
Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene

 ,
, 

Abstract
1. Introduction
2. Results
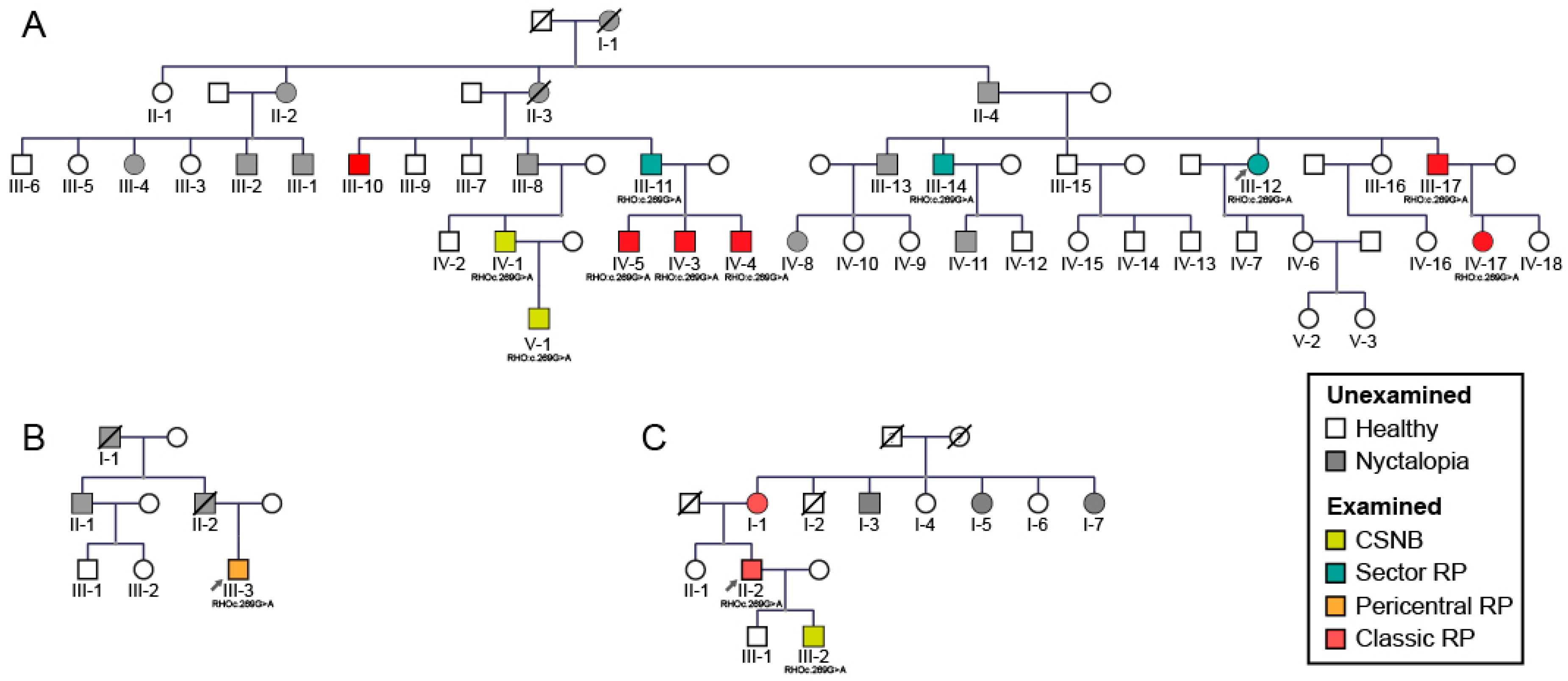
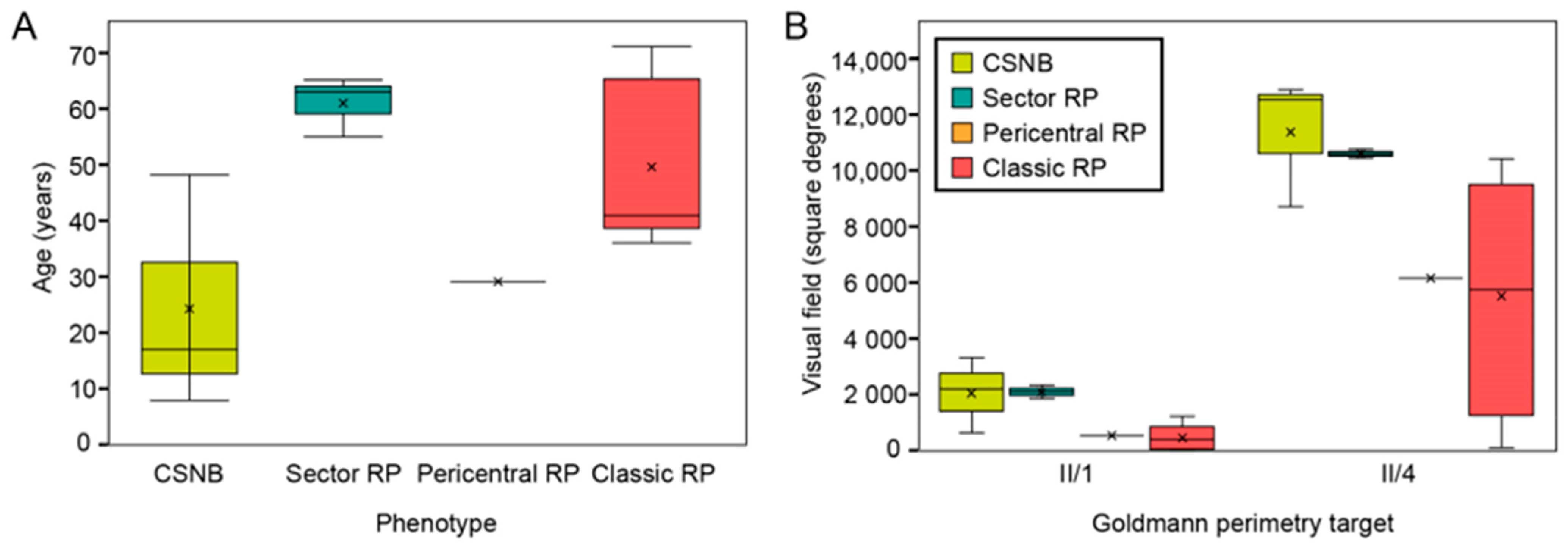
2.1. Clinical and Genetic Findings
2.2. Gender Disbalance
3. Discussion
3.1. Molecular Characteristics of RHO Mutations
3.2. Mouse Models with RHO p.G90D
3.3. Genotype–Phenotype Correlations in RHO
3.4. Phenotypes Associated with p.G90D
3.4.1. Congenital Stationary Night Blindness
3.4.2. Classic Retinitis Pigmentosa
3.4.3. Sector Retinitis Pigmentosa
3.4.4. Pericentral Retinitis Pigmentosa
3.5. Gender Disbalance
4. Materials and Methods
4.1. Patients
4.2. Genetic and Bioinformatic Analysis
4.3. Clinical Examination
4.4. Phenotype Classification
- Congenital stationary night blindness (CSNB): normal FAF and visual field;
- Classic retinitis pigmentosa (RP): concentric retinal degeneration delineated by a hyperautofluorescent ring on FAF;
- Sector RP: peripheral degeneration extending one to two quadrants on FAF;
- Pericentral RP: annular area of retinal degeneration encompassed by a double hyperautofluorescent ring on FAF.
