (IC50 = 1.03 to 7.92 μM) | [161] |
| Vaby A (110) and D (111) | Viola abyssinica | Cytotoxicity against U-937 (IC50 = 2.6 and 7.6 μM, respectively) cell. | [162] |
| Vibi G (112) and H (113) | Viola biflora | Cytotoxicity against U-937 GTB (IC50 = 0.96 and 1.6 μM, respectively) cell. | [163] |
| Vitri A (114) | Viola tricolor | Cytotoxicity against U-937 GTB (IC50 = 0.6 µM) and RPMI-8226/s (IC50 = 1 µM) cells. Cytotoxicity against U251, MDA-MB-231, A549, DU145 and BEL7402 cells (IC50 = 3.07 to 6.03 μM). | [164,165] |
| Varv A (115) and E (116) | Viola tricolor | Cytotoxicity against U-937 GTB (IC50 = 6 and 4 µM, respectively) and RPMI-8226/s (IC50 = 3 and 4 µM, respectively) cells. | [164] |
| Vitri F (117) | Viola tricolor | Cytotoxicity against U251, MDA-MB-231, A549, DU145 and BEL7402 cel ls (IC50 = 2.74 to 6.31 μM). | [165] |
4. Bacteria-Derived Anticancer Cyclopeptides
4.1. Anticancer Cyclopeptides Derived from Non-Marine Bacteria
4.2. Anticancer Cyclopeptides Derived from Marine Bacteria
4.3. Anticancer Cyclopeptides Derived from Cyanobacteria
| Name | Biological Source | Anticancer Activity | Reference |
|---|---|---|---|
| Anticancer Cyclopeptides Derived from Non-Marine Bacteria | |||
| YM753 (spiruchostatin A/OBP-801) (118) | Pseudomonas sp. | In vitro growth inhibitory ceffects on many cancer cells including WiDr, melanoma (GI50 = 5.0 nM), Calu-3 (GI50 = 1.6 nM), HOS (GI50 = 3.8 nM) cells and etc.; In vivo antitumor activity on WiDr xenograft model. | [168,169,170,171] |
| Telomestatin (119) | Streptomyces anulatus 3533-SV4 | Cytotoxicity against MiaPaCa (IC50 = 0.5 µM) cell and four neuroblastoma cells (IC50 = 0.8 to 4.0 µM). Specific telomerase inhibitor in cancer cells inhibition (inhibit cellular senescence). | [172,173,174,175,176,177,178] |
| Anticancer Cyclopeptides Derived from Marine Bacteria | |||
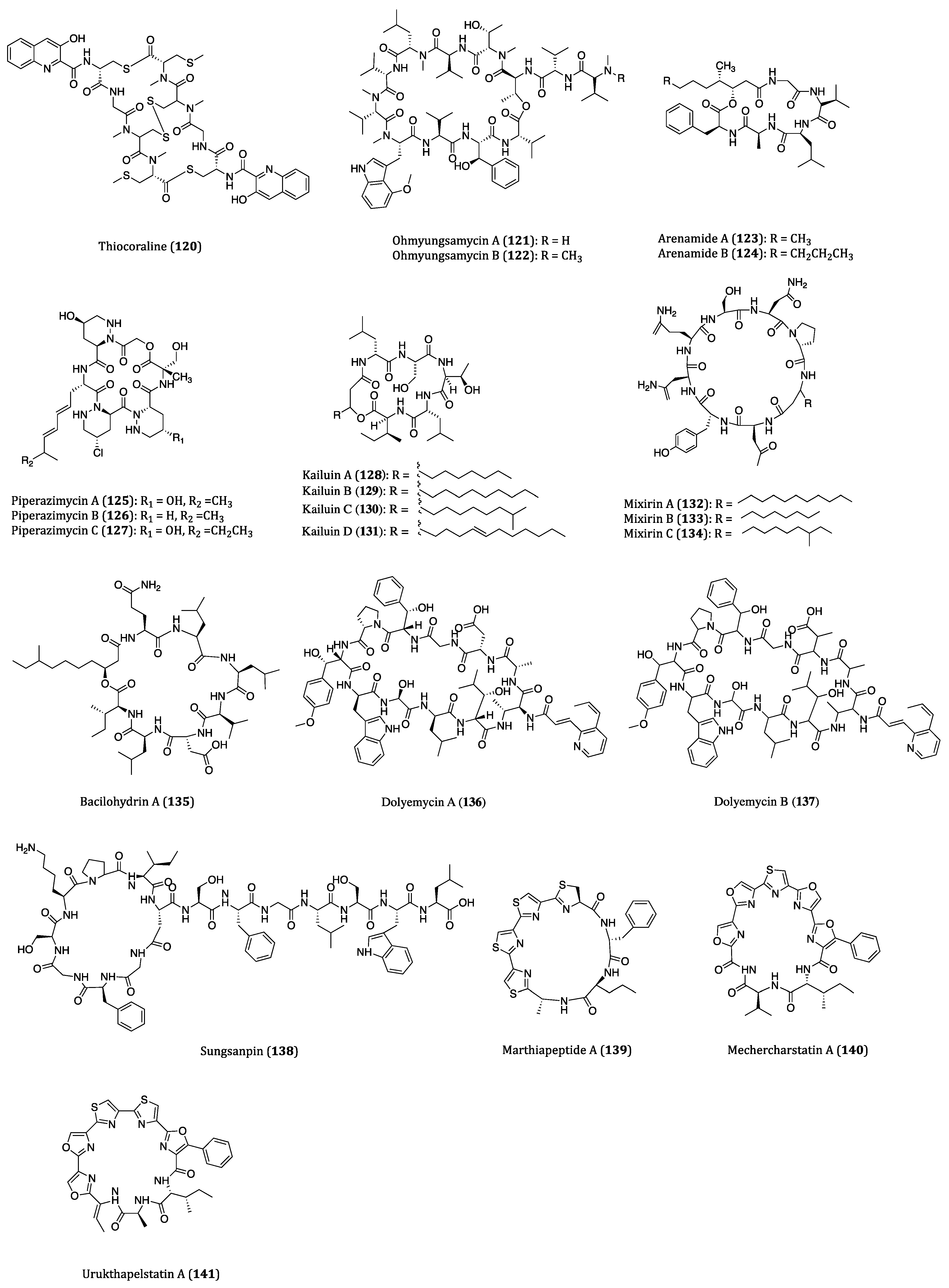
| Thiocoraline (120) | Gram-positive bacteria Micromonospora sp. ACM2-092 and Micromonospora sp. ML1 | Cytotoxicity against P388 (IC50 = 1.72 nM), A549 (IC50 = 1.72 nM), MEL28 (IC50 = 1.72 nM) and HT29 (IC50 = 8.64 nM) and MTC-TT (IC50 = 7.6 nM) cells. | [181,182,183,184,185] |
| Ohmyungsamycins A (121) and B (122) | Streptomyces sp. | Cytotoxicity against HCT116, A549, SNU-638, MDA-MB-231 and SK-HEP-1 cells. (IC50 = 359 to 816 nM and 12.4 to 16.8 µM, respectively) | [186,187] |
| Arenamides A (123) and B (124) | Salinispora arenicola | Cytotoxicity against HCT116 (IC50 = 1.97 and 2.99 μM, respectively) cell. | [188,189] |
| Piperazimycins A-C (125–127) | Streptomyces sp. | Cytotoxicity against HCT116 cell. (Average GI50 = 0.1 µM) Piperazimycin A showed a mean GI50 of 100 nM in NCI 60 cancer cell line panel screening. | [190,191] |
| Kailuins A-D (128–131) | Gram-negative bacterium BH-107 | Inhibitory activities against A549, MCF7 and HT29 cells. (IC50 = 2.66 to 5.52 µM) | [192] |
| Mixirins A-C (132–134) | Bacillus genus | Anti-proliferative activity of HCT116 (IC50 = 0.65, 1.6 and 1.26 μM, respectively) cell. | [193] |
| Bacilohydrin A (135) | Bacillus sp. SY27F | Cytotoxicity against HepG2, MCF7 and DU145 cells. (IC50 = 50.3 to 175.1 nM) | [195] |
| Dolyemycins A (136) and B (137) | Streptomyces griseus subsp. griseus HYS31 | Anti-proliferative activities against A549 cells. (IC50 = 1 and 1.2 µM, respectively) | [196] |
| Sungsanpin (138) | Streptomyces sp. | Anti-invasion effects on A549 cell. | [197] |
| Marthiapeptide A (139) | Actinomycete Marinactinospora thermotolerans | Cytotoxicity against SF-268, MCF7, NCI-H460 and HepG2 cell. (IC50 = 0.38 to 0.52 µM) | [198,199] |
| Mechercharstatin A (140) | Thermoactinomyces sp. YM3-251 | Cytotoxicity against Jurkat cell (IC50 = 4.6 × 10−8 M), A549 (IC50 = 0.03 µM), HT29 (IC50 = 0.04 µM) and MDA-MB-231 (IC50 = 0.09 µM) cells. | [198,200,201] |
| Urukthapelstatin A (141) | Mechercharimyces asporophorigenens YM11-542 | Cytotoxicity against several cancer cell lines. (Mean GI50 = 1.55 μM) | [202,203,204,205] |
| Anticancer Cyclopeptides Derived from Cyanobacteria | |||
| Microcystin-LR (142) | Mainly from Microcystis aeruginosa | Cytotoxicity against BxPC-3 (IC50 = 83.50 nM) and MIA PaCa2 (IC50 = 2.14 µM) and transfected HeLa (IC50 = 1 nM) cells. | [211,212,213,214,215,216] |
| Apratoxin A (143) | Lyngbya sp. | Cytotoxicity against KB (IC50 = 0.52 nM), LoVo (IC50 = 0.36 nM) and HCT116 (IC50 = 1.21 nM) cells. | [217,218,219,220,221] |
| Apratoxins B (144) and C (145) | Lyngbya sp. | Cytotoxicity against KB (IC50 = 21.3 and 1.0 nM, respectively) and LoVo (IC50= 10.8 and 0.73 nM, respectively) cells. | [222] |
| Apratoxin D (146) | Lyngbya majuscula and Lyngbya sordida | Cytotoxicity against H460 (IC50 = 2.6 nM) cell. | [223] |
| Apratoxin E (147) | Lyngbya bouillonii strain PS372 | Cytotoxicity against HT29 (IC50 = 21 nM), HeLa (IC50 = 72 nM) and U2OS (IC50 = 59 nM) cells. | [224] |
| Apratoxins F (148) and G (149) | Lyngbya bouillonii | Cytotoxicity against H460 (IC50 = 2 and 14 nM, respectively) and HCT116 cells. | [225] |
| Aurilide B (150) | Lyngbya majuscula | In vitro cytotoxicity against H460 (LC50 = 0.04 µM) and neuro-2a (LC50 = 0.01 µM) cells. In vivo net tumor cell killing activity | [226,227] |
| Aurilide C (151) | Lyngbya majuscula | Cytotoxicity against H460 (LC50 = 0.13 µM) and neuro-2a (LC50 = 0.05 µM) cells. | [226] |
| Lagunamide A (152) | Lyngbya majuscula | Cytotoxicity against P388, A549, PC3, HCT8 and SK-OV3 cells. (IC50 = 1.64 to 6.4 nM) | [228,229] |
| Lagunamide B (153) | Lyngbya majuscula | Cytotoxicity against P388 (IC50 = 20.5 nM) and HCT8 (IC50 = 5.2 nM) cells. | [228,229] |
| Lagunamide C (154) | Lyngbya majuscula | Cytotoxicity against P388, A549, PC3, HCT8 and SK-OV3 cells (IC50 = 2.1 to 24.2 nM). | [228,230] |
| Homodolastatin 16 (155) | Lyngbya majuscula | Cytotoxicity against WHCO1 (IC50 = 4.19 µM), WHCO6 (IC50 = 9.85 µM) and ME180 (IC50 = 8.09 µM) cells. | [231] |
| Lyngbyabellin A (156) | Lyngbya majuscula | Cytotoxicity against KB (IC50 = 0.04 µM) and LoVo (IC50 = 0.72 µM) cells. Specific cytoskeletal-disrupting effects to microfilaments in A-10 cells. | [232,233] |
| Lyngbyabellins D (157), F (158) and H (159) | Lyngbya majuscula | Cytotoxicity against H460 and KB cells. (IC50 or LC50 = 0.1 to 0.4 µM) | [234] |
| Lyngbyabellin N (160) | Moorea bouillonii | Cytotoxicity against HCT116 (IC50 = 40.9 nM) cell. | [235] |
| Palmyramide A (161) | Lyngbya majuscula | Modest cytotoxicity against H460 cell (IC50 = 39.7 µM). Blockage of the voltage-gated sodium channel in neuro-2a cell. | [236] |
| Hoiamide A (162) | Lyngbya majuscula and phormidium gracile | Cytotoxicity against H460 (IC50 = 11.2 µM) cell. | [237,238,239] |
| Hoiamide B (163) | A sample of marine cyanobacteria collected in Gallows reef, unclear species | Cytotoxicity against H460 (IC50 = 8.3 µM) cell. | [237,238] |
| Wewakazole (164) | Lyngbya majuscula | Cytotoxicity against H460 (IC50 = 10 µM) cell. | [240,241,242,243] |
| Wewakazole B (165) | Moorea peoducens | Cytotoxicity against H460 (IC50 = 1.0 µM) and MCF7 (IC50 = 0.58 µM) cells. | [241,242,243] |
| Hectochlorin (166) | Lyngbya majuscula | Average GI50 value of 5.1 µM in the tests against a panel of NCI 60 tumor cell lines. Cytotoxicity against KB (ED50 = 0.86 µM) and NCI-H187 (ED50 = 1.20 µM) cells. | [244,245,246,247] |
| Desmethoxymajusculamide C (DMMC) (167) | Lyngbya majuscula | Selectivity and cytotoxicity against HCT116 (IC50 = 0.016 µM), H460 (IC50 = 0.094 µM) and MDA-MB-435 (IC50 = 0.23 µM) cells. | [248] |
| Hantupeptin A (168) | Lyngbya majuscula | Cytotoxicity against MOLT-4 (IC50 = 32 nM) and MCF7 (IC50 = 4.0 µM) cells. | [249] |
| Hantupeptins B (169) and C (170) | Lyngbya majuscula | Cytotoxicity against MOLT-4 (IC50 = 0.2 and 3.0 µM, respectively) and MCF7 (IC50 = 0.5 and 1.0 µM, respectively) cells. | [249,250] |
| Laxaphycin A (171) | Lyngbya majuscula | Obvious biological synergism when acting against DLD1 and MDR cells in the presence of laxaphycins A and B. | [251,252] |
| Laxaphycin B (172) | Lyngbya majuscula | Cytotoxicity against CCRF-CEM (IC50 = 1.11 µM), CEM/VLB100 (IC50 = 1.02 µM) and CEM/VM-1 (IC50 = 1.37 µM) cells. Anti-proliferative effects on A549, MCF7, PA1 and PC3 cells. (IC50 = 0.19 to 1.0 µM) Obvious biological synergism when acting against DLD1 and MDR cells in the presence of laxaphycins A and B. | [251,252] |
| Laxaphycins B2 (173) and B3 (174) | Lyngbya majuscula | Moderate cytotoxicities against CCRF-CEM, CEM/VLB100 and CEM/VM-1 cells. | [252] |
| Laxaphycin B4 (175) | Hormothamnion enteromorphoides | Growth-inhibitory activity and synergistic effect with laxaphycin A when acting against HCT116 (IC50 = 1.7 µM) cell. | [253] |
| Obyanamide (176) | Lyngbya confervoides | Cytotoxicity against KB (IC50 = 0.97 µM) and LoVo (IC50 = 5.24 µM) cells. | [254,255] |
| Grassypeptolide A (177) | Lyngbya confervoides | Cytotoxicity against U2OS (IC50 = 2.2 µM), HeLa (IC50 = 1.0 µM), HT29 (IC50 = 1.5 µM) and IMR-32 (IC50 = 4.2µM) cells. | [256,257,258] |
| Grassypeptolides B (178) and C (179) | Lyngbya confervoides | Anti-proliferative effects on HT29 (IC50 = 2.97 µM and 76.7 nM, respectively) and HeLa (IC50 = 2.93 µM and 44.6 nM, respectively) cells. | [257] |
| Grassypeptolides D (180) and E (181) | Leptolyngbya sp. | Cytotoxicity against HeLa (IC50 = 335 and 192 nM, respectively) and neuro-2a (IC50 = 599 and 407 nM, respectively) cells. | [259] |
| Palauamide (182) | Lyngbya sp. | Cytotoxicity against KB (IC50 = 13 nM) cell. | [260,261,262] |
| Coibamide A (183) | Leptolyngbya sp. | Cytotoxicity against NCI-H460 and neuro-2a cells. (LC50 < 17.9 nM) | [263,264,265] |
| Ulongamides A-E (184–188) | Lyngbya sp. | Cytotoxicity against KB (IC50~1 µM) and LoVo (IC50~5 µM) cells. | [266,267,268] |
| Ulongapeptin (189) | Lyngbya sp. | Cytotoxicity against KB (IC50 = 0.63 µM) cell. | [269] |
| Odoamide (190) | Okeania sp. | Cytotoxicity against HeLa S3 (IC50 = 26.3 nM) cell. | [270,271] |
| Cryptophycin 1 (191) | Nostoc sp. | Broad-spectrum cytotoxicity against cell lines such as L1210 (IC50 = 4 pM), SKOV3 (IC50 = 7 pM), MCF7 (IC50 = 16 pM) and etc. | [272,273,274,275,276,277,278,279] |
| Largazole (192) | Symploca sp. | Cytotoxicity against MDA-MB-231 (GI50 = 122 nM, LC50 = 272 nM), NmuMG (GI50 = 7.7 nM, LC50 = 117 nM), U2OS (GI50 = 55 nM, LC50 = 94 nM), NIH3T3 (GI50 = 480 nM, LC50 > 8 µM), HCT116 (GI50 = 0.08 µM) and A549 (GI50 = 0.32 µM) cells. | [280,281,282,283,284,285] |
| Symplocamide A (193) | Symploca sp. | Cytotoxicity against H460 (IC50 = 40 nM) and neuro-2a (IC50 = 29 nM) cells. | [286,287] |
| Tasipeptins A (194) and B (195) | Symploca sp. | Cytotoxicity against KB (IC50 = 0.93 and 0.82 µM, respectively) cell. | [288,289] |
| Veraguamides A-G (196–202) | Symploca cf. hydnoides | Cytotoxicity against HT29 (IC50 = 0.84 to 49 µM) and HeLa (IC50 = 0.54 to 49 µM) cells. | [290] |
