
Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
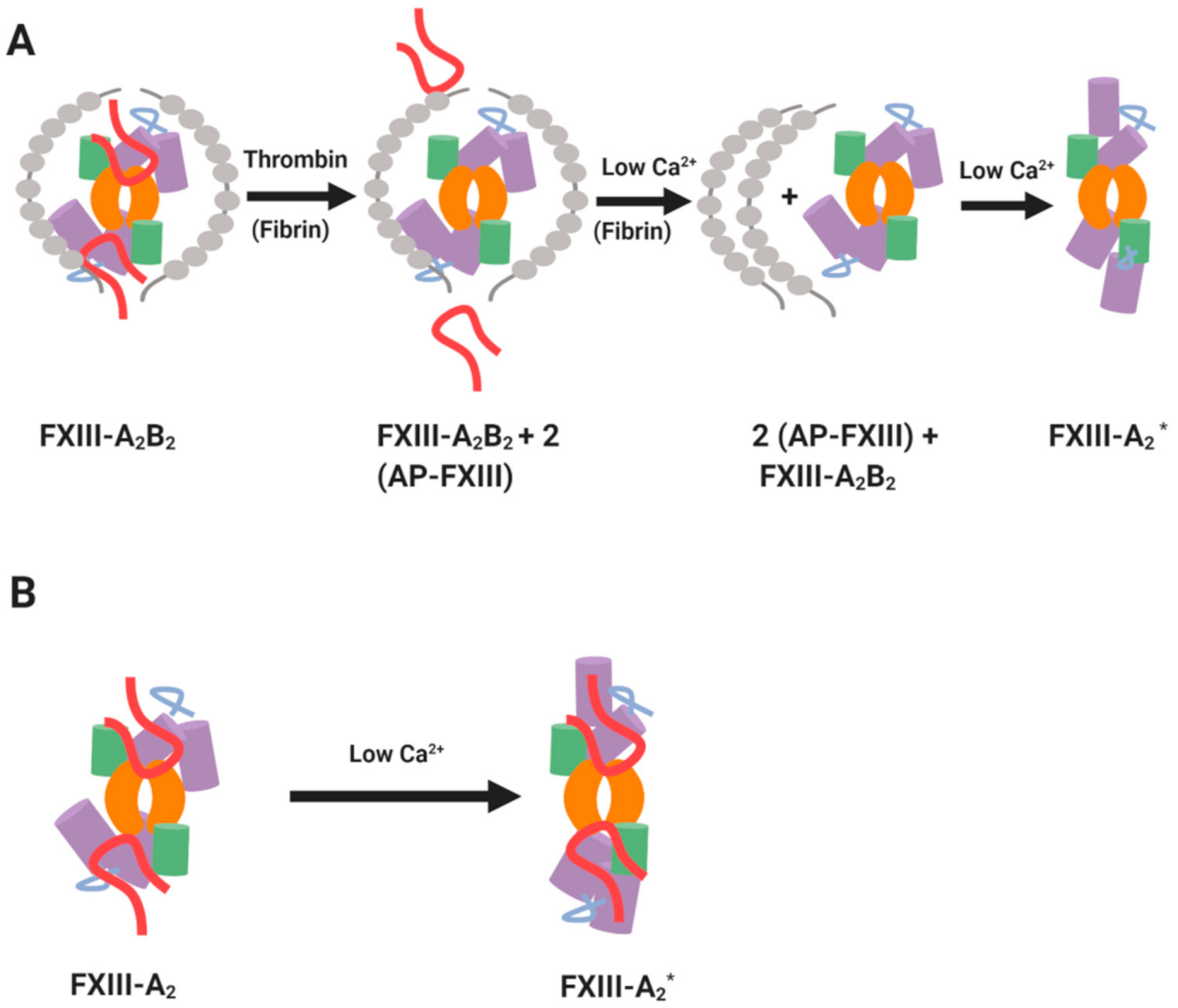
2. Structural Considerations
3. Pools of FXIII-A within the Vasculature
3.1. Platelet-Derived FXIII-A
3.2. Monocyte/Macrophage-Derived FXIII-A
4. FXIII Deficiency and Associated Complications
4.1. Congenital Deficiency
4.2. Pregnancy Complications
4.3. Acquired FXIII Deficiency
5. The Indispensable Role of FXIII-A Haemostasis and Wound Healing
5.1. FXIII in Haemostasis
5.2. FXIII in Wound Healing
6. FXIII-A Replacement Therapy and Utility as a Drug Target
7. Discussion and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bagoly, Z.; Koncz, Z.; Hársfalvi, J.; Muszbek, L. Factor XIII, clot structure, thrombosis. Thromb. Res. 2012, 129, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L. Factor XIII: Structure, Activation, and Interactions with Fibrinogen and Fibrin. Ann. N. Y. Acad. Sci. 2006, 936, 291–311. [Google Scholar] [CrossRef]
- Karimi, M.; Bereczky, Z.; Cohan, N.; Muszbek, L. Factor XIII Deficiency. Semin. Thromb. Hemost. 2009, 35, 426–438. [Google Scholar] [CrossRef]
- Yorifuji, H.; Anderson, K.; Lynch, G.W.; Van De Water, L.; McDonagh, J. B protein of Factor XIII: Differentiation between free B and complexed B. Blood 1988, 72, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Katona, É.; Pénzes, D.K.; Csapó, A.; Fazakas, F.; Udvardy, M.L.; Bagoly, Z.; Orosz, Z.Z.; Muszbek, L. Interaction of Factor XIII subunits. Blood 2014, 123, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.S.; Miraglia, C.C.; Rickles, F.R.; Shuman, M.A. Cleavage of blood coagulation Factor XIII and fibrinogen by thrombin during in vitro clotting. J. Clin. Investig. 1985, 75, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.W.; Pfueller, S.L. Thrombin-independent activation of platelet Factor XIII by endogenous platelet acid protease. Thromb. Haemost. 1988, 59, 372–377. [Google Scholar] [CrossRef]
- Ando, Y.; Imamura, S.; Yamagata, Y.; Kitahara, A.; Saji, H.; Murachi, T.; Kannagi, R. Platelet Factor XIII is activated by calpain. Biochem. Biophys. Res. Commun. 1987, 144, 484–490. [Google Scholar] [CrossRef]
- Lewis, S.D.; Janus, T.J.; Lorand, L.; Shafer, J.A. Regulation of formation of Factor XIIIa by its fibrin substrates. Biochemistry 1985, 24, 6772–6777. [Google Scholar] [CrossRef] [PubMed]
- Hornyak, T.J.; Shafer, J.A. Interactions of Factor XIII with fibrin as substrate and cofactor. Biochemistry 1992, 31, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Naski, M.C.; Shafer, J.A. A kinetic model for the alpha-thrombin-catalyzed conversion of plasma levels of fibrinogen to fibrin in the presence of antithrombin III. J. Biol. Chem. 1991, 266, 13003–13010. [Google Scholar] [CrossRef]
- Janus, T.J.; Lewis, S.D.; Lorand, L.; Shafer, J.A. Promotion of thrombin-catalyzed activation of Factor XIII by fibrinogen. Biochemistry 1983, 22, 6269–6272. [Google Scholar] [CrossRef]
- Folk, J.E.; Finlayson, J.S. The epsilon-(gamma-glutamyl)lysine crosslink and the catalytic role of transglutaminases. Adv. Protein Chem. 1977, 31, 1–133. [Google Scholar] [PubMed]
- Ádány, R.; Bárdos, H. Factor XIII subunit A as an intracellular transglutaminase. Cell. Mol. Life Sci. 2003, 60, 1049–1060. [Google Scholar] [CrossRef]
- Polgar, J.; Hidasi, V.; Muszbek, L. Non-proteolytic activation of cellular protransglutaminase (placenta macrophage Factor XIII). Biochem. J. 1990, 267, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Polgar, J.; Boda, Z. Platelet Factor XIII becomes active without the release of activation peptide during platelet acti-vation. Thromb. Haemost. 1993, 69, 282–285. [Google Scholar]
- Buluk, K. An unknown action of blood platelets; preliminary communication. Polski Tyg. Lek. 1955, 10, 191. [Google Scholar] [PubMed]
- Kiesselbach, T.H.; Wagner, R.H. Fibrin-stabilizing factor: A thrombin-labile platelet protein. Am. J. Physiol. Content 1966, 211, 1472–1476. [Google Scholar] [CrossRef]
- Luscher, E.F. Fibrin-stabilizing factor from thrombocytes. Schweiz. Med. Wochenschr. 1957, 87, 1220–1221. [Google Scholar]
- Kiesselbach, T.H.; Wagner, R.H. Demonstration of Factor XIII in human megakaryocytes by a fluorescent antibody technique. Ann. N. Y. Acad. Sci. 1972, 202, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, P.; Becker, S.; Lynch, G.; McDonagh, J. Identification of intracellular Factor XIII in human monocytes and macro-phages. J. Clin. Investig. 1985, 76, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Adány, R.; Szegedi, G.; Polgár, J.; Kávai, M. Factor XIII of blood coagulation in human monocytes. Thromb. Res. 1985, 37, 401–410. [Google Scholar] [CrossRef]
- Adany, R.; Belkin, A.; Vasilevskaya, T.; Muszbek, L. Identification of blood coagulation Factor XIII in human peritoneal macro-phages. Eur. J. Cell Biol. 1985, 38, 171–173. [Google Scholar] [PubMed]
- Nestle, F.O.; Zheng, X.G.; Thompson, C.B.; Turka, L.A.; Nickoloff, B.J. Characterization of dermal dendritic cells obtained from nor-mal human skin reveals phenotypic and functionally distinctive subsets. J. Immunol. 1993, 151, 6535–6545. [Google Scholar] [PubMed]
- Nurminskaya, M.; Linsenmayer, T.F. Identification and characterization of up-regulated genes during chondrocyte hypertrophy. Dev. Dyn. 1996, 206, 260–271. [Google Scholar] [CrossRef]
- Nurminskaya, M.; Magee, C.; Nurminsky, D.; Linsenmayer, T.F. Plasma transglutaminase in hypertrophic chondrocytes: Expres-sion and cell-specific intracellular activation produce cell death and externalization. J. Cell Biol. 1998, 142, 1135–1144. [Google Scholar] [CrossRef]
- Rosenthal, A.K.; Masuda, I.; Gohr, C.M.; Derfus, B.A.; Le, M. The transglutaminase, Factor XIIIA, is present in articular chondro-cytes. Osteoarthritis Cartil. 2001, 9, 578–581. [Google Scholar] [CrossRef][Green Version]
- Nurminskaya, M.; Kaartinen, M.T. Transglutaminases in mineralized tissues. Front. Biosci. 2006, 11, 1591–1606. [Google Scholar] [CrossRef][Green Version]
- Myneni, V.D.; Hitomi, K.; Kaartinen, M.T. Factor XIII-A transglutaminase acts as a switch between preadipocyte proliferation and differentiation. Blood 2014, 124, 1344–1353. [Google Scholar] [CrossRef]
- Souri, M.; Osaki, T.; Ichinose, A.; Hsieh, L.-S.; Tsukasa, O.; Han, G.-S.; Carman, G.M. The Non-catalytic B Subunit of Coagulation Factor XIII Accelerates Fibrin Cross-linking. J. Biol. Chem. 2015, 290, 12027–12039. [Google Scholar] [CrossRef]