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RHO | Rhodopsin gene |
| CSNB | Congenital stationary night blindness |
| RP | Retinitis pigmentosa |
| OCT | Optical coherence tomography |
| FAF | Fundus autofluorescence imaging |
| ERG | Electroretinography |
| GPCR | G protein-coupled receptor |
| PSB | Protonated Schiff base |
| CME | Cystoid macular edema |
| DA | Dark adapted |
| LA | Light adapted |
| WT | Wild type |
| UCSC | University of California Santa Cruz |
| BWA | Burrows–Wheeler aligner |
| GATK | Genome Analysis Toolkit |
| CONIFER | Copy Number Inference From Exome Reads |
| gnomAD | Genome Aggregation Database |
| ffERG | Full-field electroretinography |
| mfERG | Multifocal electroretinography |
| PERG | Pattern electroretinography |
References
- Katayama, K.; Takeyama, Y.; Enomoto, A.; Imai, H.; Kandori, H. Disruption of hydrogen-bond network in rhodopsin mutations cause night blindness. J. Mol. Biol. 2020, 432, 5378–5389. [Google Scholar] [CrossRef]
- Naash, M.I.; Wu, T.H.; Chakraborty, D.; Fliesler, S.J.; Ding, X.Q.; Nour, M.; Peachey, N.S.; Lem, J.; Qtaishat, N.; Al-Ubaidi, M.R.; et al. Retinal abnormalities associated with the G90D mutation in opsin. J. Comp. Neurol. 2004, 478, 149–163. [Google Scholar] [CrossRef]
- Kawamura, S.; Colozo, A.T.; Ge, L.; Müller, D.J.; Park, P.S. Structural, energetic, and mechanical perturbations in rhodopsin mutant that causes congenital stationary night blindness. J. Biol. Chem. 2012, 287, 21826–21835. [Google Scholar] [CrossRef] [PubMed]
- McAlear, S.D.; Kraft, T.W.; Gross, A.K. 1 rhodopsin mutations in congenital night blindness. Adv. Exp. Med. Biol. 2010, 664, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, O.G. Focus on molecules: Rhodopsin. Exp. Eye Res. 2005, 81, 366–367. [Google Scholar] [CrossRef]
- Xiao, T.; Xu, K.; Zhang, X.; Xie, Y.; Li, Y. Sector retinitis pigmentosa caused by mutations of the RHO gene. Eye 2019, 33, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’Angelo, L.; et al. Clinical and genetic analysis of a European Cohort with pericentral retinitis pigmentosa. Int. J. Mol. Sci. 2019, 21, 86. [Google Scholar] [CrossRef]
- Souied, E.; Soubrane, G.; Benlian, P.; Coscas, G.J.; Gerber, S.; Munnich, A.; Kaplan, J. Retinitis punctata albescens associated with the Arg135Trp mutation in the rhodopsin gene. Am. J. Ophthalmol. 1996, 121, 19–25. [Google Scholar] [CrossRef]
- Colozo, A.T.; Vasudevan, S.; Park, P.S. Retinal degeneration in mice expressing the constitutively active G90D rhodopsin mutant. Hum. Mol. Genet. 2020, 29, 881–891. [Google Scholar] [CrossRef]
- Park, P.S. Constitutively active rhodopsin and retinal disease. Adv. Pharmacol. 2014, 70, 1–36. [Google Scholar] [CrossRef]
- Kawamura, S.; Gerstung, M.; Colozo, A.T.; Helenius, J.; Maeda, A.; Beerenwinkel, N.; Park, P.S.; Müller, D.J. Kinetic, energetic, and mechanical differences between dark-state rhodopsin and opsin. Structure 2013, 21, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Zvyaga, T.A.; Fahmy, K.; Siebert, F.; Sakmar, T.P. Characterization of the mutant visual pigment responsible for congenital night blindness: A biochemical and Fourier-transform infrared spectroscopy study. Biochemistry 1996, 35, 7536–7545. [Google Scholar] [CrossRef] [PubMed]