| Floridamide (203) | Moorea producens | Cytotoxicity against H460 and neuro-2a cells. (EC50 = 1.89 × 10−5 µM) | [291] |
| Viequeamide A (204) | Rivularia sp. | Cytotoxicity against H460 (IC50 = 60 nM) cell. | [292,293] |
5. Fungi-Derived Anticancer Cyclopeptides
5.1. Anticancer Cyclopeptides Derived from Non-Marine Fungi
5.2. Anticancer Cyclopeptides Derived from Marine Fungi
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-tieulent, J.; Jemal, A. Global Cancer Statistics, 2012, CA a Cancer. J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Global Cancer Facts & Figures 4th Edition. Atlanta: American Cancer Society. 2018. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/global-cancer-facts-and-figures/global-cancer-facts-and-figures-4th-edition.pdf (accessed on 12 April 2021).
- Appold, K. Cancer Treatment—Top 4 Developments to Watch. 2017. Available online: https://www.managedhealthcareexecutive.com/view/cancer-treatment-developments (accessed on 12 April 2021).
- Kang, T.H.; Mao, C.P.; He, L.; Tsai, Y.C.; Liu, K.; La, V.; Wu, T.C.; Hung, C.F. Tumor-targeted delivery of IL-2 by NKG2D leads to accumulation of antigen-specific CD8+ T cells in the tumor loci and enhanced anti-tumor effects. PLoS ONE 2012, 7, e35141. [Google Scholar] [CrossRef] [PubMed]
- Amit, D.; Tamir, S.; Hochberg, A. Development of targeted therapy for a broad spectrum of solid tumors mediated by a double promoter plasmid expressing diphtheria toxin under the control of IGF2-P4 and IGF2-P3 regulatory sequences. Int. J. Clin. Exp. Med. 2013, 6, 110–118. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. NanoTechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Pawar, S.V.; Ho, J.C.H.; Yadav, G.D.; Yadav, V.G. The impending renaissance in discovery & development of natural products. Curr. Top. Med. Chem. 2017, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef]
- Wipf, P. Synthetic studies of biologically active marine cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Pomilio, A.; Battista, M.; Vitale, A. Naturally-occurring cyclopeptides: Structures and bioactivity. Curr. Org. Chem. 2006, 10, 2075–2121. [Google Scholar] [CrossRef]
- Tran, D.; Selsted, M.E.; Dorrestein, P.C.; Pavel, A. Cycloquest: Identification of cyclopeptides via database search of their mass spectra against genome databases. NIH Public Access. 2012, 10, 4505–4512. [Google Scholar] [CrossRef]
- Tan, N.H.; Zhou, J. Plant cyclopeptides. Chem. Rev. 2006, 106, 840–895. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef]
- Hur, G.H.; Vickery, C.R.; Burkart, M.D. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 2012, 29, 1074–1098. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.A.; Farias, R.N. Microcin 25, a novel antimicrobial peptide produced by Escherichia coli. J. Bacteriol. 1992. [Google Scholar] [CrossRef] [PubMed]
- Luckett, S.; Garcia, R.S.; Barker, J.J.; Konarev, A.V.; Shewry, P.R.; Clarke, A.R.; Brady, R.L. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 1999, 290, 525–533. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Ösapay, G.; Ösapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated α-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef]
- Closse, A.; Huguenin, R. Isolierung und Strukturaufklärung von Chlamydocin. Helv. Chim. Acta 1974, 57, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Stefanucci, A.; Novellino, E. Cyclotides: A natural combinatorial peptide library or a bioactive sequence player? J. Enzym. Inhib. Med. Chem. 2015, 30, 575–580. [Google Scholar] [CrossRef]
- Edman, P. Chemistry of amino acids and peptides. Annu. Rev. Biochem. 1959, 28, 69–96. [Google Scholar] [CrossRef]
- Trabi, M.; Craik, D.J. Circular proteins-No end in sight. Trends Biochem. Sci. 2002, 27, 132–138. [Google Scholar] [CrossRef]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J. Am. Chem. Soc. 2006, 128, 2510–2511. [Google Scholar] [CrossRef]
- Áron, R. Towards Targeted Photodynamic Therapy: Synthesis and Characterization of Aziridine Aldehyde-Cyclized Cancer-Targeting Peptides and Bacteriochlorin Photosensitizers. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2014. [Google Scholar]
- Malaker, A.; Ahmad, S.A.I. Therapeutic potency of anticancer peptides derived from marine organism. Int. J. Eng. Appl. Sci. 2013, 2, 53–65. [Google Scholar]
- Liu, J.; Gu, B.; Yang, L.; Yang, F.; Lin, H.; Yang, F. New anti-inflammatory cyclopeptides from a sponge-derived fungus Aspergillus violaceofuscus. Front. Chem. 2018, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Desriac, F.; Jé, C.; Balnois, E.; Brillet, B.; le Chevalier, P. Antimicrobial peptides from marine proteobacteria. Mar. Drugs 2013, 11, 3632–3660. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhen, Z.; Niu, T.; Zhu, X.; Gao, Y.; Yan, J.; Chen, Y.; Yan, X.; Chen, H. κ-Carrageenan enhances lipopolysaccharide-induced interleukin-8 secretion by stimulating the Bcl10-NF-κB pathway in HT-29 cells and aggravates C. freundii-Induced inflammation in mice. Mediat. Inflamm. 2017, 2017, 8634865. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, A.; Bifulco, G.; Giannini, C.; Bucci, M.; Cirino, G.; Gomez-paloma, L.; Ponte, V.; Salerno, F.; Ii, F.; Montesano, V.D.; et al. Halipeptins A and B: Two novel potent anti-inflammatory cyclic depsipeptides from the Vanuatu marine sponge Haliclona species. J. Am. Chem. Soc. 2001, 123, 10870–10876. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Tong, M.; Jiang, N.; Zhang, X.; Hu, H.; Pan, H.; Li, D. Antitumour bioactive peptides isolated from marine organisms. Clin. Exp. Pharmacol. Physiol. 2017, 44, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Laport, M.; Santos, O.; Muricy, G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. BioTechnol. 2009, 10, 86–105. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine Sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef]
- Kang, H.K.; Choi, M.C.; Seo, C.H.; Park, Y. Therapeutic properties and biological benefits of marine-derived anticancer peptides. Int. J. Mol. Sci. 2018, 19, 919. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, S.; Matsunaga, S.; Shindoh, N.; Terada, Y.; Nagai, K.; Yamashita, J.K.; Ganesan, A.; van Soest, R.W.M.; Fusetani, N. Azumamides A-E: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge Mycale izuensis. Angew. Chem. Int. Ed. 2006, 45, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Sriraksa, R.; Limpaiboon, T. Histone deacetylases and their inhibitors as potential therapeutic drugs for cholangiocarcinoma-cell line findings. Asian Pac. J. Cancer Prev. 2013, 14, 2503–2508. [Google Scholar] [CrossRef]
- Abdalla, M.A. Medicinal significance of naturally occurring cyclotetrapeptides. J. Nat. Med. 2016, 70, 708–720. [Google Scholar] [CrossRef]
- Maulucci, N.; Chini, M.G.; di Micco, S.; Izzo, I.; Cafaro, E.; Russo, A.; Gallinari, P.; Paolini, C.; Nardi, M.C.; Casapullo, A.; et al. Molecular insights into azumamide E histone deacetylases inhibitory activity. J. Am. Chem. Soc. 2007, 129, 3007–3012. [Google Scholar] [CrossRef]
- Wen, S.; Carey, K.L.; Nakao, Y.; Fusetani, N.; Packham, G.; Ganesan, A. Total synthesis of azumamide A and azumamide E, evaluation as histone deacetylase inhibitors, and design of a more potent analogue. Org. Lett. 2007, 9, 1105–1108. [Google Scholar] [CrossRef]
- Zhang, X.; Jacob, M.R.; Rao, R.R.; Wang, Y.H.; Agarwal, A.K.; Newman, D.J.; Khan, I.A.; Clark, A.M.; Li, X.C. Antifungal cyclic peptides from the marine sponge Microscleroderma herdmani [Corrigendum]. Res. Rep. Med. Chem. 2013, 3, 9–10. [Google Scholar] [CrossRef][Green Version]
- Bewley, C.A.; Detritus, C.; Faulkner, D.J. Microsclerodermins A and B. Antifungal cyclic peptides from the lithistid sponge Microscleroderma sp. J. Am. Chem. Soc. 1994, 116, 7631–7636. [Google Scholar] [CrossRef]
- Guzmán, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin A is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs. 2015, 33, 86–94. [Google Scholar] [CrossRef]
- Kehraus, S.; Ko, G.M.; Wright, A.D.; Bonn, D.; Woerheide, G.; Reef, B. Leucamide A: A new cytotoxic heptapeptide from the Australian sponge Leucetta microraphis leucamide. J. Org. Chem. 2002, 67, 4989–4992. [Google Scholar] [CrossRef]
- Wang, W.; Nan, F. First total synthesis of leucamide A. J. Org. Chem. 2003, 68, 1636–1639. [Google Scholar] [CrossRef]
- Afifi, A.H.; El-Desoky, A.H.; Kato, H.; Mangindaan, R.E.P.; de Voogd, N.J.; Ammar, N.M.; Hifnawy, M.S.; Tsukamoto, S. Carteritins A and B, cyclic heptapeptides from the marine sponge Stylissa carteri. Tetrahedron Lett. 2016, 57, 1285–1288. [Google Scholar] [CrossRef]
- Sun, J.; Cheng, W.; de Voogd, N.J.; Proksch, P.; Lin, W. Stylissatins B–D, cycloheptapeptides from the marine sponge Stylissa massa. Tetrahedron Lett. 2016, 57, 4288–4292. [Google Scholar] [CrossRef]
- Bonnington, L.S.; Tanaka, J.; Higa, T.; Kimura, J.; Yoshimura, Y.; Nakao, Y.; Yoshida, W.Y.; Scheuer, P.J. Cupolamide A: A cytotoxic cyclic heptapeptide from two samples of the sponge Theonella cupola. J. Org. Chem. 2002, 62, 7765–7767. [Google Scholar] [CrossRef]
- Rangel, M.; Konno, K.; Brunaldi, K.; Procopio, J.; de Freitas, J.C. Neurotoxic activity induced by a haemolytic substance in the extract of the marine sponge Geodia corticostylifera. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2005, 141, 207–215. [Google Scholar] [CrossRef]
- Rangel, M.; Prado, M.P.; Konno, K.; Naoki, H.; Freitas, J.C.; Machado-Santelli, G.M. Cytoskeleton alterations induced by Geodia corticostylifera depsipeptides in breast cancer cells. Peptides 2006, 27, 2047–2057. [Google Scholar] [CrossRef]
- Freitas, V.M.; Rangel, M.; Bisson, L.F.; Jaeger, R.G.; Machado-Santelli, G.M. The geodiamolide H, derived from Brazilian sponge Geodia corticostylifera, regulates actin cytoskeleton, migration and invasion of breast cancer cells cultured in three-dimensional environment. J. Cell. Physiol. 2008, 216, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, A.S.; Bugni, T.S.; Feng, X.; Harper, M.K.; Skalicky, J.J.; Mohammed, K.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Theopapuamide, a cyclic depsipeptide from a Papua New Guinea lithistid sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 1582–1586. [Google Scholar] [CrossRef]
- Zampella, A.; Sepe, V.; Bellotta, F.; Luciano, P.; D’Auria, M.V.; Cresteil, T.; Debitus, C.; Petek, S.; Poupat, C.; Ahond, A. Homophymines B-E and A1-E1, a family of bioactive cyclodepsipeptides from the sponge Homophymia sp. Org. Biomol. Chem. 2009, 7, 4037–4044. [Google Scholar] [CrossRef] [PubMed]
- Zampella, A.; Sepe, V.; Luciano, P.; Bellotta, F.; Monti, M.C.; D’Auria, M.V.; Jepsen, T.; Petek, S.; Adeline, M.T.; Laprévôte, O.; et al. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008, 73, 5319–5327. [Google Scholar] [CrossRef]