- Hur, W.S.; Mazinani, N.; Lu, X.J.D.; Britton, H.M.; Byrnes, J.R.; Wolberg, A.S.; Kastrup, C.J. Coagulation Factor XIIIa is inactivated by plasmin. Blood 2015, 126, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Putnam, F.W.; Takahashi, Y. Primary structure of blood coagulation Factor XIIIa (fibrinoligase, transglutaminase) from human placenta. Proc. Natl. Acad. Sci. USA 1986, 83, 8019–8023. [Google Scholar] [CrossRef]
- Bagoly, Z.; Haramura, G.; Muszbek, L. Down-regulation of activated Factor XIII by polymorphonuclear granulocyte proteases within fibrin clot. Thromb. Haemost. 2007, 98, 359–367. [Google Scholar] [CrossRef]
- Ichinose, A.; Takio, K.; Fujikawa, K. Localization of the binding site of tissue-type plasminogen activator to fibrin. J. Clin. Investig. 1986, 78, 163–169. [Google Scholar] [CrossRef]
- Yee, V.C.; Pedersen, L.C.; Le Trong, I.; Bishop, P.D.; Stenkamp, R.E.; Teller, D.C. Three-dimensional structure of a transglutaminase: Human blood coagulation Factor XIII. Proc. Natl. Acad. Sci. USA 1994, 91, 7296–7300. [Google Scholar] [CrossRef]
- Fox, B.A.; Yee, V.C.; Pedersen, L.C.; Le Trong, I.; Bishop, P.D.; Stenkamp, R.E.; Teller, D.C. Identification of the Calcium Binding Site and a Novel Ytterbium Site in Blood Coagulation Factor XIII by X-ray Crystallography. J. Biol. Chem. 1999, 274, 4917–4923. [Google Scholar] [CrossRef]
- Stieler, M.; Weber, J.; Hils, M.; Kolb, P.; Heine, A.; Büchold, C.; Pasternack, R.; Klebe, G. Structure of Active Coagulation Factor XIII Triggered by Calcium Binding: Basis for the Design of Next-Generation Anticoagulants. Angew. Chem. Int. Ed. 2013, 52, 11930–11934. [Google Scholar] [CrossRef] [PubMed]
- Komáromi, I.; Bagoly, Z.; Muszbek, L. Factor XIII: Novel structural and functional aspects. J. Thromb. Haemost. 2011, 9, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Adany, R.; Mikkola, H. Novel Aspects of Blood Coagulation Factor XIII. I. Structure, Distribution, Activation, and Function. Crit. Rev. Clin. Lab. Sci. 1996, 33, 357–421. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Freyssinet, J.M.; Holbrook, J.J. An equilibrium study of metal ion binding to human plasma coagulation Factor XIII. Biochem. J. 1978, 169, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Bányai, I.; Weiss, M.S.; Hilgenfeld, R.; Keresztessy, Z.; Muszbek, L.; Fésüs, L. Calcium Binding of Transglutaminases: A43Ca NMR Study Combined with Surface Polarity Analysis. J. Biomol. Struct. Dyn. 2001, 19, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Ariëns, R.A.; Lai, T.-S.; Weisel, J.W.; Greenberg, C.S.; Grant, P.J. Role of Factor XIII in fibrin clot formation and effects of genetic poly-morphisms. Blood 2002, 100, 743–754. [Google Scholar] [CrossRef]
- Francis, R.T.; McDonagh, J.; Mann, K.G. Factor V is a substrate for the transamidase Factor XIIIa. J. Biol. Chem. 1986, 261, 9787–9792. [Google Scholar] [CrossRef]
- Valnickova, Z.; Enghild, J.J. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases: Evidence for transglutaminase-catalyzed cross-linking to fibrin. J. Biol. Chem. 1998, 273, 27220–27224. [Google Scholar] [CrossRef]
- Sakata, Y.; Aoki, N. Cross-linking of alpha 2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. J. Clin. Investig. 1980, 65, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F. Action of fibrin-stabilizing factor on cold-insoluble globulin and alpha2-macroglobulin in clotting plasma. J. Biol. Chem. 1976, 251, 1639–1645. [Google Scholar] [CrossRef]
- Sottrup-Jensen, L.; Stepanik, T.M.; Wierzbicki, D.M.; Jones, C.M.; Lønblad, P.B.; Kristensen, T.N.; Mortensen, S.B.; Petersen, T.E.; Magnusson, S. The primary structure of alpha 2-macroglobulin and localization of a Factor XIIIa cross-linking site. Ann. N. Y. Acad. Sci. 1983, 421, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Lee, C.S.; Tae, W.-C.; Jackson, K.W.; Christiansen, V.J.; McKee, P.A. Cross-linking of Wild-type and Mutant α2-Antiplasmins to Fibrin by Activated Factor XIII and by a Tissue Transglutaminase. J. Biol. Chem. 2000, 275, 37382–37389. [Google Scholar] [CrossRef]
- Lee, K.N.; Lee, C.S.; Tae, W.C.; Jackson, K.W.; Christiansen, V.J.; McKee, P.A. Crosslinking of alpha 2-antiplasmin to fibrin. Ann. N. Y. Acad. Sci. 2001, 936, 335–339. [Google Scholar] [CrossRef]
- Skorstengaard, K.; Halkier, T.; Hojrup, P.; Mosher, D. Sequence location of a putative transglutaminase cross-linking site in hu-man vitronectin. FEBS Lett. 1990, 262, 269–274. [Google Scholar] [CrossRef]
- Wölpl, A.; Lattke, H.; Board, P.G.; Arnold, R.; Schmeiser, T.; Kubanek, B.; Robin-Winn, M.; Pichelmayr, R.; Goldmann, S.F. Coagulation Factor XIII A and B subunits in bone marrow and liver transplantation. Transplantation 1987, 43, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Pihusch, R.; Salat, C.; Schmidt, E.; Göhring, P.; Pihusch, M.; Hiller, E.; Holler, E.; Kolb, H.-J. Hemostatic complications in bone marrow transplantation: A retrospective analysis of 447 patients. Transplantation 2002, 74, 1303–1309. [Google Scholar] [CrossRef]
- Inbal, A.; Muszbek, L.; Lubetsky, A.; Katona, É.; Levi, I.; Kárpáti, L.; Nagler, A. Platelets but not monocytes contribute to the plasma levels of Factor XIII subunit A in patients undergoing autologous peripheral blood stem cell transplantation. Blood Coagul. Fibrinolysis 2004, 15, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.C.; Russell, J.A.; Low, S.; Sinclair, G.D.; Jones, A.R.; Blahey, W.; Ruether, B.A.; Hoar, D.I. Hemopoietic origin of Factor XIII A subunits in platelets, monocytes, and plasma. Evidence from bone marrow transplantation studies. J. Clin. Investig. 1989, 84, 787–792. [Google Scholar] [CrossRef]
- Cordell, P.A.; Kile, B.T.; Standeven, K.F.; Josefsson, E.C.; Pease, R.J.; Grant, P.J. Association of coagulation Factor XIII-A with Golgi pro-teins within monocyte-macrophages: Implications for subcellular trafficking and secretion. Blood 2010, 115, 2674–2681. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beckers, C.M.; Simpson, K.R.; Griffin, K.J.; Brown, J.M.; Cheah, L.T.; Smith, K.A.; Vacher, J.; Cordell, P.A.; Kearney, M.T.; Grant, P.J.; et al. Cre/lox Studies Identify Resident Macrophages as the Major Source of Circulating Coagulation Factor XIII-A. Arter. Thromb. Vasc. Biol. 2017, 37, 1494–1502. [Google Scholar] [CrossRef]
- Katona, É.; Ajzner, É.; Tóth, K.; Kárpáti, L.; Muszbek, L. Enzyme-linked immunosorbent assay for the determination of blood co-agulation Factor XIII A-subunit in plasma and in cell lysates. J. Immunol. Methods 2001, 258, 127–135. [Google Scholar] [CrossRef]