- Toledo, D.; Ramon, E.; Aguilà, M.; Cordomí, A.; Pérez, J.J.; Mendes, H.F.; Cheetham, M.E.; Garriga, P. Molecular mechanisms of disease for mutations at Gly-90 in rhodopsin. J. Biol. Chem. 2011, 286, 39993–40001. [Google Scholar] [CrossRef]
- Sieving, P.A.; Fowler, M.L.; Bush, R.A.; Machida, S.; Calvert, P.D.; Green, D.G.; Makino, C.L.; McHenry, C.L. Constitutive “light” adaptation in rods from G90D rhodopsin: A mechanism for human congenital nightblindness without rod cell loss. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 5449–5460. [Google Scholar] [CrossRef]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef]
- Sieving, P.A.; Richards, J.E.; Naarendorp, F.; Bingham, E.L.; Scott, K.; Alpern, M. Dark-light: Model for nightblindness from the human rhodopsin Gly-90-->Asp mutation. Proc. Natl. Acad. Sci. USA 1995, 92, 880–884. [Google Scholar] [CrossRef]
- Singhal, A.; Guo, Y.; Matkovic, M.; Schertler, G.; Deupi, X.; Yan, E.C.; Standfuss, J. Structural role of the T94I rhodopsin mutation in congenital stationary night blindness. EMBO Rep. 2016, 17, 1431–1440. [Google Scholar] [CrossRef]
- Neidhardt, J.; Barthelmes, D.; Farahmand, F.; Fleischhauer, J.C.; Berger, W. Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1630–1635. [Google Scholar] [CrossRef][Green Version]
- Nguyen, X.T.; Talib, M.; van Cauwenbergh, C.; van Schooneveld, M.J.; Fiocco, M.; Wijnholds, J.; Ten Brink, J.B.; Florijn, R.J.; Schalij-Delfos, N.E.; Dagnelie, G.; et al. Clinical characteristics and natural history of rho-associated retinitis pigmentosa: A long-term follow-up study. Retina 2021, 41, 213–223. [Google Scholar] [CrossRef]
- Nash, Z.A.; Naash, M.I. Light/dark translocation of alphatransducin in mouse photoreceptor cells expressing G90D mutant opsin. Adv. Exp. Med. Biol. 2006, 572, 125–131. [Google Scholar] [CrossRef]
- Dizhoor, A.M.; Woodruff, M.L.; Olshevskaya, E.V.; Cilluffo, M.C.; Cornwall, M.C.; Sieving, P.A.; Fain, G.L. Night blindness and the mechanism of constitutive signaling of mutant G90D rhodopsin. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11662–11672. [Google Scholar] [CrossRef]
- Conley, S.M.; Whalen, P.; Lewin, A.S.; Naash, M.I. Characterization of ribozymes targeting a congenital night blindness mutation in rhodopsin mutation. Adv. Exp. Med. Biol. 2016, 854, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Coussa, R.G.; Basali, D.; Maeda, A.; DeBenedictis, M.; Traboulsi, E.I. Sector retinitis pigmentosa: Report of ten cases and a review of the literature. Mol. Vis. 2019, 25, 869–889. [Google Scholar]
- Comander, J.; Weigel-DiFranco, C.; Maher, M.; Place, E.; Wan, A.; Harper, S.; Sandberg, M.A.; Navarro-Gomez, D.; Pierce, E.A. The genetic basis of pericentral retinitis pigmentosa-a form of mild retinitis pigmentosa. Genes 2017, 8, 256. [Google Scholar] [CrossRef]
- Reiff, C.; Owczarek-Lipska, M.; Spital, G.; Röger, C.; Hinz, H.; Jüschke, C.; Thiele, H.; Altmüller, J.; Nürnberg, P.; Da Costa, R.; et al. The mutation p.E113K in the Schiff base counterion of rhodopsin is associated with two distinct retinal phenotypes within the same family. Sci. Rep. 2016, 6, 36208. [Google Scholar] [CrossRef]