- Sorres, J.; Martin, M.T.; Petek, S.; Levaique, H.; Cresteil, T.; Ramos, S.; Thoison, O.; Debitus, C.; Al-Mourabit, A. Pipestelides A–C: Cyclodepsipeptides from the pacific marine sponge Pipestela candelabra. J. Nat. Prod. 2012, 75, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Pham, N.B.; Fechner, G.; Zencak, D.; Vu, H.T.; Hooper, J.N.A.; Quinn, R.J. Cytotoxic cyclic depsipeptides from the Australian marine sponge Neamphius huxleyi. J. Nat. Prod. 2012, 75, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Yamano, Y.; Arai, M.; Kobayashi, M. Neamphamide B, new cyclic depsipeptide, as an anti-dormant mycobacterial substance from a Japanese marine sponge of Neamphius sp. Bioorganic Med. Chem. Lett. 2012, 22, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Stierhof, M.; Hansen, K.Ø.; Sharma, M.; Feussner, K.; Subko, K.; Díaz-Rullo, F.F.; Isaksson, J.; Pérez-Victoria, I.; Clarke, D.; Hansen, E.; et al. New cytotoxic callipeltins from the Solomon Island marine sponge Asteropus sp. Tetrahedron 2016, 72, 6929–6934. [Google Scholar] [CrossRef]
- Coello, L.; Reyes, F.; Martín, M.J.; Cuevas, C.; Fernández, R. Isolation and structures of pipecolidepsins A and B, cytotoxic cyclic depsipeptides from the madagascan sponge Homophymia lamellosa. J. Nat. Prod. 2014, 77, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.P. Synthesis, Structural Elucidation and Biological Evaluation of Pipecolidepsin A and Synthesis, Structural Elucidation and Biological Evaluation of Pipecolidepsin A and Phakellistatin 19. Ph.D. Thesis, University of Barcelona, Barcelona, Spain, 2013. [Google Scholar]
- Molina-Guijarro, J.M.; Moneo, V.; Martinez-Leal, J.F.; Cuevas, C.; Garcia-Fernandez, L.F.; Galmarini, C.M. Pipecolidepsin A, stellatolide A and irvalec: New cyclodepsipeptides of marine origin with antitumor activity. Eur. J. Cancer 2014, 50, 24. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; García-Ramos, Y.; Martin, M.J.; Spengler, J.; Molina-Guijarro, J.M.; Munt, S.; Francesch, A.M.; Cuevas, C.; Tulla-Puche, J.; Albericio, F. The first total synthesis of the cyclodepsipeptide pipecolidepsin A. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Schlawe, D.; Kim, D.W.; Longbottom, D.A.; de Noronha, R.G.; Lizos, D.E.; Manam, R.R.; Faulkner, D.J. Total synthesis of halipeptins: Isolation of halipeptin D and synthesis of oxazoline halipeptin analogues. Chem. A Eur. J. 2005, 11, 6198–6211. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Lizos, D.E.; Kim, D.W.; Schlawe, D.; de Noronha, R.G.; Longbottom, D.A.; Rodriquez, M.; Bucci, M.; Cirino, G. Total synthesis and biological evaluation of halipeptins A and D and analogues. J. Am. Chem. Soc. 2006, 128, 4460–4470. [Google Scholar] [CrossRef] [PubMed]
- Zhan, K.; Jiao, W.; Yang, F.; Li, J.; Wang, S.; Li, Y.; Han, B.; Lin, H. Reniochalistatins A−E, cyclic peptides from the marine sponge Reniochalina stalagmitis. J. Nat. Prod. 2014, 77, 2678–2684. [Google Scholar] [CrossRef]
- Fatino, A.; Baca, G.; Weeramange, C.; Rafferty, R.J. Total synthesis of reniochalistatin E. J. Nat. Prod. 2017, 80, 3234–3240. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Edrada-Ebel, R.A.; Mohamed, G.A.; Youssef, D.T.A.; Wray, V.; Proksch, P. Callyaerin G, a new cytotoxic cyclic peptide from the marine sponge Callyspongia aerizusa. Arkivoc 2008, 12, 164–171. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Min, C.C.; Teuscher, F.; Ebel, R.; Kakoschke, C.; Lin, W.; Wray, V.; Edrada-Ebel, R.; Proksch, P. Callyaerins A-F and H, new cytotoxic cyclic peptides from the Indonesian marine sponge Callyspongia aerizusa. Bioorganic Med. Chem. 2010, 18, 4947–4956. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Raventos-Suarez, C.; Bifano, M.; Menendez, A.T.; Fairchild, C.R.; Faulkner, D.J. Scleritodermin A, a cytotoxic cyclic peptide from the Lithistid sponge Scleritoderma nodosum. J. Nat. Prod. 2004, 67, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Sellanes, D.; Manta, E.; Serra, G. Toward the total synthesis of Scleritodermin A: Preparation of the C1-N15 fragment. Tetrahedron Lett. 2007, 48, 1827–1830. [Google Scholar] [CrossRef]
- Liu, S.; Cui, Y.M.; Nan, F.J. Total synthesis of the originally proposed and revised structures of scleritodermin A. Org. Lett. 2008, 10, 3765–3768. [Google Scholar] [CrossRef]
- Kimura, M.; Wakimoto, T.; Egami, Y.; Tan, K.C.; Ise, Y.; Abe, I. Calyxamides A and B, cytotoxic cyclic peptides from the marine sponge Discodermia calyx. J. Nat. Prod. 2012, 75, 290–294. [Google Scholar] [CrossRef]
- Kobayashi, J.; Itagaki, F.; Shigemori, I.; Takao, T.; Shimonishi, Y. Keramamides E, G, H, and J, new cyclic peptides containing an oxazole or a thiazole ring from a Theonella sponge. Tetrahedron 1995, 51, 2525–2532. [Google Scholar] [CrossRef]
- Uemoto, H.; Yahiro, Y.; Shigemori, H.; Tsuda, M.; Takao, T.; Shimonishi, Y.; Kobayashi, J. Keramamides K and L, new cyclic peptides containing unusual tryptophan residue from Theonella sponge. Tetrahedron 1998, 54, 6719–6724. [Google Scholar] [CrossRef]
- Tsuda, M.; Ishiyama, H.; Masuko, K.; Takao, T.; Shimonishi, Y.; Kobayashi, J. Keramamides M and N, two new cyclic peptides with a sulfate ester from Theonella sponge. Tetrahedron 1999, 55, 12543–12548. [Google Scholar] [CrossRef]
- Junk, L.; Kazmaier, U. Total Synthesis of Keramamides A and L from a common precursor by late-stage indole synthesis and configurational revision. Angew. Chem. Int. Ed. 2018, 57, 11432–11435. [Google Scholar] [CrossRef]
- Randazzo, A.; Dal, F.; Orru, S.; Gomez-paloma, L. Axinellins A and B: New proline-containing antiproliferative cyclopeptides from the Vanuatu sponge Axinella carteri. Eur. J. Org. Chem. 1998, 3, 2659–2665. [Google Scholar] [CrossRef]
- Fairweather, K.A.; Sayyadi, N.; Roussakis, C.; Jolliffe, K.A. Synthesis of the cyclic heptapeptide axinellin A. Tetrahedron 2010, 66, 935–939. [Google Scholar] [CrossRef]
- Brennan, M.R.; Costello, C.E.; Maleknia, S.D.; Pettit, G.R.; Erickson, K.L. Stylopeptide 2, a proline-rich cyclodecapeptide from the sponge Stylotella sp. J. Nat. Prod. 2008, 71, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yamano, Y.; Fujita, M.; Setiawan, A.; Kobayashi, M. Stylissamide X, a new proline-rich cyclic octapeptide as an inhibitor of cell migration, from an Indonesian marine sponge of Stylissa sp. Bioorganic Med. Chem. Lett. 2012, 22, 1818–1821. [Google Scholar] [CrossRef]
- Huang, T.; Zou, Y.; Wu, M.C.; Zhao, Q.J.; Hu, H.G. Total synthesis of proline-rich cyclic octapeptide stylissamide X. Chem. Nat. Compd. 2015, 51, 523–526. [Google Scholar] [CrossRef]
- Bewley, C.A.; He, H.; Williams, D.H.; Faulkner, D.J. Aciculitins A–C: Cytotoxic and antifungal cyclic peptides from the lithistid sponge Aciculites orientalis. J. Am. Chem. Soc. 1996, 118, 4314–4321. [Google Scholar] [CrossRef]
- Fukuhara, K.; Takada, K.; Okada, S.; Matsunaga, S. Nazumazoles A-C, cyclic pentapeptides dimerized through a disulfide bond from the marine sponge Theonella swinhoei. Org. Lett. 2015, 17, 2646–2648. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Shaala, L.A.; Mohamed, G.A.; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R.M. Theonellamide G, a potent antifungal and cytotoxic bicyclic glycopeptide from the red sea marine sponge Theonella swinhoei. Mar. Drugs 2014, 12, 1911–1923. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Matsunaga, S.; Nakao, Y.; Furihata, K.; West, L.; Faulkner, D.J.; Fusetani, N. Koshikamide B, a cytotoxic peptide lactone from a marine sponge Theonella sp. J. Org. Chem. 2008, 73, 7889–7894. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, N.; Swalla, B.J. Global diversity of Ascidiacea. PLoS ONE 2011, 6, e20657. [Google Scholar] [CrossRef]
- Marino, A.; Rajendran, N.M. Natural products diversity of marine ascidians (Tunicates; Ascidiacea) and successful drugs in clinical development. Nat. Prod. Bioprospect 2017, 7, 1–111. [Google Scholar] [CrossRef]
- Hockless, D.C.R.; Skelton, B.W.; White, A.H. Studies of australian ascidians. IV. mollamide, a cytotoxic cyclic heptapeptide from the compound ascidian. Aust. J. Chem. 1994, 47, 61–69. [Google Scholar] [CrossRef]
- McKeever, B.; Pattenden, G. Total synthesis of the cytotoxic cyclopeptide mollamide, isolated from the sea squirt Didemnum molle. Tetrahedron 2003, 59, 2701–2712. [Google Scholar] [CrossRef]
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Patny, A.; Avery, M.; Hamann, M.T. Mollamides B and C, cyclic hexapeptides from the indonesian tunicate Didemnum molle. J. Nat. Prod. 2008, 71, 941–945. [Google Scholar] [CrossRef] [PubMed]
- McKeever, B.; Pattenden, G. Total synthesis of trunkamide A, a novel thiazoline-based prenylated cyclopeptide metabolite from Lissoclinum sp. Tetrahedron 2003, 59, 2713–2727. [Google Scholar] [CrossRef]
- Bowden, B.F.; Garcia, G.D. A Cyclic Hepta-Peptide Derivative from Colonial Ascidians, Lissoclinum sp. European Patent EP0894092B1, 3 April 2002. [Google Scholar]
- Fu, X.; Su, J.; Zeng, L. Prepatellamide A, a new cyclic peptide from the ascidian Lissoclinum patella. Sci. China Ser. B Chem. 2008, 43, 643–648. [Google Scholar] [CrossRef]
- Fernandez, A.M. Isolation and Characterization of Vitilevuamide from the Ascidians Didemnum cuculliferum and Polysyncraton lithostrotum. Ph.D. Thesis, The University of Utah, Salt Lake City, UT, USA, 1996. [Google Scholar]
- Edler, M.C.; Fernandez, A.M.; Lassota, P.; Ireland, C.M.; Barrows, L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002, 63, 707–715. [Google Scholar] [CrossRef]
- Pangestuti, R.; Kim, S.K. Bioactive peptide of marine origin for the prevention and treatment of non-communicable diseases. Mar. Drugs 2017, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, H.; Fenical, W.; Epifanio, R.D.A. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Richard, D.J.; Portonovo, P.S.; Joullie, M.M. Total Syntheses and biological investigations of tamandarins A and B and tamandarin A analogs. J. Am. Chem. Soc. 2001, 123, 4469–4474. [Google Scholar] [CrossRef]
- Rudi, A.; Aknin, M.; Gaydou, E.M.; Kashman, Y. Four new cytotoxic cyclic hexa- and heptapeptides from the marine ascidian Didemnum molle. Tetrahedron 1998, 54, 13203–13210. [Google Scholar] [CrossRef]
- Carroll, A.; Coll, J.; Bourne, D.; Macleod, J.; Zabriskie, T.; Ireland, C.; Bowden, B. Patellins 1–6 and trunkamide A: Novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust. J. Chem. 1996, 49, 659–667. [Google Scholar] [CrossRef]
- Tan, K.C.; Wakimoto, T.; Takada, K.; Ohtsuki, T.; Uchiyama, N.; Goda, Y.; Abe, I. Cycloforskamide, a cytotoxic macrocyclic peptide from the sea slug Pleurobranchus forskalii. J. Nat. Prod. 2013, 76, 1388–1391. [Google Scholar] [CrossRef]
- Rosenberg, G. A New critical estimate of named species-level diversity of the recent Mollusca. Am. Malacol. Bull. 2014, 32, 308–322. [Google Scholar] [CrossRef]
- Chakraborty, S.; Ghosh, U. Oceans: A store house of drugs-a review. J. Pharm. Res. 2010, 3, 1293–1296. [Google Scholar]