- Muszbek, L.; Yee, V.C.; Hevessy, Z. Blood coagulation Factor XIII: Structure and function. Thromb. Res. 1999, 94, 271–305. [Google Scholar] [CrossRef]
- Holme, P.A.; Brosstad, F.; Solum, N.O. The difference between platelet and plasma FXIII used to study the mechanism of platelet microvesicle formation. Thromb. Haemost. 1993, 70, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Marx, G.; Korner, G.; Mou, X.; Gorodetsky, R. Packaging zinc, fibrinogen, and Factor XIII in platelet α-granules. J. Cell. Physiol. 1993, 156, 437–442. [Google Scholar] [CrossRef]
- Kreutz, R.P.; Bitar, A.; Owens, J.; Desta, Z.; Breall, J.A.; Von Der Lohe, E.; Sinha, A.; Vatta, M.; Nystrom, P.; Jin, Y.; et al. Factor XIII Val34Leu polymorphism and recurrent myocardial infarction in patients with coronary artery disease. J. Thromb. Thromb. 2014, 38, 380–387. [Google Scholar] [CrossRef]
- Lopaciuk, S.; Lovette, K.; McDonagh, J.; Chuang, H.; McDonagh, R. Subcellular distribution of fibrinogen and Factor XIII in hu-man blood platelets. Thromb. Res. 1976, 8, 453–465. [Google Scholar] [CrossRef]
- Sixma, J.J.; Berg, A.V.D.; Schiphorst, M.; Geuze, H.J.; McDonagh, J. Immunocytochemical localization of albumin and Factor XIII in thin cryo sections of human blood platelets. Thromb. Haemost. 1984, 51, 388–391. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Lionikiene, A.S.; Fraser, S.R.; Whyte, C.S.; Booth, N.A.; Mutch, N.J. Functional Factor XIII-A is exposed on the stimulated platelet surface. Blood 2014, 124, 3982–3990. [Google Scholar] [CrossRef]
- Nurden, A.T.; Kunicki, T.J.; Dupuis, D.; Soria, C.; Caen, J.P. Specific protein and glycoprotein deficiencies in platelets isolated from two patients with the gray platelet syndrome. Blood 1982, 59, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Joist, J.H.; Niewiarowski, S. Retention of platelet fibrin stabilizing factor during the platelet release reaction and clot retraction. Thromb. Diath. Haemorrh. 1973, 29, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Agbani, E.O.; Hers, I.; Poole, A.W. Temporal contribution of the platelet body and balloon to thrombin generation. Haematologica 2017, 102, e379–e381. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Podoplelova, N.A.; Sveshnikova, A.N.; Kotova, Y.N.; Eckly, A.; Receveur, N.; Nechipurenko, D.Y.; Obydennyi, S.I.; Kireev, I.I.; Gachet, C.; Ataullakhanov, F.I.; et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood 2016, 128, 1745–1755. [Google Scholar] [CrossRef]
- Bale, M.D.; Mosher, D.F. Effects of thrombospondin on fibrin polymerization and structure. J. Biol. Chem. 1986, 261, 862–868. [Google Scholar] [CrossRef]
- Lynch, G.W.; Slayter, H.; Miller, B.; McDonagh, J. Characterization of thrombospondin as a substrate for Factor XIII transglutaminase. J. Biol. Chem. 1987, 262, 1772–1778. [Google Scholar] [CrossRef]
- Whyte, C.S.; Swieringa, F.; Mastenbroek, T.G.; Lionikiene, A.S.; Lance, M.D.; van der Meijden, P.E.; Heemskerk, J.W.; Mutch, N.J. Plas-minogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood 2015, 125, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Abaeva, A.A.; Canault, M.; Kotova, Y.N.; Obydennyy, S.I.; Yakimenko, A.O.; Podoplelova, N.A.; Kolyadko, V.N.; Chambost, H.; Mazurov, A.V.; Ataullakhanov, F.I.; et al. Procoagulant Platelets Form an α-Granule Protein-covered “Cap” on Their Surface That Promotes Their Attachment to Aggregates. J. Biol. Chem. 2013, 288, 29621–29632. [Google Scholar] [CrossRef]
- Kotova, Y.N.; Podoplelova, N.A.; Obydennyy, S.I.; Kostanova, E.A.; Ryabykh, A.A.; Demyanova, A.S.; Biriukova, M.I.; Rosenfeld, M.A.; Sokolov, A.V.; Chambost, H.; et al. Binding of Coagulation Factor XIII Zymogen to Activated Platelet Subpopulations: Roles of Integrin αIIbβ3 and Fibrinogen. Thromb. Haemost. 2019, 119, 906–915. [Google Scholar] [CrossRef]
- Mattheij, N.J.; Swieringa, F.; Mastenbroek, T.G.; Berny-Lang, M.A.; May, F.; Baaten, C.C.; van der Meijden, P.E.; Henskens, Y.M.; Beckers, E.A.; Suylen, D.P. Coated platelets function in platelet-dependent fibrin formation via integrin αIIbβ3 and transglutaminase Factor XIII. Haematologica 2016, 101, 427. [Google Scholar] [CrossRef]
- Heemskerk, J.W.M.; Mattheij, N.J.A.; Cosemans, J.M.E.M. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Arachiche, A.; Kerbiriou-Nabias, D.; Garcin, I.; Letellier, T.; Dachary-Prigent, J. Rapid Procoagulant Phosphatidylserine Exposure Relies on High Cytosolic Calcium Rather Than on Mitochondrial Depolarization. Arter. Thromb. Vasc. Biol. 2009, 29, 1883–1889. [Google Scholar] [CrossRef]
- Abbasian, N.; Millington-Burgess, S.L.; Chabra, S.; Malcor, J.-D.; Harper, M.T. Supramaximal calcium signaling triggers procoagu-lant platelet formation. Blood Adv. 2020, 4, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Stalker, T.J.; Welsh, J.D.; Tomaiuolo, M.; Wu, J.; Colace, T.V.; Diamond, S.L.; Brass, L.F. A systems approach to hemostasis: Thrombus consolidation regulates intrathrombus solute transport and local thrombin activity. Blood 2014, 124, 1824–1831. [Google Scholar] [CrossRef]
- Carr, M.E.; Angchaisuksiri, P.; Carr, S.L.; Martin, E.J. Effect of Non-Heparin Thrombin Antagonists on Thrombin Generation, Platelet Function, and Clot Structure in Whole Blood. Cell Biophys. 2003, 39, 89–100. [Google Scholar] [CrossRef]
- Lam, W.A.; Chaudhuri, O.; Crow, A.; Webster, K.D.; Li, T.-D.; Kita, A.; Huang, J.; Fletcher, D.A. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat. Mater. 2010, 10, 61–66. [Google Scholar] [CrossRef]
- Muthard, R.W.; Diamond, S.L. Blood clots are rapidly assembled hemodynamic sensors: Flow arrest triggers intraluminal throm-bus contraction. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2938–2945. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Kaneda, M.; Miki, T.; Iida, K.; Sekino-Suzuki, N.; Kawashima, I.; Suzuki, H.; Shimonaka, M.; Arai, M.; Ohno-Iwashita, Y.; et al. Clot retraction is mediated by Factor XIII-dependent fibrin-alphaIIbbeta3-myosin axis in platelet sphingomyelin-rich membrane rafts. Blood 2013, 122, 3340–3348. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, I.; Leisner, T.M.; Lam, S.C. Direct binding of the platelet integrin alphaIIbbeta3 (GPIIb-IIIa) to talin. Evidence that in-teraction is mediated through the cytoplasmic domains of both alphaIIb and beta3. J. Biol. Chem. 1996, 271, 16416–16421. [Google Scholar] [CrossRef]