- Marmor, M.F.; Zeitz, C. Riggs-type dominant congenital stationary night blindness: ERG findings, a new GNAT1 mutation and a systemic association. Doc. Ophthalmol. Adv. Ophthalmol. 2018, 137, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Jobling, A.I.; Vessey, K.A.; Waugh, M.; Mills, S.A.; Fletcher, E.L. A naturally occurring mouse model of achromatopsia: Characterization of the mutation in cone transducin and subsequent retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3350–3359. [Google Scholar] [CrossRef]
- Mastey, R.R.; Georgiou, M.; Langlo, C.S.; Kalitzeos, A.; Patterson, E.J.; Kane, T.; Singh, N.; Vincent, A.; Moore, A.T.; Tsang, S.H.; et al. Characterization of retinal structure in ATF6-associated achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2631–2640. [Google Scholar] [CrossRef]
- Hirji, N.; Georgiou, M.; Kalitzeos, A.; Bainbridge, J.W.; Kumaran, N.; Aboshiha, J.; Carroll, J.; Michaelides, M. Longitudinal assessment of retinal structure in achromatopsia patients with long-term follow-up. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5735–5744. [Google Scholar] [CrossRef]
- Georgiou, M.; Litts, K.M.; Kalitzeos, A.; Langlo, C.S.; Kane, T.; Singh, N.; Kassilian, M.; Hirji, N.; Kumaran, N.; Dubra, A.; et al. Adaptive optics retinal imaging in CNGA3-associated achromatopsia: Retinal characterization, interocular symmetry, and intrafamilial variability. Investig. Ophthalmol. Vis. Sci. 2019, 60, 383–396. [Google Scholar] [CrossRef]
- Thiadens, A.A.; Somervuo, V.; van den Born, L.I.; Roosing, S.; van Schooneveld, M.J.; Kuijpers, R.W.; van Moll-Ramirez, N.; Cremers, F.P.; Hoyng, C.B.; Klaver, C.C. Progressive loss of cones in achromatopsia: An imaging study using spectral-domain optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5952–5957. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Robson, A.G.; Singh, N.; Pontikos, N.; Kane, T.; Hirji, N.; Ripamonti, C.; Rotsos, T.; Dubra, A.; Kalitzeos, A.; et al. Deep phenotyping of PDE6C-associated achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5112–5123. [Google Scholar] [CrossRef]
- Grøndahl, J.; Riise, R.; Heiberg, A.; Leren, T.; Christoffersen, T.; Bragadottir, R. Autosomal dominant retinitis pigmentosa in Norway: A 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol. Scand. 2007, 85, 287–297. [Google Scholar] [CrossRef]
- Takahashi, V.K.L.; Takiuti, J.T.; Carvalho-Jr, J.R.L.; Xu, C.L.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Fundus autofluorescence and ellipsoid zone (EZ) line width can be an outcome measurement in RHO-associated autosomal dominant retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Von Graefes Arch. Fur Klin. Und Exp. Ophthalmol. 2019, 257, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Nilsson, J.; Li, S.; Jalali, S.; Fulton, A.B.; Tormene, A.P.; Holder, G.E.; Brodie, S.E. ISCEV guide to visual electrodiagnostic procedures. Doc. Ophthalmol. Adv. Ophthalmol. 2018, 136, 1–26. [Google Scholar] [CrossRef]
- Schuster, A.; Weisschuh, N.; Jägle, H.; Besch, D.; Janecke, A.R.; Zierler, H.; Tippmann, S.; Zrenner, E.; Wissinger, B. Novel rhodopsin mutations and genotype-phenotype correlation in patients with autosomal dominant retinitis pigmentosa. Br. J. Ophthalmol. 2005, 89, 1258–1264. [Google Scholar] [CrossRef]