- Wesson, K.J.; Hamann, M.T. Keenamide A, a bioactive cyclic peptide from the marine mollusk Pleurobranchus forskalii. J. Nat. Prod. 1996, 59, 629–631. [Google Scholar] [CrossRef]
- Suenaga, K.; Mutou, T.; Shibata, T.; Itoh, T.; Fujita, T.; Takada, N.; Hayamizu, K.; Takagi, M.; Irifune, T. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron 2004, 60, 8509–8527. [Google Scholar] [CrossRef]
- Sato, S.I.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the opa1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.P.; Hogan, F.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Cerny, R.L.; Boyd, M.R. Isolation and structure of the human cancer cell growth inhibitory cyclodepsipeptide dolastatin 16. J. Nat. Prod. 1997, 60, 752–754. [Google Scholar] [CrossRef]
- Ishiwata, H.; Nemoto, T.; Ojika, M.; Yamada, K. Isolation and stereostructure of doliculide, a cytotoxic cyclodepsipeptide from the Japanese sea hare Dolabella auricularia. J. Org. Chem. 1994, 59, 4710–4711. [Google Scholar] [CrossRef]
- Ishiwata, H.; Sone, H.; Kigoshi, H.; Yamada, K. Total synthesis of doliculide, a potent cytotoxic cyclodepsipeptide from the Japanese sea hare Dolabella auricularia. J. Org. Chem. 1994, 59, 4712–4713. [Google Scholar] [CrossRef]
- Matcha, K.; Madduri, A.V.R.; Roy, S.; Ziegler, S.; Waldmann, H.; Hirsch, A.K.H.; Minnaard, A.J. Total synthesis of (-)-doliculide, structure-activity relationship studies and its binding to F-actin. ChemBioChem 2012, 13, 2537–2548. [Google Scholar] [CrossRef]
- Foerster, F.; Braig, S.; Chen, T.; Altmann, K.H.; Vollmar, A.M. Pharmacological characterization of actin-binding (-)-doliculide. Bioorganic Med. Chem. 2015, 22, 5117–5122. [Google Scholar] [CrossRef]
- Kimura, J.; Takada, Y.; Inayoshi, T.; Nakao, Y.; Goetz, G.; Yoshida, W.Y.; Scheuer, P.J. Kulokekahilide-1, a cytotoxic depsipeptide from the cephalaspidean mollusk Philinopsis speciosa. J. Org. Chem. 2002, 67, 1760–1767. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Negishi, T.; Tashiro, T.; Nakao, Y.; Kimura, J. Structure-related cytotoxic activity of derivatives from kulokekahilide-2, a cyclodepsipeptide in Hawaiian marine mollusk. Bioorganic Med. Chem. Lett. 2012, 22, 7422–7425. [Google Scholar] [CrossRef]
- Umehara, M.; Negishi, T.; Maehara, Y.; Nakao, Y.; Kimura, J. Stereochemical analysis and cytotoxicity of kulokekahilide-2 and its analogues. Tetrahedron 2013, 69, 3045–3053. [Google Scholar] [CrossRef]
- Von Schwarzenberg, K.; Vollmar, A.M. Targeting apoptosis pathways by natural compounds in cancer: Marine compounds as lead structures and chemical tools for cancer therapy. Cancer Lett. 2013, 332, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Martín-Algarra, S.; Espinosa, E.; Rubió, J.; López, J.J.L.; Manzano, J.L.; Carrión, L.A.; Plazaola, A.; Tanovic, A.; Paz-Ares, L. Phase II study of weekly kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer 2009, 45, 732–735. [Google Scholar] [CrossRef]
- Gao, J.; Hamann, M.T. Chemistry and biology of lipids. Chem. Rev. 2009, 53, 35–43. [Google Scholar] [CrossRef]
- El Gamal, A.A. Biological importance of marine algae. Saudi Pharm. J. 2010, 18, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.J.; Liao, X.J.; Xu, S.H.; Diao, J.Z.; Du, B.; Zhou, X.L.; Pan, S.S. Isolation, structure determination, and synthesis of galaxamide, a rare cytotoxic cyclic pentapeptide from a marine algae Galaxaura filamentosa. Org. Lett. 2008, 10, 4569–4572. [Google Scholar] [CrossRef]
- Lunagariya, J.; Liao, X.; Long, W.; Zhong, S.; Bhadja, P.; Li, H.; Zhao, B.; Xu, S. Cytotoxicity study of cyclopentapeptide analogues of marine natural product galaxamide towards human breast cancer cells. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Allkin, B. Useful Plants–Medicines: At Least 28,187 Plant Species are Currently Recorded as Being of Medicinal Use. In State of the World’s Plants 201; Royal Botanic Gardens, Kew: London, UK, 2017. [Google Scholar] [PubMed]
- Wu, D.; Gao, Y.; Qi, Y.; Chen, L.; Ma, Y.; Li, Y. Peptide-based cancer therapy: Opportunity and challenge. Cancer Lett. 2014, 351, 13–22. [Google Scholar] [CrossRef]
- Wélé, A.; Zhang, Y.; Ndoye, I.; Brouard, J.P.; Pousset, J.L.; Bodo, B. A cytotoxic cyclic heptapeptide from the seeds of Annona cherimola. J. Nat. Prod. 2004, 67, 1577–1579. [Google Scholar] [CrossRef]
- Wélé, A.; Ndoye, I.; Zhang, Y.; Brouard, J.P.; Bodo, B. Cherimolacyclopeptide D, a novel cycloheptapeptide from the seeds of Annona cherimola. Phytochemistry 2005, 66, 693–696. [Google Scholar] [CrossRef]
- Wélé, A.; Zhang, Y.; Brouard, J.P.; Pousset, J.L.; Bodo, B. Two cyclopeptides from the seeds of Annona cherimola. Phytochemistry 2005, 66, 2376–2380. [Google Scholar] [CrossRef]
- Idrissa, N.; Adama, D.; Mamadou, B.; Rokhaya, S.G.; Yoro, T. Novel cytotoxic cycloheptapeptide from the latex of Jatropha integerrima. J. Chem. Pharm. Res. 2016, 8, 135–139. [Google Scholar]
- Mongkolvisut, W.; Sutthivaiyakit, S.; Leutbecher, H.; Mika, S.; Klaiber, I.; Möller, W.; Rösner, H.; Beifuss, U.; Conrad, J. Integerrimides A and B, cyclic heptapeptides from the latex of Jatropha integerrima. J. Nat. Prod. 2006, 69, 1435–1441. [Google Scholar] [CrossRef]
- Goyal, A.; Sharma, V.; Upadhyay, N.; Gill, S.; Sihag, M. Flax and flaxseed oil: An ancient medicine & modern functional food. J. Food Sci. Technol. 2014, 51, 1633–1653. [Google Scholar] [CrossRef]
- Okinyo-Owiti, D.P.; Dong, Q.; Ling, B.; Jadhav, P.D.; Bauer, R.; Maley, J.M.; Reaney, M.J.T.; Yang, J.; Sammynaiken, R. Evaluating the cytotoxicity of flaxseed orbitides for potential cancer treatment. Toxicol. Rep. 2015, 2, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Lomchoey, N.; Panseeta, P.; Boonsri, P.; Apiratikul, N.; Prabpai, S.; Kongsaeree, P.; Suksamrarn, S. New bioactive cyclopeptide alkaloids with rare terminal unit from the root bark of: Ziziphus cambodiana. RSC Adv. 2018, 8, 18204–18215. [Google Scholar] [CrossRef]
- Hsieh, P.-W.; Chang, F.-R.; Wu, C.-C.; Li, C.-M.; Wu, K.-Y.; Chen, S.-L.; Yen, H.-F.; Wu, Y.-C. Longicalycinin A, a New cytotoxic cyclic peptide from Dianthus superbus var. longicalycinus (MAXIM.) WILL. Chem. Pharm. Bull. 2005, 53, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W. Solid-phase total synthesis of cyclic pentapeptide longicalycinin A, by using 2-chlorotrityl chloride resin. J. Cancer Res. Exp. Oncol. 2014, 5, 8–19. [Google Scholar] [CrossRef]
- HoushdarTehrani, M.H.; Bamoniri, A.; Mirjalili, B.B.F.; Gholibeikian, M. Synthesis of linear and cyclic disulfide heptapeptides of longicalycinin a and evaluation of toxicity on cancerous cells HepG2 and HT-29. Iran. J. Pharm. Res. 2018, 17, 956–963. [Google Scholar] [PubMed]
- Hsieh, P.W.; Chang, F.R.; Wu, C.C.; Wu, K.Y.; Li, C.M.; Chen, S.L.; Wu, Y.C. New cytotoxic cyclic peptides and dianthramide from Dianthus superbus. J. Nat. Prod. 2004, 67, 1522–1527. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Young, L.W.; Arnison, P.G.; Gilding, E.; Reaney, M.J.T. Proposed systematic nomenclature for orbitides. J. Nat. Prod. 2015, 78, 645–652. [Google Scholar] [CrossRef]
- Beirigo, P.J.S.D.; Torquato, H.F.V.; Santos, C.H.C.D.; de Carvalho, M.G.; Castro, R.N.; Paredes-Gamero, E.J.; de Sousa, P.T.; Jacinto, M.J.; da Silva, V.C. [1-8-NαlC]-Zanriorb A1, a proapoptotic orbitide from leaves of Zanthoxylum riedelianum. J. Nat. Prod. 2016, 79, 1454–1458. [Google Scholar] [CrossRef]
- Lv, J.P.; Yang, S.; Dong, J.X.; Jin, H. New cyclopeptide alkaloids from the whole plant of Justicia procumbens L. Nat. Prod. Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, J.; Xiao, S.; Wang, D. Structural characterization of ginseng cyclopeptides and detection of capability to induce apoptosis in gastrointestinal cancer cells. RSC Adv. 2019, 9, 29847–29855. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Cemazar, M.; Wang, C.K.L.; Daly, N.L. The cyclotide family of circular miniproteins: Nature’s combinatorial peptide teplate. Biopolymer 2006, 84, 250–266. [Google Scholar] [CrossRef]
- Zhao, S.M.; Kuang, B.; Fan, J.T.; Yan, H.; Xu, W.Y.; Tan, N.H. Antitumor cyclic hexapeptides from Rubia plants: History, chemistry, and mechanism (2005–2011). Chim. Int. J. Chem. 2011, 65, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, P.O.; Matos, M.D.F.C.; Perdomo, R.T.; Kato, W.H.; Barros, M.V.G.O.; Garcez, F.R.; Garcez, W.S. Rubiaceae-type cyclopeptides from Galianthe thalictroides. J. Nat. Prod. 2016, 79, 1165–1169. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, S.M.; Zhao, L.M.; Chen, X.Q.; Zeng, G.Z.; Tan, N.H. Rubipodanin A, the first natural ndesmonomethyl rubiaceae-type cyclopeptide from rubia podantha, indicating an important role of the n9- methyl group in the conformation and bioactivity. PLoS ONE 2015, 10, e0144950. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Feng, L.; Wang, J.; Zhang, X.J.; Wang, Z.; Tan, N.H. Rubipodanin B, a new cytotoxic cyclopeptide from Rubia podantha. Chem. Biodivers. 2019, 16, 16–21. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, D.; He, J.; Song, L.; Chen, H.; Zhang, Z.; Tan, N. Inhibition of fatty acid synthesis arrests colorectal neoplasm growth and metastasis: Anti-cancer therapeutical effects of natural cyclopeptide RA-XII. Biochem. Biophys. Res. Commun. 2019, 512, 819–824. [Google Scholar] [CrossRef]
- Leung, H.W.; Zhao, S.M.; Yue, G.G.L.; Lee, J.K.M.; Fung, K.P.; Leung, P.C.; Tan, N.H.; Lau, C.B.S. RA-XII inhibits tumour growth and metastasis in breast tumour-bearing mice via reducing cell adhesion and invasion and promoting matrix degradation. Sci. Rep. 2015, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, Z.; Wang, Y.; Guo, D.; Yang, J.; Chen, L.; Tan, N. Natural cyclopeptide RA-XII, a new autophagy inhibitor, suppresses protective autophagy for enhancing apoptosis through AMPK/mTOR/P70S6K pathways in HepG2 Cells. Molecules 2017, 22, 1934. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Li, L.; Feng, L.; Wang, Y.R.; Wang, Z.; Tan, N.H. RA-XII, a bicyclic hexapeptidic glucoside isolated from Rubia yunnanensis Diels, exerts antitumor activity by inhibiting protective autophagy and activating Akt-mTOR pathway in colorectal cancer cells. J. Ethnopharmacol. 2021, 266, 113438. [Google Scholar] [CrossRef] [PubMed]
- Hitotsuyanagi, Y.; Hirai, M.; Odagiri, M.; Komine, M.; Hasuda, T.; Fukaya, H.; Takeya, K. RA-XXV and RA-XXVI, Bicyclic hexapeptides from Rubia cordifolia L.: Structure, synthesis, and conformation. Chem. Asian J. 2019, 14, 205–215. [Google Scholar] [CrossRef]
- Ding, X.; Bai, D.; Qian, J. Novel cyclotides from Hedyotis biflora inhibit proliferation and migration of pancreatic cancer cell in vitro and in vivo. Med. Chem. Res. 2014, 23, 1406–1413. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Zhang, S.; Nguyen, N.T.K.; Nguyen, P.Q.T.; Chiu, M.S.; Hardjojo, A.; Tam, J.P. Discovery and characterization of novel cyclotides originated from chimeric precursors consisting of albumin-1 chain a and cyclotide domains in the Fabaceae family. J. Biol. Chem. 2011, 286, 24275–24287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiao, K.Z.; Jin, J.; Zhang, Y.; Zhou, W. Chemosensitizing activities of cyclotides from clitoria ternatea in paclitaxel-resistant lung cancer cells. Oncol. Lett. 2013, 5, 641–644. [Google Scholar] [CrossRef]