- Shattil, S.J. Signaling through platelet integrin alpha IIb beta 3: Inside-out, outside-in, and sideways. Thromb. Haemost. 1999, 82, 318–325. [Google Scholar]
- Cohen, I.; Gerrard, J.M.; White, J.G. Ultrastructure of clots during isometric contraction. J. Cell Biol. 1982, 93, 775–787. [Google Scholar] [CrossRef]
- Jelenska, M.; Kopeć, M.; Breddin, K. On the Retraction of Collagen and Fibrin Induced by Normal, Defective and Modified Platelets. Pathophysiol. Haemost. Thromb. 1985, 15, 169–175. [Google Scholar] [CrossRef]
- Niewiarowski, S.; Markiewicz, M.; Nath, N. Inhibition of the platelet-dependent fibrin retraction by the fibrin stabilizing factor (FSF, factor 13). J. Lab. Clin. Med. 1973, 81, 641–650. [Google Scholar]
- Rao, K.M.K.; Newcomb, T.F. Clot Retraction in a Factor XIII Free System. Scand. J. Haematol. 2009, 24, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Souri, M.; Kaneda, M.; Miki, T.; Yamamoto, N.; Ichinose, A. Impaired clot retraction in Factor XIII A subunit–deficient mice. Blood 2010, 115, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Kattula, S.; Byrnes, J.R.; Martin, S.M.; Holle, L.A.; Cooley, B.C.; Flick, M.J.; Wolberg, A.S. Factor XIII in plasma, but not in platelets, me-diates red blood cell retention in clots and venous thrombus size in mice. Blood Adv. 2018, 2, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Jackson, S.P. Platelet Factor XIII and Calpain Negatively Regulate Integrin αIIbβ3 Adhesive Function and Thrombus Growth. J. Biol. Chem. 2004, 279, 30697–30706. [Google Scholar] [CrossRef] [PubMed]
- Kradin, R.L.; Lynch, G.W.; Kurnick, J.T.; Erikson, M.; Colvin, R.B.; McDonagh, J. Factor XIII A is synthesized and expressed on the surface of U937 cells and alveolar macrophages. Blood 1987, 69, 778–785. [Google Scholar] [CrossRef]
- Muszbek, L.; Adány, R.; Kávai, M.; Boda, Z.; Lopaciuk, S. Monocytes of patients congenitally deficient in plasma Factor XIII lack Factor XIII subunit a antigen and transglutaminase activity. Thromb. Haemost. 1988, 59, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Kradin, R.L.; Boyle, L.A.; Preffer, F.I.; Callahan, R.J.; Barlai-Kovach, M.; Strauss, H.W.; Dubinett, S.; Kurnick, J.T. Tumor-derived inter-leukin-2-dependent lymphocytes in adoptive immunotherapy of lung cancer. Cancer Immunol. Immunother. 1987, 24, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Conkling, P.R.; Achyuthan, K.E.; Greenberg, C.S.; Newcomb, T.F.; Weinberg, J.B. Human mononuclear phagocyte transglutaminase activity cross-links fibrin. Thromb. Res. 1989, 55, 57–68. [Google Scholar] [CrossRef]
- Akimov, S.S.; Belkin, A.M. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fi-bronectin. Blood 2001, 98, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Akagi, A.; Tajima, S.; Ishibashi, A.; Matsubara, Y.; Takehana, M.; Kobayashi, S.; Yamaguchi, N. Type XVI Collagen is Expressed in Factor XIIIa+ Monocyte-Derived Dermal Dendrocytes and Constitutes a Potential Substrate for Factor XIIIa. J. Investig. Dermatol. 2002, 118, 267–274. [Google Scholar] [CrossRef]
- Piercy-Kotb, S.A.; Mousa, A.; Al-Jallad, H.F.; Myneni, V.D.; Chicatun, F.; Nazhat, S.N.; Kaartinen, M.T. Factor XIIIA transglutaminase expression and secretion by osteoblasts is regulated by extracellular matrix collagen and the MAP kinase signaling pathway. J. Cell. Physiol. 2012, 227, 2936–2946. [Google Scholar] [CrossRef]
- Ádány, R.; Bárdos, H.; Antal, M.; Módis, L.; Sárváry, A.; Szücs, S.; Balogh, I. Factor XIII of blood coagulation as a nuclear crosslink-ing enzyme. Thromb. Haemost. 2001, 85, 845–851. [Google Scholar] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E., Jr. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef]
- Pabst, M.J.; Pabst, K.M.; Handsman, D.B.; Beranova-Giorgianni, S.; Giorgianni, F. Proteome of monocyte priming by lipopolysaccharide, including changes in interleukin-1beta and leukocyte elastase inhibitor. Proteome Sci. 2008, 6, 13. [Google Scholar] [CrossRef]
- Töröcsik, D.; Szeles, L.; Paragh, G.; Rákosy, Z.; Bárdos, H.; Nagy, L.; Balázs, M.; Inbal, A.; Ádány, R. Factor XIII-A is involved in the regulation of gene expression in alternatively activated human macrophages. Thromb. Haemost. 2010, 104, 709–717. [Google Scholar] [CrossRef]
- Chaitidis, P.; O’Donnell, V.; Kuban, R.J.; Bermudez-Fajardo, A.; Ungethuem, U.; Kuhn, H. Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and -13. Cytokine 2005, 30, 366–377. [Google Scholar] [CrossRef]
- Gratchev, A.; Kzhyshkowska, J.; Utikal, J.; Goerdt, S. Interleukin-4 and dexamethasone counterregulate extracellular matrix re-modelling and phagocytosis in type-2 macrophages. Scand. J. Immunol. 2005, 61, 10–17. [Google Scholar] [CrossRef]
- May, R.C.; Machesky, L.M. Phagocytosis and the actin cytoskeleton. J. Cell Sci. 2001, 114, 1061–1077. [Google Scholar] [PubMed]
- Cohen, I.; Blankenberg, T.A.; Borden, D.; Kahn, D.R.; Veis, A. Factor XIIIa-catalyzed cross-linking of platelet and muscle actin. Regulation by nucleotides. Biochim. Biophys. Acta 1980, 628, 365–375. [Google Scholar] [CrossRef]
- Cohen, I.; Glaser, T.; Veis, A.; Bruner-Lorand, J. Ca2+-dependent cross-linking processes in human platelets. Biochim. Biophys. Acta 1981, 676, 137–147. [Google Scholar] [CrossRef]
- Serrano, K.; Devine, D.V. Intracellular Factor XIII crosslinks platelet cytoskeletal elements upon platelet activation. Thromb. Haemost. 2002, 88, 315–320. [Google Scholar] [CrossRef]
- Sarvary, A.; Szucs, S.; Balogh, I.; Becsky, A.; Bardos, H.; Kavai, M.; Seligsohn, U.; Egbring, R.; Lopaciuk, S.; Muszbek, L.; et al. Possible role of Factor XIII subunit A in Fcgamma and complement receptor-mediated phagocytosis. Cell Immunol. 2004, 228, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kavai, M.; Adany, R.; Pasti, G.; Suranyi, P.; Szucs, G.; Muszbek, L.; Bojan, F.; Szegedi, G. Marker profile, enzyme activity, and func-tion of a human myelomonocytic leukemia cell line. Cell Immunol. 1992, 139, 531–540. [Google Scholar] [CrossRef]
- Jayo, A.; Conde, I.; Lastres, P.; Jimenez-Yuste, V.; Gonzalez-Manchon, C. Possible role for cellular FXIII in monocyte-derived den-dritic cell motility. Eur. J. Cell Biol. 2009, 88, 423–431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higaki, S.; Nakano, K.; Onaka, S.; Amano, A.; Tanioka, Y.; Harada, K.; Hashimoto, S.; Sakaida, I.; Okita, K. Clinical significance of measuring blood coagulation Factor XIIIA regularly and continuously in patients with Crohn’s disease. J. Gastroenterol. Hepatol. 2006, 21, 1407–1411. [Google Scholar] [CrossRef]