- Georgiou, M.; Grewal, P.S.; Narayan, A.; Alser, M.; Ali, N.; Fujinami, K.; Webster, A.R.; Michaelides, M. Sector retinitis pigmentosa: Extending the molecular genetics basis and elucidating the natural history. Am. J. Ophthalmol. 2021, 221, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Ramon, E.; Cordomí, A.; Aguilà, M.; Srinivasan, S.; Dong, X.; Moore, A.T.; Webster, A.R.; Cheetham, M.E.; Garriga, P. Differential light-induced responses in sectorial inherited retinal degeneration. J. Biol. Chem. 2014, 289, 35918–35928. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Gaudio, A.R.; Berson, E.L. Disease course of patients with pericentral retinitis pigmentosa. Am. J. Ophthalmol. 2005, 140, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ramon, E.; del Valle, L.J.; Garriga, P. Unusual thermal and conformational properties of the rhodopsin congenital night blindness mutant Thr-94 --> Ile. J. Biol. Chem. 2003, 278, 6427–6432. [Google Scholar] [CrossRef] [PubMed]
- Al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81. [Google Scholar] [CrossRef]
- Runhart, E.H.; Khan, M.; Cornelis, S.S.; Roosing, S.; Del Pozo-Valero, M.; Lamey, T.M.; Liskova, P.; Roberts, L.; Stöhr, H.; Klaver, C.C.W.; et al. Association of sex with frequent and mild ABCA4 alleles in Stargardt disease. JAMA Ophthalmol. 2020, 138, 1035–1042. [Google Scholar] [CrossRef]
- Li, B.; Gografe, S.; Munchow, A.; Lopez-Toledano, M.; Pan, Z.H.; Shen, W. Sex-related differences in the progressive retinal degeneration of the rd10 mouse. Exp. Eye Res. 2019, 187, 107773. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Meynert, A.M.; Ansari, M.; FitzPatrick, D.R.; Taylor, M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinform. 2014, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. Adv. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. Adv. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef]
- Bach, M.; Brigell, M.G.; Hawlina, M.; Holder, G.E.; Johnson, M.A.; McCulloch, D.L.; Meigen, T.; Viswanathan, S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc. Ophthalmol. Adv. Ophthalmol. 2013, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Sex | Age at Last Visit (Years) | Problems with Daytime Vision (Age at Onset) | BCVA (Snellen) | Color Vision (Ishihara Plates Read; ≤9/15 = Abnormal) | Visual Field—II/1 Isopter Field Area (deg2) | Bone Spicule Pigmentation | Fundus Autofluorescence Pattern | CME | ERG | Phenotype Classification | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | RE | LE | |||||||||
| A:III-10 | M | 71 | no | 1.0 | 0.9 | 12/15 | 12/15 | 610 | 385 | Yes | Hyperautofluorescent ring | No | BE: mfERG mildly reduced ffERG undetectable DA 0.01 ERG, normal LA amplitudes with prolong peak times | Classic RP |
| A:III-11 | M | 64 | no | 1.0 | 1.0 | 15/15 | 15/15 | 2045 | 1592 | Yes | Sectoral degeneration | No | BE: mfERG normal, ffERG undetectable DA 0.01 ERG, normal LA responses, undetectable S-cone ERG | Sector RP |
| A:III-12 | F | 64 | yes (63) | 0.7 | 0.6 | 1/15 | 1/15 | N/A | N/A | Yes | Sectoral degeneration | No | N/A | Sector RP |
| A:III-14 | M | 55 | no | 1.0 | 1.0 | N/A | N/A | 2298 | 2309 | Yes | Sectoral degeneration | No | N/A | Sector RP |
| A:III-17 | M | 65 | no | 0.9 | 0.8 | 4/15 | 2/15 | 342 | 81 | Yes | Hyperautofluorescent ring | No | BE: mfERG reduced, ffERG undetectable DA 0.01 ERG, reduced LA responses, undetectable S-cone ERG | Classic RP |
| A:IV-1 | M | 48 | no | 1.0 | 1.0 | 15/15 | 15/15 | 2073 | 2233 | No | Normal | No | BE: mfER mildly reduced, ffERG undetectable DA 0.01 ERG, normal to slightly reduced LA responses with normal peak times, reduced S-cone ERG | CSNB |
