
Strongyloides stercoralis Infestation in a Child: How a Nematode Can Affect Gut Microbiota



Abstract
1. Introduction
2. Results
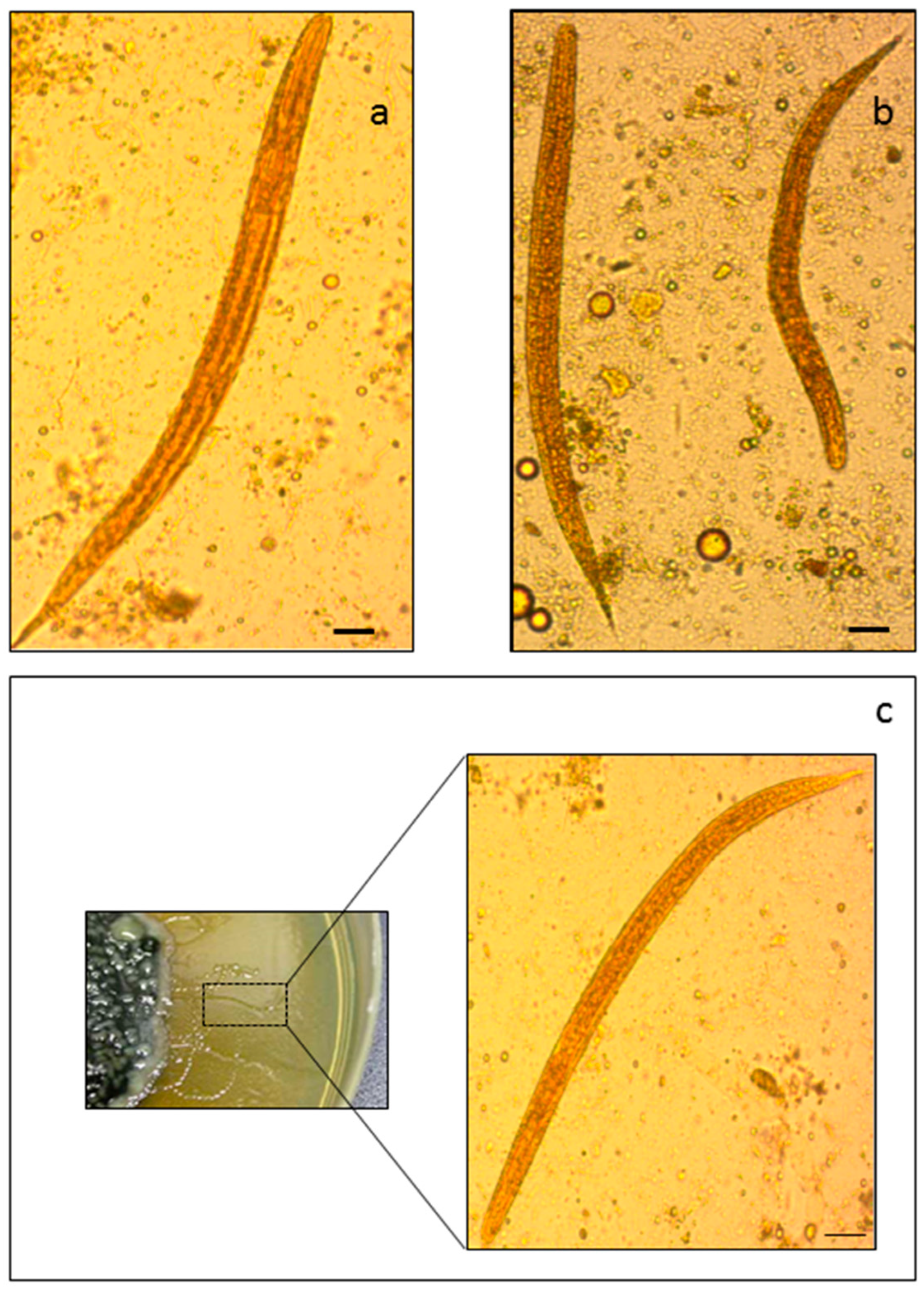
2.1. Morphology-Based and PCR-Based Methods for the Identification of S. stercoralis
2.2. Gut Microbiota Profiling Associated with S. stercoralis Infection
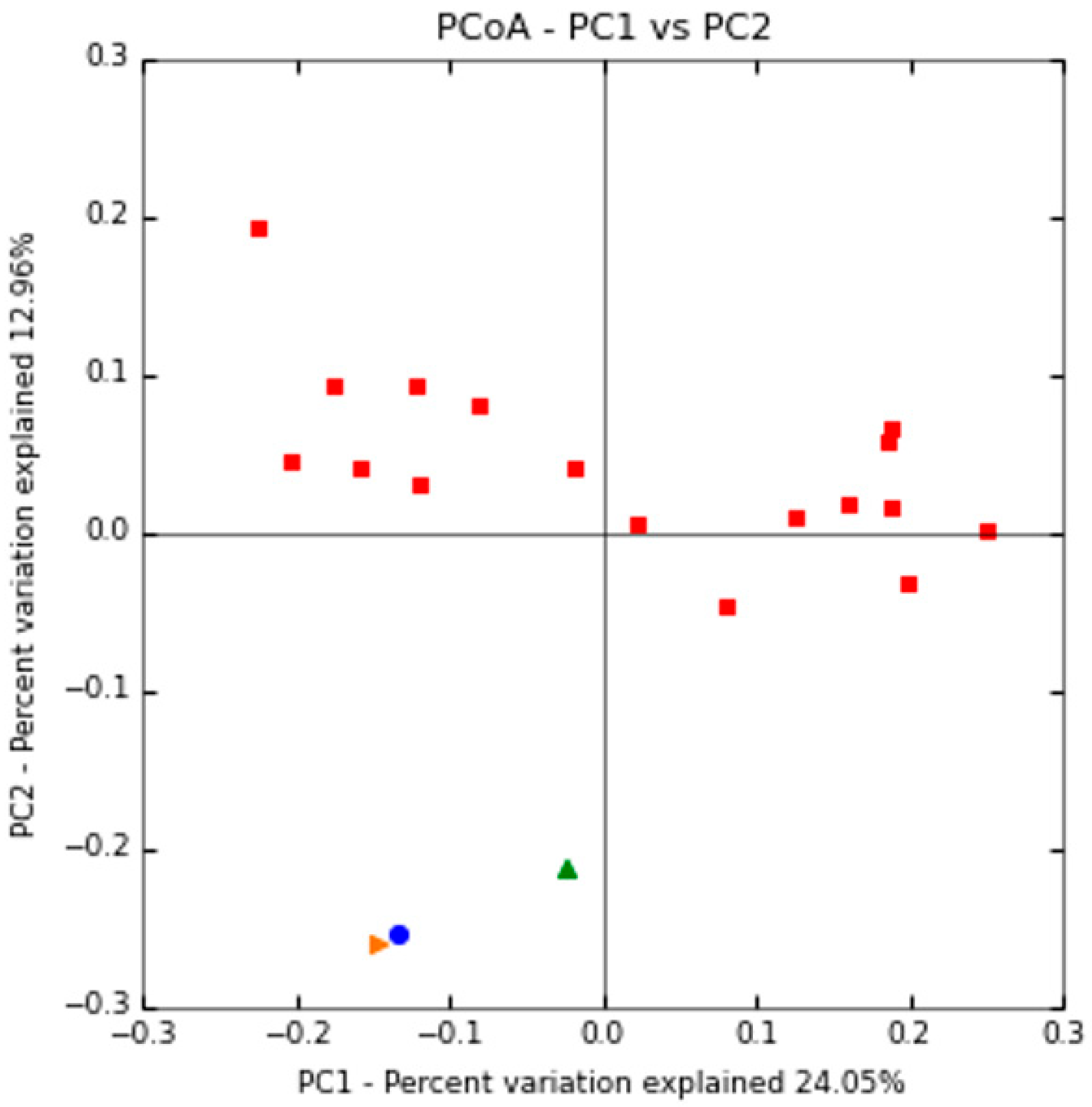
2.2.1. Shaping of Patient’s Gut Microbiota Ecology
2.2.2. Metabolic Prediction of the Patient’s Gut Microbiota during S. stercoralis Infection
3. Discussion
4. Case Presentation and Laboratory Diagnosis
4.1. Patient’s Characteristics
4.2. Laboratory Diagnosis of Strongyloidiasis
4.2.1. Optical Microscopy
4.2.2. Stool Agar Culture
4.2.3. Immunological Methods
4.2.4. DNA Extraction and PCR Amplification for S. stercoralis Detection
4.3. Gut Microbiota Profiling by 16S rRNA Targeted-Metagenomics Sequencing
4.3.1. Bacterial DNA Extraction from Stool Samples
4.3.2. Bioinformatics Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bisoffi, Z.; Buonfrate, D.; Montresor, A.; Requena-Mendez, A.; Munoz, J.; Krolewiecki, A.J.; Gotuzzo, E.; Mena, M.A.; Chiodini, P.L.; Anselmi, M.; et al. Strongyloides stercoralis: A plea for action. PLoS Neglect. Trop. D 2013, 7, e2214. [Google Scholar] [CrossRef]
- Nutman, T.B. Human infection with Strongyloides stercoralis and other related Strongyloides species. Parasitology 2017, 144, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Buonfrate, D.; Baldissera, M.; Abrescia, F.; Bassetti, M.; Caramaschi, G.; Giobbia, M.; Mascarello, M.; Rodari, P.; Scattolo, N.; Napoletano, G.; et al. Epidemiology of Strongyloides stercoralis in northern Italy: Results of a multicentre case-control study, February 2013 to July 2014. Eurosurveillance 2016, 21, 30310. [Google Scholar] [CrossRef] [PubMed]