- Gerlach, U.; Burman, S.L.; Debasis Monda, R.; Goransson, D. Characterization and bioactivity of cyclotides from Psychotria leptothyrsa (Rubiaceae). J. Nat. Prod. 2010, 73, 1207–1213. [Google Scholar] [CrossRef]
- Hashempour, H.; Koehbach, J.; Daly, N.L.; Ghassempour, A.; Gruber, C.W. Characterizing circular peptides in mixtures: Sequence fragment assembly of cyclotides from a violet plant by MALDI-TOF/TOF mass spectrometry. Amino Acids. 2013, 44, 581–595. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Abagheri-Mahabadi, N.; Hashempour, H.; Farhadpour, M.; Gruber, C.W.; Ghassempour, A. Viola plant cyclotide vigno 5 induces mitochondria-mediated apoptosis via cytochrome C release and caspases activation in cervical cancer cells. Fitoterapia 2016, 109, 162–168. [Google Scholar] [CrossRef]
- Herrmann, A.; Svangård, E.; Claeson, P.; Gullbo, J.; Bohlin, L.; Göransson, U. Key role of glutamic acid for the cytotoxic activity of the cyclotide cycloviolacin O2. Cell. Mol. Life Sci. 2006, 63, 235–245. [Google Scholar] [CrossRef]
- Burman, R.; Svedlund, E.; Felth, J.; Hassan, S.; Herrmann, A.; Clark, R.J.; Craik, D.J.; Bohlin, L.; Claeson, P.; Göransson, U.; et al. Evaluation of toxicity and antitumor activity of cycloviolacin O2 in mice. Biopolymers 2010, 94, 626–634. [Google Scholar] [CrossRef]
- Svangård, E.; Burman, R.; Gunasekera, S.; Lövborg, H.; Gullbo, J.; Göransson, U. Mechanism of action of cytotoxic cyclotides: Cycloviolacin O2 disrupts lipid membranes. J. Nat. Prod. 2007, 70, 643–647. [Google Scholar] [CrossRef]
- Gerlach, S.L.; Rathinakumar, R.; Chakravarty, G.; Göransson, U.; Wimley, W.C.; Darwin, S.P.; Mondal, D. Anticancer and chemosensitizing abilities of cycloviolacin 02 from Viola odorata and psyle cyclotides from Psychotria leptothyrsa. Biopolymers 2010, 94, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, S.L.; Yeshak, M.; Göransson, U.; Roy, U.; Izadpanah, R.; Mondal, D. Cycloviolacin O2 (CyO2) suppresses productive infection and augments the antiviral efficacy of nelfinavir in HIV-1 infected monocytic cells. Biopolymers 2013, 100, 471–479. [Google Scholar] [CrossRef]
- He, W.; Yue, L.; Zeng, G.; Daly, N.L.; Craik, D.J.; Tan, N. Peptides Isolation and characterization of cytotoxic cyclotides from Viola philippica. Peptides 2011, 32, 1719–1723. [Google Scholar] [CrossRef]
- Yeshak, M.Y.; Burman, R.; Asres, K.; Göransson, U. Cyclotides from an extreme habitat: Characterization of cyclic peptides from Viola abyssinica of the Ethiopian highlands. J. Nat. Prod. 2011, 74, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Burman, R.; Mylne, J.S.; Karlsson, G.; Gullbo, J.; Craik, D.J.; Clark, R.J.; Göransson, U. The alpine violet, Viola biflora, is a rich source of cyclotides with potent cytotoxicity. Phytochemistry 2008, 69, 939–952. [Google Scholar] [CrossRef]
- Svangård, E.; Göransson, U.; Hocaoglu, Z.; Gullbo, J.; Larsson, R.; Claeson, P.; Bohlin, L. Cytotoxic cyclotides from Viola tricolor. J. Nat. Prod. 2004, 67, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.; Wang, C.K.; Pan, X.; Yan, H.; Zeng, G.; Xu, W.; He, W.; Chan, L.Y.; Zeng, G.; Daly, N.L.; et al. Isolation and characterization of cytotoxic cyclotides from Viola tricolor. Peptides 2010, 32, 1719–1723. [Google Scholar] [CrossRef]
- Karoum, F.; Commissiong, J.W.; Neff, N.H.; Wyatt, R.J. Biochemical evidence for uncrossed and crossed locus coeruleus projections to the spinal cord. Brain Res. 1980, 196, 237–241. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. NeuroSci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Masuoka, Y.; Nagai, A.; Shin-ya, K.; Furihata, K.; Nagai, K.; Suzuki, K.; Hayakawa, Y.; Seto, H. Spiruchostatins A and B, novel gene expression-enhancing substances produced by Pseudomonas sp. Tetrahedron Lett. 2001, 42, 41–44. [Google Scholar] [CrossRef]
- Shindoh, N.; Mori, M.; Terada, Y.; Oda, K.; Amino, N.; Kita, A.; Taniguchi, M.; Sohda, K.Y.; Nagai, K.; Sowa, Y.; et al. YM753, a novel histone deacetylase inhibitor, exhibits antitumor activity with selective, sustained accumulation of acetylated histones in tumors in the WiDr xenograft model. Int. J. Oncol. 2008, 32, 545–555. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toriyama, S.; Horinaka, M.; Yasuda, S.; Taniguchi, T.; Aono, Y.; Takamura, T.; Morioka, Y.; Miki, T.; Ukimura, O.; Sakai, T. A histone deacetylase inhibitor, OBP-801, and celecoxib synergistically inhibit the cell growth with apoptosis via a DR5-dependent pathway in bladder cancer cells. Mol. Cancer Ther. 2016, 15, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Takamura, T.; Horinaka, M.; Yasuda, S.; Toriyama, S.; Aono, Y.; Sowa, Y.; Miki, T.; Ukimura, O.; Sakai, T. FGFR inhibitor BGJ398 and HDAC inhibitor OBP-801 synergistically inhibit cell growth and induce apoptosis in bladder cancer cells. Oncol. Rep. 2018, 39, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K. Telomestatin, a Novel Telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef]
- Liu, W.; Sun, D.; Hurley, L.H. Binding of G-quadruplex-interactive agents to distinct G-quadruplexes induces different biological effects in MiaPaCa cells. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1801–1815. [Google Scholar] [CrossRef]
- Binz, N.; Shalaby, T.; Rivera, P.; Shin-Ya, K.; Grotzer, M.A. Telomerase inhibition, telomere shortening, cell growth suppression and induction of apoptosis by telomestatin in childhood neuroblastoma cells. Eur. J. Cancer 2005, 41, 2873–2881. [Google Scholar] [CrossRef]
- Amagai, K.; Ikeda, H.; Hashimoto, J.; Kozone, I.; Izumikawa, M.; Kudo, F.; Eguchi, T.; Nakamura, T.; Osada, H.; Takahashi, S.; et al. Identification of a gene cluster for telomestatin biosynthesis and heterologous expression using a specific promoter in a clean host. Sci. Rep. 2017, 7, 3382. [Google Scholar] [CrossRef]
- Arndt, G.M.; MacKenzie, K.L. New prospects for targeting telomerase beyond the telomere. Nat. Rev. Cancer. 2016, 16, 508–524. [Google Scholar] [CrossRef]
- Doi, T.; Yoshida, M.; Shin-ya, K.; Takahashi, T. Total synthesis of (R)-telomestatin. Org. Lett. 2006, 8, 4165–4167. [Google Scholar] [CrossRef]
- Ducklow, H.W. Bacterioplankton. Encycl. Ocean. Sci. 2001, 217–224. [Google Scholar] [CrossRef]
- Joseph, A.; Guedes, A.C.; Malcata, F.X. Biotechnological and pharmacological applications of biotoxins and other bioactive molecules from dinoflagellates. Mar. Drug. 2017, 15, 393. [Google Scholar] [CrossRef]
- Lombó, F.; Velasco, A.; Castro, A.; de la Calle, F.; Braña, A.F.; Sánchez-Puelles, J.M.; Méndez, C.; Salas, J.A. Deciphering the biosynthesis pathway of the antitumor thiocoraline from a marine actinomycete and its expression in two Streptomyces species. ChemBioChem 2006, 7, 366–376. [Google Scholar] [CrossRef]
- Romero, F.; Espliego, F.; Baz, J.P.; de Quesada, T.G.; Grávalos, D.; la Calle, F.D.; Fernández-Puentes, J.L. Thiocoraline, a New depsipeptide with antitumor activity produced by a marine Micromonospora. I. taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Erba, E.; Bergamaschi, D.; Ronzoni, S.; Faretta, M.; Taverna, S.; Bonfanti, M.; Catapano, C.V.; Faircloth, G.; Jimeno, J.; D’Incalci, M. Mode of action at thiocoraline, a natural marine compound with anti-tumour activity. Br. J. Cancer. 1999, 80, 971–980. [Google Scholar] [CrossRef]
- Sohn, J.A.; Zarebczan, B.; Wyche, T.P.; Bugni, T.S.; Kunnimalaiyaan, M.; Jaskula-Sztul, R.; Chen, H. Thiocoraline regulates neuroendocrine phenotype and inhibits proliferation in carcinoid tumor cells. J. Surg. Res. 2012, 172, 237. [Google Scholar] [CrossRef]
- Tesfazghi, S.; Eide, J.; Dammalapati, A.; Korlesky, C.; Wyche, T.P.; Bugni, T.S.; Chen, H.; Jaskula-Sztul, R. Thiocoraline alters neuroendocrine phenotype and activates the Notch pathway in MTC-TT cell line. Cancer Med. 2013, 2, 734–743. [Google Scholar] [CrossRef]
- Um, S.; Choi, T.J.; Kim, H.; Kim, B.Y.; Kim, S.H.; Lee, S.K.; Oh, K.B.; Shin, J.; Oh, D.C. Ohmyungsamycins A and B: Cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Jang, J.; Sim, J.; Son, W.S.; Ahn, H.C.; Kim, T.S.; Shin, Y.H.; Lim, C.; Lee, S.; An, H.; et al. Conformation-enabled total syntheses of ohmyungsamycins A and B and structural revision of ohmyungsamycin B. Angew. Chem. Int. Ed. 2018, 57, 3069–3073. [Google Scholar] [CrossRef]
- Asolkar, R.N.; Freel, K.C.; Jensen, P.R.; Fenical, W.; Kondratyuk, E.-J.; Park, T.P.; Pezzuto, J.M. Arenamides A-C, cytotoxic NF-κB inhibitors from the marine Actinomycete salinispora. J. Nat. Prod. 2009, 72, 396–402. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Pavankumarreddy, G.; Sathish, K. Total synthesis of arenamide A and its diastereomer. Tetrahedron Lett. 2009, 50, 6851–6854. [Google Scholar] [CrossRef]
- Miller, E.D.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Piperazimycins: Cytotoxic hexadepsipeptides from a marine-derived bacterium of the genus Streptomyces. J. Org. Chem. 2007, 72, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gan, J.; Ma, D. Total synthesis of piperazimycin a: A cytotoxic cyclic hexadepsipeptide. Angew. Chem. Int. Ed. 2009, 48, 8891–8895. [Google Scholar] [CrossRef]
- Harrigan, G.G.; Harrigan, B.L.; Davidson, B.S. Kailuins A-D, new cyclic acyldepsipeptides from cultures of a marine-derived bacterium. Tetrahedron 1997, 53, 1577–1582. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hua, H.M.; Pei, Y.H.; Yao, X.S. Three new cytotoxic cyclic acylpeptides from marine Bacillus sp. Chem. Pharm. Bull. 2004, 52, 1029–1030. [Google Scholar] [CrossRef] [PubMed]
- Rehman, N.U.; Abed, R.M.M.; Hussain, H.; Khan, H.Y.; Khan, A.; Khan, A.L.; Ali, M.; Al-Nasri, A.; Al-Harrasi, K.; Al-Rawahi, A.N.; et al. Anti-proliferative potential of cyclotetrapeptides from Bacillus velezensis RA5401 and their molecular docking on G-protein-coupled receptors. Microb. Pathog. 2018, 123, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; He, Y.; Tian, Y.; Cong, B.; Yang, H. Bacilohydrin A, a new cytotoxic cyclic lipopeptide of surfactins class produced by bacillus sp. sy27f from the indian ocean hydrothermal vent. Nat. Prod. Commun. 2019, 14, 141–146. [Google Scholar] [CrossRef]
- Liu, X.d.; Gu, K.b.; Xia, S.S.; Zhang, D.J.; Li, Y.G. Dolyemycins A and B, two novel cyclopeptides isolated from Streptomyces griseus subsp. griseus HYS31. J. Antibiot. 2018, 71, 838–845. [Google Scholar] [CrossRef]