| A:IV-3 | M | 42 | no | 0.9 | 1.0 | 15/15 | 15/15 | 1220 | 1147 | Yes | Hyperautofluorescent ring | No | N/A | Classic RP |
| A:IV-4 | M | 39 | no | 1.0 | 1.0 | 15/15 | 15/15 | 876 | 927 | Yes | Hyperautofluorescent ring | No | N/A | Classic RP |
| A:IV-5 | M | 36 | no | 1.0 | 1.0 | 15/15 | 15/15 | 537 | 553 | Yes | Hyperautofluorescent ring | No | N/A | Classic RP |
| A:IV-17 | F | 38 | yes (15) | 0.6 | 0.4 | 1/15 | 1/15 | 0 | 0 | Yes | Hyperautofluorescent ring | No | BE: mfERG undetectable, ffERG undetectable | Classic RP |
| A:V-1 | M | 17 | no | 1.0 | 1.0 | 15/15 | 15/15 | 3189 | 3377 | no | Sectoral degeneration | No | BE: mfERG normal, ffERG undetectable DA 0.01 ERG, normal to slightly reduced LA amplitudes with normal peak times, normal S-cone ERG | CSNB |
| B:III-3 | M | 29 | no | 1.0 | 1.0 | 10/15 | 13/15 | 511 | 443 | Yes | Double hyperautofluorescent ring | no | BE: mfERG reduced, ffERG reduced, DA 0.01 ERG, reduced LA amplitudes with prolonged peak times | Pericentral RP |
| C:I-1 | F | 66 | yes (46) | 0.1 | 0.1 | 1/15 | 1/15 | 0 | 0 | Yes | Hyperautofluorescent ring | yes | BE: ffERG: undetectable DA 0.01 ERG, undetectable LA ERG amplitudes, undetectable S-cone ERG, PERG significantly reduced | Classic RP |
| C:II-2 | M | 41 | yes (37) | 0.2 | 0.1 | 0/15 | 0/15 | 91 | 107 | Yes | Hyperautofluorescent ring | yes | BE: mfERG: undetectable in the foveolar region and reduced towards periphery; ffERG: undetectable DA 0.01 ERG, reduced LA ERG amplitudes with severely prolonged peak times, undetectable S-cone ERG | Classic RP |
| C:III-2 | M | 8 | no | 1.0 | 1.0 | 15/15 | 15/15 | 933 | 252 | No | Normal | no | BE: ffERG: undetectable DA 0.01 ERG, borderline LA ERG amplitudes with borderline prolonged peak times | CSNB |
| Potential Carriers | Affected | Unaffected | |
|---|---|---|---|
| Male | 34 (57%) | 22 (69%) | 12 (43%) |
| Female | 26 (43%) | 10 (31%) | 16 (57%) |
| Total | 60 | 32 | 28 |
| Potential Carriers | Affected | Unaffected | |
|---|---|---|---|
| Male | 35 (54%) | 22 (65%) | 13 (42%) |
| Female | 30 (46%) | 12 (35%) | 18 (58%) |
| Total | 65 | 34 | 31 |
| Potential Carriers | Affected | Unaffected | |
|---|---|---|---|
| Male | 69 (55%) | 44 (67%) | 25 (42%) |
| Female | 56 (45%) | 22 (33%) | 34 (58%) |
| Total | 125 | 66 | 59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobal, N.; Krašovec, T.; Šuštar, M.; Volk, M.; Peterlin, B.; Hawlina, M.; Fakin, A. Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene. Int. J. Mol. Sci. 2021, 22, 2133. https://doi.org/10.3390/ijms22042133
Kobal N, Krašovec T, Šuštar M, Volk M, Peterlin B, Hawlina M, Fakin A. Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene. International Journal of Molecular Sciences. 2021; 22(4):2133. https://doi.org/10.3390/ijms22042133
Chicago/Turabian StyleKobal, Nina, Tjaša Krašovec, Maja Šuštar, Marija Volk, Borut Peterlin, Marko Hawlina, and Ana Fakin. 2021. "Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene" International Journal of Molecular Sciences 22, no. 4: 2133. https://doi.org/10.3390/ijms22042133
APA StyleKobal, N., Krašovec, T., Šuštar, M., Volk, M., Peterlin, B., Hawlina, M., & Fakin, A. (2021). Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene. International Journal of Molecular Sciences, 22(4), 2133. https://doi.org/10.3390/ijms22042133