- Chamouard, P.; Grunebaum, L.; Wiesel, M.L.; Sibilia, J.; Coumaros, G.; Wittersheim, C.; Baumann, R.; Cazenave, J.P. Significance of diminished Factor XIII in Crohn’s disease. Am. J. Gastroenterol. 1998, 93, 610–614. [Google Scholar] [CrossRef]
- Hudson, M.; Wakefield, A.J.; Hutton, R.A.; Sankey, E.A.; Dhillon, A.P.; More, L.; Sim, R.; Pounder, R.E. Factor XIIIA subunit and Crohn’s disease. Gut 1993, 34, 75–79. [Google Scholar] [CrossRef][Green Version]
- Inbal, A.; Muszbek, L. Coagulation Factor Deficiencies and Pregnancy Loss. Semin. Thromb. Hemost. 2003, 29, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Anwar, R.; Miloszewski, K.J.A. FACTOR XIII DEFICIENCY. Br. J. Haematol. 1999, 107, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Kohler, H.; Ichinose, A.; Seitz, R.; Ariens, R.; Muszbek, L.; Factor XIII and Fibrinogen SSC Subcommittee of the ISTH. Diagnosis and classification of Factor XIII deficiencies. J. Thromb. Haemost. 2011, 9, 1404–1406. [Google Scholar] [CrossRef]
- Durda, M.A.; Wolberg, A.S.; Kerlin, B.A. State of the art in Factor XIII laboratory assessment. Transfus. Apher. Sci. 2018, 57, 700–704. [Google Scholar] [CrossRef]
- Beckman, J.D.; Kasthuri, R.S.; Wolberg, A.S.; Ma, A.D. Challenges in diagnosis and management of acquired Factor XIII (FXIII) in-hibitors. Haemoph. Off. J. World Fed. Hemoph. 2018, 24, e417. [Google Scholar] [CrossRef] [PubMed]
- Kohler, H.P. Novel treatment for congenital FXIII deficiency. Blood 2012, 119, 5060–5061. [Google Scholar] [CrossRef][Green Version]
- Board, P.; Lososky, M.; Miloszewski, K. Factor XIII: Inherited and acquired deficiency. Blood Rev. 1993, 7, 229–242. [Google Scholar] [CrossRef]
- Asahina, T.; Kobayashi, T.; Takeuchi, K.; Kanayama, N. Congenital blood coagulation Factor XIII deficiency and successful deliv-eries: A review of the literature. Obstetr. Gynecol. Surv. 2007, 62, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Koseki-Kuno, S.; Yamakawa, M.; Dickneite, G.; Ichinose, A. Factor XIII A subunit-deficient mice developed severe uterine bleed-ing events and subsequent spontaneous miscarriages. Blood 2003, 102, 4410–4412. [Google Scholar] [CrossRef]
- Adány, R.; Muszbek, L. Immunohistochemical detection of Factor XIII subunit a in histiocytes of human uterus. Histochem. Cell Biol. 1989, 91, 169–174. [Google Scholar]
- Adány, R.; Glukhova, M.A.; Kabakov, A.Y.; Muszbek, L. Characterisation of connective tissue cells containing Factor XIII subunit a. J. Clin. Pathol. 1988, 41, 49–56. [Google Scholar] [CrossRef]
- Kobayashi, T.; Asahina, T.; Okada, Y.; Terao, T. Studies on the localization of adhesive proteins associated with the development of extravillous cytotrophoblast. Placenta 1999, 20, 35–53. [Google Scholar] [CrossRef]
- Asahina, T.; Kobayashi, T.; Okada, Y.; Goto, J.; Terao, T. Maternal Blood Coagulation Factor XIII is Associated with the Development of Cytotrophoblastic Shell. Placenta 2000, 21, 388–393. [Google Scholar] [CrossRef]
- Hsieh, L.; Nugent, D. Factor XIII deficiency. Haemophilia 2008, 14, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Stirling, Y.; Woolf, L.; North, W.R.; Seghatchian, M.J.; Meade, T.W. Haemostasis in normal pregnancy. Thromb. Haemost. 1984, 52, 176–182. [Google Scholar] [CrossRef]
- Coopland, A.; Alkjaersig, N.; Fletcher, A.P. Reduction in plasma Factor XIII (fibrin stabilizing factor) concentration during preg-nancy. J. Lab. Clin. Med. 1969, 73, 144–153. [Google Scholar] [PubMed]
- Nossel, H.L.; Lanzkowsky, P.; Levy, S.; Mibashan, R.S.; Hansen, J.D. A study of coagulation factor levels in women during labour and in their newborn infants. Thromb. Diath. Haemorrh. 1966, 16, 185–197. [Google Scholar] [CrossRef]
- Mercelina-Roumans, P.; Ubachs, J.; van Wersch, J. Smoking and pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 75, 113–114. [Google Scholar]
- Ogasawara, M.S.; Aoki, K.; Katano, K.; Ozaki, Y.; Suzumori, K. Factor XII but not protein C, protein S, antithrombin III, or Factor XIII is a predictor of recurrent miscarriage. Fertil. Steril. 2001, 75, 916–919. [Google Scholar] [CrossRef]
- Pasquier, E.; Martin, L.D.S.; Bohec, C.; Chauleur, C.; Bretelle, F.; Marhic, G.; Le Gal, G.; Debarge, V.; LeComte, F.; Denoual-Ziad, C.; et al. Enoxaparin for prevention of unexplained recurrent miscarriage: A multicenter randomized double-blind placebo-controlled trial. Blood 2015, 125, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Bagoly, Z.; Cairo, A.; Peyvandi, F. Novel aspects of Factor XIII deficiency. Curr. Opin. Hematol. 2011, 18, 366–372. [Google Scholar] [CrossRef]
- Soendergaard, C.; Kvist, P.H.; Seidelin, J.B.; Nielsen, O.H. Tissue-regenerating functions of coagulation Factor XIII. J. Thromb. Haemost. 2013, 11, 806–816. [Google Scholar] [CrossRef]
- Ajzner, É.; Schlammadinger, Á.; Kerényi, A.; Bereczky, Z.; Katona, É.; Haramura, G.; Boda, Z.; Muszbek, L. Severe bleeding complica-tions caused by an autoantibody against the B subunit of plasma Factor XIII: A novel form of acquired Factor XIII deficiency. Blood J. Am. Soc. Hematol. 2009, 113, 723–725. [Google Scholar]
- Ballerini, G.; Gemmati, D.; Moratelli, S.; Morelli, P.; Serino, M.L. A photometric method for the dosage of Factor XIII applied to the study of chronic hepatopathies. Thromb. Res. 1995, 78, 451–456. [Google Scholar] [CrossRef]
- Nikolajsen, C.L.; Dyrlund, T.F.; Poulsen, E.T.; Enghild, J.J.; Scavenius, C. Coagulation Factor XIIIa substrates in human plasma: Identification and incorporation into the clot. J. Chem. 2014, 289, 6526–6534. [Google Scholar] [CrossRef] [PubMed]
- Mutch, N.J.; Engel, R.; De Willige, S.U.; Philippou, H.; Ariëns, R.A.S. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood 2010, 115, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.R.; Booth, N.A.; Mutch, N.J. The antifibrinolytic function of Factor XIII is exclusively expressed through alpha-antiplasmin cross-linking. Blood 2011, 117, 6371–6374. [Google Scholar] [CrossRef] [PubMed]
- Robbie, L.A.; Kinghorn, S.; Exley, R.; Booth, N.A.; Ritchie, H. Monocyte Plasminogen Activator Inhibitor 2 (PAI-2) Inhibits u-PA-mediated Fibrin Clot Lysis and Is Cross-linked to Fibrin. Thromb. Haemost. 1999, 81, 96–103. [Google Scholar] [CrossRef]
- Kolev, K.; Tenekedjiev, K.; Komorowicz, E.; Machovich, R. Functional Evaluation of the Structural Features of Proteases and Their Substrate in Fibrin Surface Degradation. J. Biol. Chem. 1997, 272, 13666–13675. [Google Scholar] [CrossRef]