- Albarqi, M.M.Y.; Stoltzfus, J.D.; Pilgrim, A.A.; Nolan, T.J.; Wang, Z.; Kliewer, S.A.; Mangelsdorf, D.J.; Lok, J.B. Regulation of Life Cycle Checkpoints and Developmental Activation of Infective Larvae in Strongyloides stercoralis by Dafachronic Acid. PLoS Pathog 2016, 12, e1005358. [Google Scholar] [CrossRef]
- Csermely, L.; Jaafar, H.; Kristensen, J.; Castella, A.; Gorka, W.; Chebli, A.A.; Trab, F.; Alizadeh, H.; Hunyady, B. Strongyloides hyper-infection causing life-threatening gastrointestinal bleeding. World J. Gastroenterol. 2006, 12, 6401–6404. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-E.; Chuah, S.-K.; Tai, W.-C.; Lee, C.-H. Upper gastrointestinal bleeding caused by Strongyloides stercoralis: Highlighting a neglected parasitic infection. Kaohsiung J. Med. Sci. 2017, 33, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Marcos, L.A.; Terashima, A.; Dupont, H.L.; Gotuzzo, E. Strongyloides hyperinfection syndrome: An emerging global infectious disease. Trans. R. Soc. Trop Med. Hyg. 2008, 102, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.A.; Pacheco, F.T.F.; Souza, J.N.; Silva, M.L.S.; Ines, E.J.; Soares, N.M. Strongyloides stercoralis Infection in Alcoholic Patients. Biomed. Res. Int. 2016, 2016, 4872473. [Google Scholar] [CrossRef]
- Jenkins, T.P.; Formenti, F.; Castro, C.; Piubelli, C.; Perandin, F.; Buonfrate, D.; Otranto, D.; Griffin, J.L.; Krause, L.; Bisoffi, Z.; et al. A comprehensive analysis of the faecal microbiome and metabolome of Strongyloides stercoralis infected volunteers from a non-endemic area. Sci. Rep.