- Um, S.; Kim, Y.J.; Kwon, H.; Wen, H.; Kim, S.H.; Kwon, H.C.; Park, S.; Shin, J.; Oh, D.C. Sungsanpin, a lasso peptide from a deep-sea Streptomycete. J. Nat. Prod. 2013, 76, 873–879. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, H.; Chen, Y.; Tan, J.; Song, Y.; Zou, J.; Tian, X.; Hua, Y.; Ju, J. Marthiapeptide A, an anti-infective and cytotoxic polythiazole cyclopeptide from a 60 L scale fermentation of the deep sea-derived Marinactinospora thermotolerans SCSIO 00652. J. Nat. Prod. 2012, 75, 2251–2255. [Google Scholar] [CrossRef]
- Zhang, Y.; Islam, M.A.; McAlpine, S.R. Synthesis of the natural product marthiapeptide A. Org. Lett. 2015, 17, 5149–5151. [Google Scholar] [CrossRef]
- Kanoh, K.; Matsuo, Y.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Mechercharmycins A and B, cytotoxic substances from marine-derived Thermoactinomyces sp. YM3-251. J. Antibiot. 2005, 58, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Hernández, D.; Altuna, M.; Cuevas, C.; Aligué, R.; Albericio, F.; Álvarez, M. Synthesis and activity of cytotoxic mechercharmycin A analogs. J. Med. Chem. 2008, 51, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kanoh, K.; Imagawa, H.; Adachi, K.; Nishizawa, M.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542: II. Physico-chemical properties and structural elucidation. J. Antibiot. 2007, 60, 256–260. [Google Scholar] [CrossRef]
- Schwenk, S.; Ronco, C.; Oberheide, A.; Arndt, H.-D. Biomimetic synthesis of urukthapelstatin A by Aza-Wittig ring contraction. J. Subst. Abuse Treat. 2016, 13, 287–288. [Google Scholar] [CrossRef]
- Pan, C.M.; Lin, C.C.; Kim, S.J.; Sellers, R.P.; McAlpine, S.R. Progress toward the synthesis of urukthapelstatin A and two analogues. Tetrahedron Lett. 2012, 53, 4065–4069. [Google Scholar] [CrossRef]
- Lin, C.; Tantisantisom, W.; Mcalpine, S.R. Total synthesis and biological activity of natural product urukthapelstatin A. Org. Lett. 2013, 15, 3574–3577. [Google Scholar] [CrossRef]
- Hamilton, T.L.; Bryant, D.A.; Macalady, J.L. The role of biology in planetary evolution: Cyanobacterial primary production in low-oxygen Proterozoic oceans. Environ. Microbiol. 2016, 18, 325–340. [Google Scholar] [CrossRef]
- Sainis, I.; Fokas, D.; Vareli, K.; Tzakos, A.G.; Kounnis, V.; Briasoulis, E. Cyanobacterial cyclopeptides as lead compounds to novel targeted cancer drugs. Mar. Drugs 2010, 8, 629–657. [Google Scholar] [CrossRef]
- Janssen, E.M. Cyanobacterial peptides beyond microcystins e A review on co-occurrence, toxicity, and challenges for risk assessment. Water Res. 2019, 151, 488–499. [Google Scholar] [CrossRef]
- Falconer, I.R. Health Risk Assessment of Cyanobacterial (Blue-green Algal) Toxins in health risk assessment of cyanobacterial (blue-green algal) toxins in drinking water. Int. J. Environ. Res. Public Health 2005, 2, 43–50. [Google Scholar] [CrossRef]
- Dawson, R.M. The toxicology of microcystins. Toxicon 1998, 36, 953–962. [Google Scholar] [CrossRef]
- Lone, Y.; Bhide, M.; Koiri, R.K. Microcystin-LR induced immunotoxicity in mammals. J. Toxicol. 2016, 2016. [Google Scholar] [CrossRef]
- Piyathilaka, M.A.P.C.; Pathmalal, M.M.; Tennekoon, K.H.; de Silva, B.G.D.N.K.; Samarakoon, S.R.; Chanthirika, S. Microcystin-LR-induced cytotoxicity and apoptosis in human embryonic kidney and human kidney adenocarcinoma cell lines. Microbiology 2015, 161, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Physiology, C. Chronic microcystin-LR exposure induces hepatocarcinogenesis via increased gankyrin in vitro and in vivo. Cell. Physiol. Biochem. 2018, 49, 1420–1430. [Google Scholar] [CrossRef]
- Kounnis, V.; Chondrogiannis, G.; Mantzaris, M.D.; Tzakos, A.G.; Fokas, D.; Papanikolaou, N.A.; Galani, V.; Sainis, I.; Briasoulis, E. Microcystin lr shows cytotoxic activity against pancreatic cancer cells expressing the membrane OATP1B1 and OATP1B3 transporters. Anticancer Res. 2015, 35, 5857–5865. [Google Scholar] [PubMed]
- Niedermeyer, T.H.J.; Daily, A.; Swiatecka-hagenbruch, M.; Moscow, J.A. Selectivity and potency of microcystin congeners against OATP1B1 and OATP1B3 expressing cancer cells. PLoS ONE 2014, 9, e91476. [Google Scholar] [CrossRef]
- Monks, N.R.; Moscow, J.A. Microcystins as Agents for Treatment of Cancer. U.S. Patent US9006173B2, 14 April 2015. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Belmonte, J.C.I.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Law, B.K.; Luesch, H. Apratoxin A aeversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 2009, 76, 91–104. [Google Scholar] [CrossRef]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin Kills Cells by Direct Blockade of the Sec61 protein translocation channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.-M.; et al. Apratoxin A shows novel pancreas-targeting activity through the binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T. Apratoxin D, a potent cytotoxic cyclodepsipeptide from Papua New Guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef]
- Matthew, S.; Schupp, P.J.; Luesch, H. Apratoxin E, a Cytotoxic peptolide from a Guamanian collection of the marine cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [Google Scholar] [CrossRef]
- Tidgewell, K.; Engene, N.; Byrum, T.; Media, J.; Doi, T.; Valeriote, F.A.; Gerwick, W.H. Evolved diversification of a modular natural product pathway: Apratoxins F and G, two cytotoxic cyclic depsipeptides from a Palmyra collection of Lyngbya bouillonii. ChemBioChem 2012, 11, 1458–1466. [Google Scholar] [CrossRef]
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Kurokawa, R.; Arai, D.; Kanto, K.K.; Okino, T.; Nakao, Y.; Kushiro, T.; Yoshida, M.; Matsumoto, K. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci. Rep. 2017, 7, 2002. [Google Scholar] [CrossRef]
- Huang, X.; Huang, W.; Li, L.; Sun, X.; Song, S.; Xu, Q.; Zhang, L.; Wei, B.; Deng, X. Structure determinants of lagunamide A for anticancer activity and its molecular mechanism of mitochondrial apoptosis. Mol. Pharm. 2016, 13, 3756–3763. [Google Scholar] [CrossRef]
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Ping, K.; Chen, D.Y.; Tong, L. Phytochemistry lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef]
- Davies-Coleman, M.T.; Dzeha, T.M.; Gray, C.A.; Hess, S.; Pannell, L.K.; Hendricks, D.T.; Arendse, C.E. Isolation of homodolastatin 16, a new cyclic depsipeptide from a Kenyan collection of Lyngbya majuscula. J. Nat. Prod. 2003, 66, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Mooberry, S.L.; Yoshida, W.Y.; Luesch, H.; Paul, V.J. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2002, 63, 611–615. [Google Scholar] [CrossRef]
- Yokokawa, F.; Sameshima, H.; Shioiri, T. Total synthesis of lyngbyabellin A, a potent cytotoxic metabolite from the marine cyanobacterium Lyngbya majuscula. Tetrahedron Lett. 2001, 63, 1440–1443. [Google Scholar] [CrossRef]
- Gogineni, V.; Hamann, M.T. Marine natural product peptides with therapeutic potential: Chemistry, biosynthesis, and pharmacology. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 81–196. [Google Scholar] [CrossRef]
- Choi, H.; Mevers, E.; Byrum, T.; Valeriote, F.A.; Gerwick, W.H. Lyngbyabellins K-N from two Palmyra atoll collections of the marine cyanobacterium Moorea bouillonii. Eur. J. Org. Chem. 2012, 2012, 5141–5150. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nunnery, J.K.; Engene, N.; Esquenazi, E.; Byrum, T.; Dorrestein, P.C.; Gerwick, W.H. Palmyramide A, a Cyclic depsipeptide from a Palmyra atoll collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two Papua New Guinea cyanobacteria. Chem. Biol. 2009, 16, 893–906. [Google Scholar] [CrossRef]
- Mangoni, A.; Shuman, C.F.; Engene, N.; Pereira, A.R.; Gerwick, W.H.; Cao, Z.; Murray, T.F.; Byrum, T.; Choi, H.; Matainaho, T. The Hoiamides, structurally intriguing neurotoxic lipopeptides from Papua New Guinea marine cyanobacteria. J. Nat. Prod. 2010, 73, 1411–1421. [Google Scholar] [CrossRef]
- Cao, Z.; Li, X.; Zou, X.; Greenwood, M.; Gerwick, W.H.; Murray, T.F. Involvement of JNK and caspase activation in hoiamide A-induced neurotoxicity in neocortical neurons. Mar. Drugs 2015, 13, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Nogle, L.M.; Marquez, B.L.; Gerwick, W.H. Wewakazole, a novel cyclic dodecapeptide from a papua new guinea Lyngbya majuscula. Org. Lett. 2003, 5, 3–6. [Google Scholar] [CrossRef]
- Lopez, J.A.V.; Al-Lihaibi, S.S.; Alarif, W.M.; Abdel-Lateff, A.; Nogata, Y.; Washio, K.; Morikawa, M.; Okino, T. Wewakazole B, a cytotoxic cyanobactin from the cyanobacterium Moorea producens collected in the Red Sea. J. Nat. Prod. 2016, 79, 1213–1218. [Google Scholar] [CrossRef]
- Nayani, K.; Hussaini, S.D.A. Synthesis of cytotoxic cyanobactin, Wewakazole B. Tetrahedron Lett. 2017, 58, 1166–1169. [Google Scholar] [CrossRef]
- Inman, M.; Dexter, H.L.; Moody, C.J. Total synthesis of the cyclic dodecapeptides wewakazole and wewakazole B. Org. Lett. 2017, 19, 3454–3457. [Google Scholar] [CrossRef]
- Marquez, B.L.; Watts, K.S.; Yokochi, A.; Roberts, M.A.; Verdier-Pinard, P.; Jimenez, J.I.; Hamel, E.; Scheuer, P.J.; Gerwick, W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002, 65, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Suntornchashwej, S.; Chaichit, N.; Isobe, M.; Suwanborirux, K. Hectochlorin and morpholine derivatives from the Thai sea hare, Bursatella leachii. J. Nat. Prod. 2005, 68, 951–955. [Google Scholar] [CrossRef]
- Ramaswamy, A.V.; Sorrels, C.M.; Gerwick, W.H. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2007, 70, 1977–1986. [Google Scholar] [CrossRef]
- Cetusic, J.R.P.; Green, F.R.; Graupner, P.R.; Oliver, M.P. Total synthesis of hectochlorin. Org. Lett. 2002, 4, 1307–1310. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Nogle, L.M.; Media, J.; Valeriote, F.A.; Mooberry, S.L.; Gerwick, W.H. Desmethoxymajusculamide C, a cyanobacterial depsipeptide with potent cytotoxicity in both cyclic and ring-opened forms. J. Nat. Prod. 2009, 72, 1011–1016. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.F.; Lik, T.T. Hantupeptin A, a cytotoxic cyclic depsipeptide from a Singapore collection of Lyngbya majuscula. J. Nat. Prod. 2009, 72, 29–32. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.F.; Tan, L.T. Hantupeptins B and C, cytotoxic cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2010, 71, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, I.; Rolland, M.; Francisco, C.; Banaigs, B. Total structure and biological properties of laxaphycins A and B, cyclic lipopeptides from the marine cyanobacterium Lyngbya majuscula. Int. J. Pept. Res. Ther. 1997, 4, 289–292. [Google Scholar] [CrossRef]
- Bonnard, I.; Rolland, M.; Salmon, J.M.; Debiton, E.; Barthomeuf, C.; Banaigs, B. Total structure and inhibition of tumor cell proliferation of laxaphycins. J. Med. Chem. 2007, 50, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Matthew, S.; Chen, Q.Y.; Paul, V.J.; Luesch, H. Discovery of new A- and B-type laxaphycins with synergistic anticancer activity. Bioorganic Med. Chem. 2018, 26, 2310–2319. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure determination of obyanamide, a novel cytotoxic cyclic depsipeptide from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2002, 65, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ding, N.; Li, Y. Synthesis and biological evaluation of analogues of the marine cyclic depsipeptide obyanamide. J. Pept. Sci. 2011, 17, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Rocca, J.R.; Abboud, K.A.; Paul, V.J.; Luesch, H. Total structure determination of grassypetolide, a new marine cyanobacterical cytotoxin. Org. Lett. 2008, 10, 789–792. [Google Scholar] [CrossRef]