- Sobel, J.H.; Gawinowicz, M.A. Identification of the α Chain Lysine Donor Sites Involved in Factor XIIIa Fibrin Cross-linking. J. Biol. Chem. 1996, 271, 19288–19297. [Google Scholar] [CrossRef]
- Hethershaw, E.L.; La Corte, A.L.C.; Duval, C.; Ali, M.; Grant, P.J.; Ariëns, R.A.S.; Philippou, H. The effect of blood coagulation Factor XIII on fibrin clot structure and fibrinolysis. J. Thromb. Haemost. 2014, 12, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Park, D.; Lesty, C.; Soria, J.; Soria, C.; Montalescot, G.; Weisel, J.W. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: Dynamic and structural approaches by confocal microscopy. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1354–1361. [Google Scholar] [CrossRef]
- Dunn, E.J.; Philippou, H.; Ariëns, R.A.S.; Grant, P.J. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetoloia 2006, 49, 1071–1080. [Google Scholar] [CrossRef]
- Longstaff, C.; Thelwell, C.; Williams, S.C.; Silva, M.M.; Szabo, L.; Kolev, K. The interplay between tissue plasminogen activator do-mains and fibrin structures in the regulation of fibrinolysis: Kinetic and microscopic studies. Blood 2011, 117, 661–668. [Google Scholar] [CrossRef]
- Mutch, N.J.; Koikkalainen, J.S.; Fraser, S.R.; Duthie, K.M.; Griffin, M.; Mitchell, J.; Watson, H.G.; Booth, N.A. Model thrombi formed un-der flow reveal the role of Factor XIII-mediated cross-linking in resistance to fibrinolysis. J. Thromb. Haemost. JTH 2010, 8, 2017–2024. [Google Scholar] [CrossRef]
- Minkema, J.; Bouma, B.; Jansen, J. Cross-linking of alpha 2-antiplasmin to fibrin is a key factor in regulating blood clot lysis: Species differences. Blood Coagul. Fibrinol. Int. J. Haemost. Thromb. 1993, 4, 869–875. [Google Scholar]
- Jensen, P.H.; Lorand, L.; Ebbesen, P.; Gliemann, J. Type-2 plasminogen-activator inhibitor is a substrate for trophoblast transglu-taminase and Factor XIIIa: Transglutaminase-catalyzed cross-linking to cellular and extracellular structures. Eur. J. Biochem. 1993, 214, 141–146. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Chung, K.H.; McKee, P.A. A novel plasma proteinase potentiates α2-antiplasmin inhibi-tion of fibrin digestion. Blood 2004, 103, 3783–3788. [Google Scholar] [CrossRef]
- Bangert, K.; Johnsen, A.H.; Christensen, U.; Thorsen, S. Different N-terminal forms of α2-plasmin inhibitor in human plasma. Biochem. J. 1993, 291, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Sumi, Y.; Ichikawa, Y.; Nakamura, Y.; Miura, O.; Aoki, N. Expression and Characterization of Pro α2-Plasmin Inhibitor. J. Biochem. 1989, 106, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Rijken, D.; Abdul, S.; Malfliet, J.J.M.C.; Leebeek, F.W.G.; De Willige, S.U. Compaction of fibrin clots reveals the antifibrinolytic effect of Factor XIII. J. Thromb. Haemost. 2016, 14, 1453–1461. [Google Scholar] [CrossRef][Green Version]
- Reed, G.L.; Matsueda, G.R.; Haber, E. Fibrin-fibrin and alpha 2-antiplasmin-fibrin cross-linking by platelet Factor XIII increases the resistance of platelet clots to fibrinolysis. Trans. Assoc. Am. Physicians 1991, 104, 21–28. [Google Scholar] [PubMed]
- Reed, G.L.; Matsueda, G.R.; Haber, E. Platelet Factor XIII increases the fibrinolytic resistance of platelet-rich clots by accelerating the crosslinking of alpha 2-antiplasmin to fibrin. Thromb. Haemost. 1992, 68, 315–320. [Google Scholar] [PubMed]
- Francis, C.W.; Marder, V.J. Rapid formation of large molecular weight alpha-polymers in cross-linked fibrin induced by high Factor XIII concentrations. Role of platelet Factor XIII. J. Clin. Investig. 1987, 80, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Hevessy, Z.; Haramura, G.; Boda, Z.; Udvardy, M.; Muszbek, L. Promotion of the crosslinking of fibrin and alpha 2-antiplasmin by platelets. Thromb. Haemost. 1996, 75, 161–167. [Google Scholar] [PubMed]
- Rubens, F.D.; Perry, D.W.; Hatton, M.W.; Bishop, P.D.; Packham, M.A.; Kinlough-Rathbone, R.L. Platelet accumulation on fibrin-coated polyethylene: Role of platelet activation and Factor XIII. Thromb. Haemost. 1995, 73, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Aleman, M.M.; Byrnes, J.R.; Wang, J.G.; Tran, R.; Lam, W.A.; Di Paola, J.; Mackman, N.; Degen, J.L.; Flick, M.J.; Wolberg, A.S. Factor XIII activity mediates red blood cell retention in venous thrombi. J. Clin. Investig. 2014, 124, 3590–3600. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Duval, C.; Wang, Y.; Hansen, C.E.; Ahn, B.; Mooberry, M.J.; Clark, M.A.; Johnsen, J.M.; Lord, S.T.; Lam, W.A.; et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain cross-linking. Blood 2015, 126, 1940–1948. [Google Scholar] [CrossRef]
- Paragh, L.; Törőcsik, D. Factor XIII Subunit A in the Skin: Applications in Diagnosis and Treatment. BioMed Res. Int. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Seitz, R.; Kohler, H.P.; Schroeder, V.; Muszbek, L.; Ariens, R.A.S.; Seifried, E.; Oldenburg, J.; Ivaskevicius, V. International Registry on Factor XIII Deficiency: A basis formed mostly on European data. Thromb. Haemost. 2007, 97, 914–921. [Google Scholar] [CrossRef]
- Seitz, R.; Duckert, F.; Lopaciuk, S.; Muszbek, L.; Rodeghiero, F.; Seligsohn, U. ETRO Working Party on Factor XIII Questionnaire on Congenital Factor XIII Deficiency in Europe: Status and Perspectives. Semin. Thromb. Hemost. 1996, 22, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Inbal, A.; Lubetsky, A.; Krapp, T.; Castel, D.; Shaish, A.; Dickneitte, G.; Módis, L.; Muszbek, L.; Inbal, A. Impaired wound healing in Factor XIII deficient mice. Thromb. Haemost. 2005, 94, 432–437. [Google Scholar] [CrossRef]
- D’Argenio, G.; Grossman, A.; Cosenza, V.; Della Valle, N.; Mazzacca, G.; Bishop, P.D. Recombinant Factor XIII Improves Established Experimental Colitis in Rats. Dig. Dis. Sci. 2000, 45, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komáromi, I.; Katona, É. Factor XIII: A coagulation factor with multiple plasmatic and cellu-lar functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef]
- Magwenzi, S.G.; Ajjan, R.A.; Standeven, K.F.; Parapia, L.A.; Naseem, K.M. Factor XIII supports platelet activation and enhances thrombus formation by matrix proteins under flow conditions. J. Thromb. Haemost. 2011, 9, 820–833. [Google Scholar] [CrossRef]
- Lanir, N.; Ciano, P.S.; Van De Water, L.; McDonagh, J.; Dvorak, A.M.; Dvorak, H.F. Macrophage migration in fibrin gel matrices. II. Effects of clotting Factor XIII, fibronectin, and glycosaminoglycan content on cell migration. J. Immunol. 1988, 140, 2340–2349. [Google Scholar] [PubMed]