- Kwan, J.C.; Ratnayake, R.; Abboud, K.A.; Paul, V.J.; Luesch, H. Grassypeptolides A-C, cytotoxic bis-thiazoline containing marine cyclodepsipeptides. J. Org. Chem. 2010, 75, 8012–8023. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Wang, Z.; Xing, X.; Maguire, A.R.; Luesch, H.; Zhang, H.; Xu, Z.; Ye, T. Total synthesis and biological evaluation of grassypeptolide A. Chem. A Eur. J. 2013, 19, 6774–6784. [Google Scholar] [CrossRef]
- Thornburg, C.C.; Thimmaiah, M.; Shaala, L.A.; Hau, A.M.; Malmo, J.M.; Ishmael, J.E.; Youssef, D.T.A.; McPhail, K.L. Cyclic Depsipeptides, grassypeptolides D and E and Ibu-epidemethoxylyngbyastatin 3, from a Red Sea Leptolyngbya cyanobacterium. J. Nat. Prod. 2011, 74, 1677–1685. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Quon, M.K.; Moore, R.E.; Paul, V.J. The Structure of Palau’ amide, a potent cytotoxin from a species of the marine cyanobacterium Lyngbya. J. Nat. Prod. 2003, 66, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Watanabe, A.; Teruya, T.; Suenaga, K. Synthesis of Palau’amide and its diastereomers: Confirmation of its stereostructure. Tetrahedron Lett. 2009, 50, 7343–7345. [Google Scholar] [CrossRef]
- Zou, B.; Long, K.; Ma, D. Total synthesis and cytotoxicity studies of a cyclic depsipeptide with proposed structure of Palau’ amide. Org. Lett. 2005, 7, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef]
- Hau, A.M.; Greenwood, J.A.; Löhr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [PubMed]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Investig. New Drugs. 2016, 34, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A-F, new β-amino acid-containing cyclodepsipeptides from Palauan collections of the marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000. [Google Scholar] [CrossRef]
- Alvarado, C.; Hernández, G.; Díaz, E.; Soano, J.D.; Vilchis-Reyes, M.A.; Martínez-Urbina, M.A.; Guzmán, A. Synthesis of O-Me ulongamide B and O-Me ulongamide C, natural modified cyclodepsipeptides. Synth. Commun. 2013, 43. [Google Scholar] [CrossRef]
- Alvarado, C.; Díaz, E.; Guzmán, Á. Total synthesis of ulongamide A, a cyclic depsipeptide isolated from marine cyanobacteria Lyngbya sp. Tetrahedron Lett. 2007, 48, 603–607. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Quon, M.K.; Moore, R.E.; Paul, V.J. Ulongapeptin, a Cytotoxic cyclic depsipeptide from a Palauan marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2003, 4, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Kaneda, M.; Kawaguchi, S.; Fujii, N.; Ohno, H.; Oishi, S. Structure–activity relationship study on odoamide: Insights into the bioactivities of aurilide-family hybrid peptide–polyketides. ACS Med. Chem. Lett. 2018, 9, 365–369. [Google Scholar] [CrossRef]
- Shih, C.; Teicher, B.A. Cryptophycins: A novel class of potent antimitotic antitumor depsipeptides. Curr. Pharm. Des. 2001, 7, 1259–1276. [Google Scholar] [CrossRef]
- Georg, G.I. Halohydrin analogues of cryptophycin 1: Synthesis and biological activity. Bioorg. Med. Chem. Lett. 1998, 8, 1959–1962. [Google Scholar] [CrossRef]
- Cells, D.; Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.L.; Moore, R.E. Cryptophycin: A new antimicrotubule agent active against drug-resistant cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar]
- Panda, D.; Himes, R.H.; Moore, R.E.; Wilson, L.; Jordan, M.A. Mechanism of action of the unusually potent microtubule inhibitor. Biochemistry 1997, 21, 12948–12953. [Google Scholar] [CrossRef] [PubMed]
- Kerksiek, K.; Mejillano, M.R.; Schwartz, R.E.; Georg, G.I.; Himes, R.H. Interaction of cryptophycin 1 with tubulin and microtubules. FEBS Lett. 1995, 377, 59–61. [Google Scholar] [CrossRef]
- Bai, R.; Schwartz, R.E.; Kepler, J.A.; Pettit, G.R.; Hamel, E. Characterization of the interaction of cryptophycin 1 with tubulin: Binding in the vinca domain, competitive inhibition of dolastatin 10 binding, and an unusual aggregation reaction. Cancer Res. 1996, 56, 4398–4406. [Google Scholar] [PubMed]
- Brenda, J.; Richard, A.; Frederick, A. Cryptophycin 1 cellular levels and effects in vitro using L1210 cells. Investig. New. Drugs 1999, 16, 199–204. [Google Scholar]
- Ilson, L.E.W. Antiproliferative mechanism of action of cryptophycin-52: Kinetic stabilization of microtubule dynamics by high-affinity binding to microtubule ends. Proc. Natl. Acad. Sci. USA 1998, 95, 9313–9318. [Google Scholar] [CrossRef]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Bradner, J.E.; Schreiber, S.L.; West, N.; Williams, R.M.; Taunton, J.; Bowers, A. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J. Total synthesis and biological evaluation of largazole and derivatives. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef]
- Byeon, S.; Law, B.K.; Liu, Y.; Hong, J.; Salvador, L.A.; Ying, Y.; Kwan, J.C.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef]
- Wang, W.; Parker, S.J.; Zhang, G.; Kuchta, R.D.; Ungermannova, D.; Nasveschuk, C.G.; Quade, B.; Liu, X.; Phillips, A.J. Largazole and its derivatives selectively inhibit ubiquitin activating enzyme (E1). PLoS ONE 2012, 7, e29208. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide and protease inhibitor symplocamide A. Chem. Commun. 2010, 46, 8857–8859. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Tasipeptins A and B: New cytotoxic depsipeptides from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2003, 66, 620–624. [Google Scholar] [CrossRef]
- Köcher, S.; Rey, J.; Bongard, J.; Tiaden, A.N.; Meltzer, M.; Richards, P.J.; Ehrmann, M.; Kaiser, M. Tailored Ahp-cyclodepsipeptides as potent non-covalent serine protease inhibitors. Angew. Chem. Int. Ed. 2017, 56, 8555–8558. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Biggs, J.S.; Paul, V.J.; Luesch, H. Veraguamides A-G, cyclic hexadepsipeptides from a dolastatin 16-producing cyanobacterium Symploca cf. hydnoides from Guam. J. Nat. Prod. 2011, 74, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Sabry, O.M.; Goeger, D.E.; Gerwick, W.H. Biologically active new metabolites from a Florida collection of Moorea producens. Nat. Prod. Res. 2017, 25, 555–561. [Google Scholar] [CrossRef]
- Boudreau, P.D.; Byrum, T.; Liu, W.T.; Dorrestein, P.C.; Gerwick, W.H. Viequeamide a, a cytotoxic member of the kulolide superfamily of cyclic depsipeptides from a marine button cyanobacterium. J. Nat. Prod. 2012, 75, 1560–1570. [Google Scholar] [CrossRef]
- Wang, D.; Song, S.; Tian, Y.; Xu, Y.; Miao, Z.; Zhang, A. Total synthesis of the Marine cyclic depsipeptide viequeamide A. J. Nat. Prod. 2013. [Google Scholar] [CrossRef]
- Lücking, D.L.H.R. Fungal diversity revisited: 2.2 to 3.8 million species. Neurol. India. 2015, 63, 1004–1005. [Google Scholar] [CrossRef]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism-From biochemistry to genomics. Nat. Rev. Microbiol. 2005. [Google Scholar] [CrossRef]
- Du, L.; Risinger, A.L.; King, J.B.; Powell, D.R.; Cichewicz, R.H. A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J. Nat. Prod. 2014, 77, 1753–1757. [Google Scholar] [CrossRef]
- Guo, H.; Kreuzenbeck, N.B.; Otani, S.; Garcia-Altares, M.; Dahse, H.M.; Weigel, C.; Aanen, D.K.; Hertweck, C.; Poulsen, M.; Beemelmanns, C. Pseudoxylallemycins A-F, cyclic tetrapeptides with rare allenyl modifications isolated from Pseudoxylaria sp. X802: A competitor of fungus-growing termite cultivars. Org. Lett. 2016, 18, 3338–3341. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Liu, H.; Liu, X.; Che, Y. Cycloaspeptides F and G, cyclic pentapeptides from a Cordyceps-colonizing isolate of Isaria farinosa. J. Nat. Prod. 2009, 72, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, M.; Wang, D.; Li, X.; Wang, W.; Lou, H.; Yuan, H. Malformin A1 promotes cell death through induction of apoptosis, necrosis and autophagy in prostate cancer cells. Cancer Chemother. Pharmacol. 2016, 77, 63–75. [Google Scholar] [CrossRef]
- Park, S.Y.; Oh, H.H.; Park, Y.L.; Yu, H.M.; Myung, D.S.; Cho, S.B.; Lee, W.S.; Park, D.; Joo, Y.E. Malformin A1 treatment alters invasive and oncogenic phenotypes of human colorectal cancer cells through stimulation of the p38 signaling pathway. Int. J. Oncol. 2017, 51, 959–966. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Notarte, K.; Nakao, Y.; Yaguchi, T.; Bungihan, M.; Suganuma, K.; dela Cruz, T.E. Trypanocidal activity, cytotoxicity and histone modifications induced by malformin A1 isolated from the marine-derived fungus Aspergillus tubingensis IFM 63452. Mycosphere 2017, 8, 111–120. [Google Scholar] [CrossRef]
- Ma, Y.M.; Liang, X.A.; Zhang, H.C.; Liu, R. Cytotoxic and antibiotic cyclic pentapeptide from an endophytic Aspergillus tamarii of Ficus carica. J. Agric. Food Chem. 2016, 64, 3789–3793. [Google Scholar] [CrossRef]
- Song, H.H.; Lee, H.S.; Lee, C. A new cytotoxic cyclic pentadepsipeptide, neo-N-methylsansalvamide produced by Fusarium solani KCCM90040, isolated from potato. Food Chem. 2011, 126, 472–478. [Google Scholar] [CrossRef]
- Lee, H.S.; Phat, C.; Choi, S.U.; Lee, C. Synergistic effect of a novel cyclic pentadepsipeptide, neo N-methylsansalvamide, and paclitaxel on human multidrug resistance cancer cell lines. Anticancer Drugs 2013, 24, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Erosa, G.; Sterner, O.; Anke, T. Cylindrocyclin A, a new cytotoxic cyclopeptide from Cylindrocarpon sp. J. Antibiot. 2006, 59, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Ványolós, A.; Dékány, M.; Kovács, B.; Krámos, B.; Bérdi, P.; Zupkó, I.; Hohmann, J.; Béni, Z. Gymnopeptides A and B, Cyclic octadecapeptides from the mushroom Gymnopus fusipes. Org. Lett. 2016, 18, 2688–2691. [Google Scholar] [CrossRef]
- Pan, Z.; Wu, C.; Wang, W.; Cheng, Z.; Yao, G.; Liu, K.; Li, H.; Fang, L.; Su, W. Total synthesis and stereochemical assignment of gymnopeptides A and B. Org. Lett. 2017, 19, 4420–4423. [Google Scholar] [CrossRef]
- Schloß, S.; Hackl, T.; Herz, C.; Lamy, E.; Koch, M.; Rohn, S.; Maul, R. Detection of a toxic methylated derivative of phomopsin A produced by the legume-infesting fungus Diaporthe toxica. J. Nat. Prod. 2017, 80, 1930–1934. [Google Scholar] [CrossRef]
- Chen, C.H.; Lang, G.; Mitova, M.I.; Murphy, A.C.; Cole, A.L.J.; Din, L.B.; Blunt, J.W.; Munro, M.H.G. Pteratides I-IV, new cytotoxic cyclodepsipeptides from the Malaysian basidiomycete Pterula sp. J. Org. Chem. 2006, 71, 7947–7951. [Google Scholar] [CrossRef]
- He, F.; Bao, J.; Zhang, X.Y.; Tu, Z.C.; Shi, Y.M.; Qi, S.H. Asperterrestide A, a cytotoxic cyclic tetrapeptide from the marine-derived fungus Aspergillus terreus SCSGAF0162. J. Nat. Prod. 2013, 76, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Sugai, M.; Zhang, L.; Masuda, Y.; Yoshida, M.; Doi, T. Total synthesis and structural revision of cyclotetrapeptide asperterrestide A. J. Org. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Srisanoh, U.; Lartpornmatulee, N.; Boonruangprapa, T. ES-242 derivatives and cycloheptapeptides from Cordyceps sp. strains BCC 16173 and BCC 16176. J. Nat. Prod. 2007, 70, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Rukachaisirikul, V.; Chantaruk, S.; Tansakul, C.; Saithong, S.; Chaicharernwimonkoon, L.; Pakawatchai, C.; Isaka, M.; Intereya, K. A cyclopeptide from the insect pathogenic fungus Cordyceps sp. BCC 1788. J. Nat. Prod. 2006, 69, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C-E, from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Solution phase synthesis and bioevaluation of cordyheptapeptide B. Bull. Pharm. Res. 2011, 1, 1–10. [Google Scholar]
- Oh, D.C.; Jensen, P.R.; Fenical, W. Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 2006, 47, 8625–8628. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, F.; Zhang, Y.; Liu, J.O.; Ma, D. Synthesis and antitumor activity of cyclodepsipeptide zygosporamide and its analogues. Bioorganic Med. Chem. Lett. 2008, 18, 4385–4387. [Google Scholar] [CrossRef]
- Belofsky, G.N.; Jensen, P.R.; Fenical, W. Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999, 40, 2913–2916. [Google Scholar] [CrossRef]
- Heiferman, M.J.; Salabat, M.R.; Ujiki, M.B.; Strouch, M.J.; Cheon, E.C.; Silverman, R.B.; Bentrem, D.J. Sansalvamide induces pancreatic cancer growth arrest through changes in the cell cycle. Anticancer Res. 2010, 30, 73–78. [Google Scholar] [PubMed]
- Hwang, Y.; Rowley, D.; Rhodes, D.; Gertsch, J.; Fenical, W.; Bushman, F. Mechanism of inhibition of a poxvirus topoisomerase by the marine natural product sansalvamide A. Mol. Pharmacol. 1999, 55, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, M.; Zhou, Y.; Liu, H.; Jiang, H. Ionic-liquid-supported total synthesis of sansalvamide A peptide. Synth. Commun. 2008, 38, 239–248. [Google Scholar] [CrossRef]
- Carroll, C.L.; Johnston, J.V.C.; Kekec, A.; Brown, J.D.; Parry, E.; Cajica, J.; Medina, I.; Cook, K.M.; Corral, R.; Pan, P.S.; et al. Synthesis and cytotoxicity of novel sansalvamide A derivatives. Org. Lett. 2005, 7, 3481–3484. [Google Scholar] [CrossRef]
- Pan, P.S.; Vasko, R.C.; Lapera, S.A.; Johnson, V.A.; Sellers, R.P.; Lin, C.C.; Pan, C.M.; Davis, M.R.; Ardi, V.C.; McAlpine, S.R. A comprehensive study of sansalvamide A derivatives: The structure-activity relationships of 78 derivatives in two pancreatic cancer cell lines. Bioorganic Med. Chem. 2009, 17, 5806–5825. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ye, P.; Chen, C.T.A.; Wang, K.; Liu, P.; He, S.; Wu, X.; Gan, L.; Ye, Y.; Wu, B. Two novel hepatocellular carcinoma cycle inhibitory cyclodepsipeptides from a hydrothermal vent crab-associated fungus Aspergillus clavatus C2WU. Mar. Drugs 2013, 11, 4761–4772. [Google Scholar] [CrossRef]
- Chettu, S.K.; Konidena, L.N.S.; Korupolu, R.B.; Rao, N.S.K.; Doddipalla, R.; Gandham, H.B.; Guduru, R. Ring opening of benzoxazinones: An improved and efficient synthesis of clavatustides A & B. Tetrahedron Lett. 2017, 58, 3418–3420. [Google Scholar] [CrossRef]
- Liao, Y.; Hong, X.; Yang, J.; Qin, J.J.; Zhang, B.; Lin, J.; Shao, Z.; Wang, W. Structure elucidation of a novel cyclic tripeptide from the marine-derived fungus Aspergillus ochraceopetaliformis DSW-2. Nat. Prod. Res. 2020, 1–7. [Google Scholar] [CrossRef]
- Yu, Z.; Lang, G.; Kajahn, I.; Schmaljohann, R.; Imhoff, J.F. Scopularides A and B, cyclodepsipeptides from a marine sponge-derived fungus, Scopulariopsis brevicaulis. J. Nat. Prod. 2008, 71, 1052–1054. [Google Scholar] [CrossRef]
- Cruz, L.J.; Insua, M.M.; Baz, J.P.; Trujillo, M.; Rodriguez-Mias, R.A.; Oliveira, E.; Giralt, E.; Albericio, F.; Cañedo, L.M. IB-01212, a new cytotoxic cyclodepsipeptide isolated from the marine fungus Clonostachys sp. ESNA-A009. J. Org. Chem. 2006, 71, 3335–3338. [Google Scholar] [CrossRef]
- Luque-Ortega, J.R.; Cruz, L.J.; Albericio, F.; Rivas, L. The antitumoral depsipeptide IB-01212 kills Leishmania through an apoptosis-like process involving intracellular targets. Mol. Pharm. 2010, 7, 1608–1617. [Google Scholar] [CrossRef]
- Nabika, R.; Oishi, S.; Misu, R.; Ohno, H.; Fujii, N. Synthesis of IB-01212 by multiple N-methylations of peptide bonds. Bioorganic Med. Chem. 2014, 22, 6156–6162. [Google Scholar] [CrossRef]
- Cruz, L.J.; Francesch, A.; Cuevas, C.; Albericio, F. Synthesis and structure-activity relationship of cytotoxic marine cyclodepsipeptide IB-01212 analogues. ChemMedChem 2007, 2, 1076–1084. [Google Scholar] [CrossRef]
- Gu, W.; Cueto, M.; Jensen, P.R.; Fenical, W.; Silverman, R.B. Microsporins A and B: New histone deacetylase inhibitors from the marine-derived fungus Microsporum cf. gypseum and the solid-phase synthesis of microsporin A. Tetrahedron 2007, 63, 6535–6541. [Google Scholar] [CrossRef]
- Mukherjee, J.P.; Sil, S.; Chattopadhyay, S.K. A modular approach to cyclic tetrapeptides related to histone deacetylase inhibition: Synthesis of epi-microsporin A. Tetrahedron Lett. 2016, 57, 739–742. [Google Scholar] [CrossRef]
- Swaroop, P.S.; Tripathy, S.; Jachak, G.; Reddy, D.S. Efforts towards the synthesis of microsporin B: Ready access to both the enantiomers of the key amino acid fragment. Tetrahedron Lett. 2014, 55, 4777–4779. [Google Scholar] [CrossRef]














| Name | Biological Source | Anticancer Activity | Reference |
|---|---|---|---|
| Anticancer Cyclopeptides Derived from Non-Marine Fungi | |||
| 1-alaninechlamydocin (205) | Tolypocladium sp. | Cytotoxicity against MIA PaCa-2 (GI50 = 5.3 nM, LC50 = 22 nM) cell. | [296] |
| Pseudoxylallemycin C (206) | Pseudoxylaria sp. X802 | Anti-proliferative effect to K562 (GI50 = 6.8 µM) cell. Cytotoxicity against HeLa (CC50 = 16.68 µM) cell. | [297] |
| Cycloaspeptides F (207) and G (208) | Isaria farinosa | Inhibitory activities toward MCF7 (GI50 = 18.7 and 15.2 µM, respectively) cell. | [298] |
| Malformin A1 (MA1) (209) | Aspergillus niger | Cytotoxicity against PC3 (IC50 = 130 nM), LNCaP (IC50 = 90 nM), HeLa (IC50 = 9.48 nM) and P388 (IC50 = 132.9 nM) cells. | [299,300,301] |
| Malformin E (210) | Aspergillus tamarii | Cytotoxicity against MCF7 (IC50 = 0.65 µM) and A549 (IC50 = 2.42 µM) cells. | [302] |
| Neo-N-methylsansalvamide (211) | Fusarium solani KCCM90040 | Cytotoxicity against A549, SK-OV-3, SK-MEL-2 and MES-SA cells. (EC50 = 10.0 to 14.74 µM) Synergistic effect of neo-N-methylsansalvamide and paclitaxel when acting against MES-SA, HCT15, MES-SA/DX5 and HCT15/CL05 cells. | [303,304] |
| Cylindrocyclin A (212) | Cyclindrocarpon sp. strain A101-96 | Cytotoxicity against COLO-320, HL-60, L1210 and human T lymphocyte Jurkat cells. (IC50~11 µM) | [305] |
| Gymnopeptides A (213) and B (214) | Mushroom, Gymnopus fusipes | Anti-proliferative activities against HeLa, A431, T47D, MCF7 and MDA-MB-231 cells. (IC50 = 18.0 to 88.4 nM and 14.0 to 44.3 nM, respectively) | [306,307] |
| Phomopsin F (215) | Diaporthe toxica | Impact on viability and vitability of HepG2 (IC50 = 6.1 µM) cell. | [308] |
| Pteratides I-IV (216–219) | Pterula sp. | Cytotoxicity against P388 (IC50 = 41 nM, 40 nM, 7.4 µM and 2.9 µM, respectively) cell. | [309] |
| Anticancer Cyclopeptides Derived from Marine Fungi | |||
| Asperterrestide A (220) | Aspergillus terreus SCSGAF0162 | Cytotoxicity against U937 (IC50 = 6.4 μM) and MOL-4 (IC50 = 6.2 μM) cells. | [310,311] |
| Cordyheptapeptide A (221) | Cordyceps sp. BCC 1788 | Anti-proliferative activities to KB (IC50 = 0.78 μM), BC (IC50 = 0.2 μM), NCI-H187 (IC50 = 0.18 μM) and Vero (IC50 = 14 μM) cells. | [312,313] |
| Cordyheptapeptide B (222) | Cordyceps sp. BCC 16173 | Anti-proliferative activities to KB (IC50 = 2.0 μM), BC (IC50 = 0.66 μM), NCI-H187 (IC50 = 3.1 μM) and Vero (IC50 = 1.6 μM) cells. | [312,313] |
| Cordyheptapeptide C (223) | Acremonium persicinum | Cytotoxicity against SF-268 (IC50 = 3.7 μM) and MCF7 (IC50 = 3.0 μM) cells. | [314] |
| Cordyheptapeptide E (224) | Acremonium persicinum | Cytotoxicity against SF-268 (IC50 = 3.2 μM), MCF7 (IC50 = 2.7 μM) and NCI-H460 (IC50 = 4.5 μM) cells. | [314] |
| Scytalidamides A (225) and B (226) | Scytalidium sp. | Cytotoxicity against HCT116 (IC50 = 2.7 and 11.0 µM, respectively) cell. Moderate cytotoxicitty in NCI 60 cell line panel. (Mean GI50 = 7.9 and 4.1 µM, respectively) | [315] |
| Zygosporamide (227) | Zygosporium masonii | Cytotoxicity against SF-268 (IC50 = 6.5 nM) and RXF393 (IC50 ≤ 5.0 nM) cells. | [316,317] |
| Sansalvamide A (228) | Fusarium sp. | Cytotoxicity against HCT116, COLO 205 and SK-MEL-2 (IC50 = 9.7, 3.46 and 5.84 μM, respectively) cells. | [318,319,320,321,322,323] |
| Clavatustide A (229) | Aspergillus clavatus | Anti-proliferative activity against Hep G2, SMMC-7721 and Bel-7402 cells. (IC50~32 μM) | [324] |
| Clavatustide B (230) | Aspergillus clavatus | Anti-proliferative activity against Hep G2, SMMC-7721 and Bel-7402 cells. (IC50~32 μM) Cell growth inhibitory activity against PANC-1 and PC3 cells. | [324,325] |
| Sclerotiotide M (231) | Aspergillus ochraceopetaliformis DSW-2 | Cytotoxicity against HPAC and BXPC3 cells. (IC50 > 20 μM) | [326] |
| Scopularides A (232) and B (233) | Scopulariopsis brevicaulis | Viability inhibition of several cancer cell lines including Colo357, Panc89 and HT29 cells at a concentration of 14.87 μM. | [327] |
| IB-01212 (234) | Clonostachys sp. ESNA-A009 | Inhibitory activity against several cancer cell lines including LNCaP, SKBR3, HT29 and HeLa with the GI50 values on the order of 10−8 M. | [328,329,330,331] |
| Microsporins A (235) and B (236) | Microsporum cf. gypseum | Cytotoxicity especially against HCT116 cell. (IC50 = 1.12 and 15.82 μM, respectively) | [332,333,334] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.-N.; Xia, Y.-X.; Zhang, H.-J. Natural Cyclopeptides as Anticancer Agents in the Last 20 Years. Int. J. Mol. Sci. 2021, 22, 3973. https://doi.org/10.3390/ijms22083973
Zhang J-N, Xia Y-X, Zhang H-J. Natural Cyclopeptides as Anticancer Agents in the Last 20 Years. International Journal of Molecular Sciences. 2021; 22(8):3973. https://doi.org/10.3390/ijms22083973
Chicago/Turabian StyleZhang, Jia-Nan, Yi-Xuan Xia, and Hong-Jie Zhang. 2021. "Natural Cyclopeptides as Anticancer Agents in the Last 20 Years" International Journal of Molecular Sciences 22, no. 8: 3973. https://doi.org/10.3390/ijms22083973
APA StyleZhang, J.-N., Xia, Y.-X., & Zhang, H.-J. (2021). Natural Cyclopeptides as Anticancer Agents in the Last 20 Years. International Journal of Molecular Sciences, 22(8), 3973. https://doi.org/10.3390/ijms22083973