- Grinnell, F.; Feld, M.; Minter, D. Fibroblast adhesion to fibrinogen and fibrin substrata: Requirement for cold-insoluble globulin (plasma fibronectin). Cell 1980, 19, 517–525. [Google Scholar] [CrossRef]
- Dardik, R.; Shenkman, B.; Tamarin, I.; Eskaraev, R.; Hársfalvi, J.; Varon, D.; Inbal, A. Factor XIII mediates adhesion of platelets to endothelial cells through αvβ3 and glycoprotein IIb/IIIa integrins. Thromb. Res. 2002, 105, 317–323. [Google Scholar] [CrossRef]
- Törőcsik, D.; Bárdos, H.; Nagy, L.; Ádány, R. Identification of Factor XIII-A as a marker of alternative macrophage activation. Cell. Mol. Life Sci. 2005, 62, 2132–2139. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Yamamoto, T.; Matsubara, S.; Kukita, I.; Takeya, M.; Miyauchi, Y.; Kambara, T. Factor XIII-dependent generation of 5th complement component(C5)-derived monocyte chemotactic factor coinciding with plasma clotting. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1992, 1138, 53–61. [Google Scholar] [CrossRef]
- Semba, U.; Chen, J.; Ota, Y.; Jia, N.; Arima, H.; Nishiura, H.; Yamamoto, T. A plasma protein indistinguishable from ribosomal pro-tein S19: Conversion to a monocyte chemotactic factor by a Factor XIIIa-catalyzed reaction on activated platelet membrane phosphatidylserine in association with blood coagulation. Am. J. Pathol. 2010, 176, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Noll, T.; Wozniak, G.; McCarson, K.; Hajimohammad, A.; Metzner, H.J.; Inserte, J.; Kummer, W.; Hehrlein, F.W.; Piper, H.M. Effect of Factor XIII on Endothelial Barrier Function. J. Exp. Med. 1999, 189, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Shinbo, K.; Takahashi, M.; Matsuishi, T. Suppressive effect of human blood coagulation Factor XIII on the vascular permeability induced by anti-guinea pig endothelial cell antiserum in guinea pigs. Thromb. Res. 1993, 71, 139–148. [Google Scholar] [CrossRef]
- Wozniak, G.; Noll, T.; Akintürk, H.; Thul, J.; Müller, M. Factor XIII prevents development of myocardial edema in children under-going surgery for congenital heart disease. Ann. N. Y. Acad. Sci. 2001, 936, 617–620. [Google Scholar] [CrossRef]
- Schroth, M.; Meissner, U.; Cesnjevar, R.; Weyand, M.; Singer, H.; Rascher, W.; Klinge, J. Plasmatic [corrected] Factor XIII reduces severe pleural effusion in children after open-heart surgery. Pediatr. Cardiol. 2006, 27, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Solomon, A.; Loscalzo, J.; Eskaraev, R.; Bialik, A.; Goldberg, I.; Schiby, G.; Inbal, A. Novel Proangiogenic Effect of Factor XIII Associated with Suppression of Thrombospondin 1 Expression. Arter. Thromb. Vasc. Biol. 2003, 23, 1472–1477. [Google Scholar] [CrossRef]
- Dardik, A.; Chen, L.; Frattini, J.; Asada, H.; Aziz, F.; Kudo, F.A.; Sumpio, B.E. Differential effects of orbital and laminar shear stress on endothelial cells. J. Vasc. Surg. 2005, 41, 869–880. [Google Scholar] [CrossRef]
- Auerbach, W.; Auerbach, R. Angiogenesis inhibition: A review. Pharmacol. Ther. 1994, 63, 265–311. [Google Scholar] [CrossRef]
- Nugent, D.; Ashley, C.; Garcia-Talavera, J.; Lo, L.; Mehdi, A.; Mangione, A. Pharmacokinetics and safety of plasma-derived Factor XIII concentrate (human) in patients with congenital Factor XIII deficiency. Haemophilia 2015, 21, 95–101. [Google Scholar] [CrossRef]
- Suzuki, H.; Kaneda, T. Tooth extraction in two patients who had a congenital deficiency of Factor XIII. J. Oral Maxillofac. Surg. 1985, 43, 221–224. [Google Scholar] [CrossRef]
- Key, N.S.; Negrier, C. Coagulation factor concentrates: Past, present, and future. Lancet 2007, 370, 439–448. [Google Scholar] [CrossRef]
- Brackmann, H.H.; Egbring, R.; Ferster, A.; Fondu, P.; Girardel, J.M.; Kreuz, W.; Masure, R.; Miloszewski, K.; Stibbe, J.; Zimmermann, R.; et al. Pharmacokinetics and tolerability of Factor XIII concentrates prepared from human placenta or plasma: A crossover ran-domised study. Thromb. Haemost. 1995, 74, 622–625. [Google Scholar] [CrossRef]
- Lusher, J.; Pipe, S.W.; Alexander, S.; Nugent, D. Prophylactic therapy with Fibrogammin P is associated with a decreased inci-dence of bleeding episodes: A retrospective study. Haemophilia 2010, 16, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Katona, É.; Muszbek, L. Diagnosis and Management of Congenital and Acquired FXIII Deficiencies. Semin. Thromb. Hemost. 2016, 42, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, A. Physiopathology and Regulation of Factor XIII. Thromb. Haemost. 2001, 86, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L. Sol Sherry Lecture in Thrombosis: Research on clot stabilization provides clues for improving thrombolytic thera-pies. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2–9. [Google Scholar] [CrossRef]
- Nugent, D. Corifact/Fibrogammin(R) P in the prophylactic treatment of hereditary Factor XIII deficiency: Results of a prospec-tive, multicenter, open-label study. Thromb. Res. 2012, 130 (Suppl. S2), S12–S14. [Google Scholar] [CrossRef]
- Lovejoy, A.E.; Reynolds, T.C.; Visich, J.E.; Butine, M.D.; Young, G.; Belvedere, M.A.; Blain, R.C.; Pederson, S.M.; Ishak, L.M.; Nugent, D.J. Safety and pharmacokinetics of recombinant Factor XIII-A2 administration in patients with congenital Factor XIII deficiency. Blood 2006, 108, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Carcao, M.; Altisent, C.; Castaman, G.; Fukutake, K.; Kerlin, B.A.; Kessler, C.; Lassila, R.; Nugent, D.; Oldenburg, J.; Garly, M.-L.; et al. Recombinant FXIII (rFXIII-A2) Prophylaxis Prevents Bleeding and Allows for Surgery in Patients with Congenital FXIII A-Subunit Deficiency. Thromb. Haemost. 2018, 118, 451–460. [Google Scholar] [CrossRef]
- Naderi, M.; Dorgalaleh, A.; Alizadeh, S.; Tabibian, S.; Hosseini, S.; Shamsizadeh, M.; Bamedi, T. Clinical manifestations and man-agement of life-threatening bleeding in the largest group of patients with severe Factor XIII deficiency. Int. J. Hematol. 2014, 100, 443–449. [Google Scholar] [CrossRef]
- Abdel-Samad, N. Treatment with Recombinant Factor XIII (Tretten) in a Pregnant Woman with Factor XIII Deficiency. Am. J. Case Rep. 2017, 18, 436–439. [Google Scholar] [CrossRef]
- Ichinose, A.; Japanese Collaborative Research Group on AH. Autoimmune acquired Factor XIII deficiency due to anti-Factor XIII/13 antibodies: A summary of 93 patients. Blood Rev. 2017, 31, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Osaki, T.; Ichinose, A. Anti-Factor XIII A subunit (FXIII-A) autoantibodies block FXIII-A2 B2 assembly and steal FXIII-A from native FXIII-A2 B. J. Thromb. Haemost. 2015, 13, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Tone, K.J.; James, T.E.; Fergusson, D.A.; Tinmouth, A.; Tay, J.; Avey, M.T.; Kilty, S.; Lalu, M.M. Acquired Factor XIII Inhibitor in Hospi-talized and Perioperative Patients: A Systematic Review of Case Reports and Case Series. Transfus. Med. Rev. 2016, 30, 123–131. [Google Scholar] [CrossRef]
- Hiippala, S.T.; Myllyla, G.J.; Vahtera, E.M. Hemostatic factors and replacement of major blood loss with plasma-poor red cell con-centrates. Anesth. Analg. 1995, 81, 360–365. [Google Scholar] [PubMed]
- Floccard, B.; Rugeri, L.; Faure, A.; Saint Denis, M.; Boyle, E.M.; Peguet, O.; Levrat, A.; Guillaume, C.; Marcotte, G.; Vulliez, A. Early co-agulopathy in trauma patients: An on-scene and hospital admission study. Injury 2012, 43, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hagemo, J.S.; Stanworth, S.; Juffermans, N.P.; Brohi, K.; Cohen, M.J.; Johansson, P.I.; Røislien, J.; Eken, T.A.; Næss, P.; Gaarder, C. Prevalence, predictors and outcome of hypofibrinogenaemia in trauma: A multicentre observational study. Crit. Care 2014, 18, R52. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Stanworth, S.J.; Curry, N. Do we still need cryoprecipitate? Cryoprecipitate and fibrinogen concentrate as treatments for major hemorrhage—How do they compare? Expert Rev. Hematol. 2018, 11, 351–360. [Google Scholar] [CrossRef]
- Rourke, C.; Curry, N.; Khan, S.; Taylor, R.; Raza, I.; Davenport, R.; Stanworth, S.; Brohi, K. Fibrinogen levels during trauma hemor-rhage, response to replacement therapy, and association with patient outcomes. J. Thromb. Haemost. 2012, 10, 1342–1351. [Google Scholar] [CrossRef]
- Morrison, J.J.; Ross, J.D.; Dubose, J.J.; Jansen, J.O.; Midwinter, M.J.; Rasmussen, T.E. Association of cryoprecipitate and tranexamic acid with improved survival following wartime injury: Findings from the MATTERs II Study. JAMA Surg. 2013, 148, 218–225. [Google Scholar] [CrossRef]
- Schlimp, C.J.; Voelckel, W.; Inaba, K.; Maegele, M.; Ponschab, M.; Schöchl, H. Estimation of plasma fibrinogen levels based on hemoglobin, base excess and Injury Severity Score upon emergency room admission. Crit. Care 2013, 17, R137. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.; Fries, D.; Velik-Salchner, C.; Reif, C.; Klingler, A.; Innerhofer, P. The In Vitro Effects of Fibrinogen Concentrate, Factor XIII and Fresh Frozen Plasma on Impaired Clot Formation After 60% Dilution. Anesth. Analg. 2008, 106, 1360–1365. [Google Scholar] [CrossRef]
- Wettstein, P.; Haeberli, A.; Stutz, M.; Rohner, M.; Corbetta, C.; Gabi, K.; Schnider, T.; Korte, W. Decreased Factor XIII Availability for Thrombin and Early Loss of Clot Firmness in Patients with Unexplained Intraoperative Bleeding. Anesth. Analg. 2004, 99, 1564–1569. [Google Scholar] [CrossRef]
- Chandler, W.L.; Patel, M.A.; Gravelle, L.; Soltow, L.O.; Lewis, K.; Bishop, P.D.; Spiess, B.D. Factor XIIIA and clot strength after cardio-pulmonary bypass. Blood Coagul. Fibrinolysis 2001, 12, 101–108. [Google Scholar] [CrossRef]
- Shainoff, J.R.; Estafanous, F.G.; Yared, J.P.; DiBello, P.M.; Kottke-Marchant, K.; Loop, F.D. Low Factor XIIIA levels are associated with increased blood loss after coronary artery bypass grafting. J. Thorac. Cardiovasc. Surg. 1994, 108, 437–445. [Google Scholar] [CrossRef]
- Korte, W.C.; Szadkowski, C.; Gahler, A.; Gabi, K.; Kownacki, E.; Eder, M.; Degiacomi, P.; Zoller, N.; Devay, J.; Lange, J.; et al. Factor XIII substitution in surgical cancer patients at high risk for intraoperative bleeding. Anesthesiology 2009, 110, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Shebuski, R.J.; Sitko, G.R.; Claremon, D.A.; Baldwin, J.J.; Remy, D.C.; Stern, A.M. Inhibition of Factor XIIIa in a canine model of coro-nary thrombosis: Effect on reperfusion and acute reocclusion after recombinant tissue-type plasminogen activator. Blood 1990, 75, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Finney, S.; Seale, L.; Sawyer, R.T.; Wallis, R.B. Tridegin, a new peptidic inhibitor of Factor XIIIa, from the blood-sucking leech Haementeria ghilianii. Biochem. J. 1997, 324, 797–805. [Google Scholar] [CrossRef][Green Version]
- Schmitz, T.; Paul George, A.A.; Nubbemeyer, B.; Bauml, C.A.; Steinmetzer, T.; Ohlenschlager, O.; Biswas, A.; Imhof, D. NMR-Based Structural Characterization of a Two-Disulfide-Bonded Analogue of the FXIIIa Inhibitor Tridegin: New Insights into Structure-Activity Relationships. Int. J. Mol. Sci. 2021, 22, 880. [Google Scholar] [CrossRef]
- Avery, C.A.; Pease, R.J.; Smith, K.; Boothby, M.; Buckley, H.M.; Grant, P.J.; Fishwick, C.W. (±) cis-bisamido epoxides: A novel series of potent FXIII-A inhibitors. Eur. J. Med. Chem. 2015, 98, 49–53. [Google Scholar] [CrossRef]
- Pasternack, R.; Büchold, C.; Jähnig, R.; Pelzer, C.; Sommer, M.; Heil, A.; Florian, P.; Nowak, G.; Gerlach, U.; Hils, M. Novel inhibitor ZED3197 as potential drug candidate in anticoagulation targeting coagulation FXIIIa (F13a). J. Thromb. Haemost. 2020, 18, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Strilchuk, A.W.; Meixner, S.C.; Leung, J.; Safikhan, N.; Kulkarni, J.A.; Russell, H.M.; Van Der Meel, R.; Sutherland, M.R.; Iii, A.P.O.; Palumbo, J.; et al. Sustained Depletion of FXIII-A by Inducing Acquired FXIII-B Deficiency. Blood 2020, 136, 2946–2954. [Google Scholar] [CrossRef]
- Dull, K.; Fazekas, F.; Törőcsik, D. Factor XIII-A in Diseases: Role Beyond Blood Coagulation. Int. J. Mol. Sci. 2021, 22, 1459. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshehri, F.S.M.; Whyte, C.S.; Mutch, N.J. Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. Int. J. Mol. Sci. 2021, 22, 3055. https://doi.org/10.3390/ijms22063055
Alshehri FSM, Whyte CS, Mutch NJ. Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. International Journal of Molecular Sciences. 2021; 22(6):3055. https://doi.org/10.3390/ijms22063055
Chicago/Turabian StyleAlshehri, Fahad S. M., Claire S. Whyte, and Nicola J. Mutch. 2021. "Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing" International Journal of Molecular Sciences 22, no. 6: 3055. https://doi.org/10.3390/ijms22063055
APA StyleAlshehri, F. S. M., Whyte, C. S., & Mutch, N. J. (2021). Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. International Journal of Molecular Sciences, 22(6), 3055. https://doi.org/10.3390/ijms22063055