-UK 2018, 8, 15651. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Jackson, M.A.; Pallister, T.; Steves, C.J.; Spector, T.D.; Valdes, A.M. Gut microbiome diversity and high-fibre intake are related to lower long-term weight gain. Int. J. Obes. 2017, 41, 1099–1105. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Rapin, A.; Lebon, L.; Dubey, L.K.; Mosconi, I.; Sarter, K.; Piersigilli, A.; Menin, L.; Walker, A.W.; Rougemont, J.; et al. The Intestinal Microbiota Contributes to the Ability of Helminths to Modulate Allergic Inflammation. Immunity 2015, 43, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Djuardi, Y.; Sartono, E.; Rosa, B.A.; Supali, T.; Mitreva, M.; Houwing-Duistermaat, J.J.; Yazdanbakhsh, M. Dynamic changes in human-gut microbiome in relation to a placebo-controlled anthelminthic trial in Indonesia. PLoS Neglect. Trop. D 2018, 12, e0006620. [Google Scholar] [CrossRef]
- Ramanan, D.; Bowcutt, R.; Lee, S.C.; Tang, M.S.; Kurtz, Z.D.; Ding, Y.; Honda, K.; Gause, W.C.; Blaser, M.J.; Bonneau, R.A.; et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 2016, 352, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunology 2016, 5, e73. [Google Scholar] [CrossRef]
- Liu, S.; Pan, J.; Meng, X.; Zhu, J.; Zhou, J.; Zhu, X. Trichinella spiralis infection decreases the diversity of the intestinal flora in the infected mouse. J. Microbiol. Immunol. Infect. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brosschot, T.P.; Reynolds, L.A. The impact of a helminth-modified microbiome on host immunity. Mucosal. Immunol. 2018, 11, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.B.; Sorobetea, D.; Kiilerich, P.; Ramayo-Caldas, Y.; Estelle, J.; Ma, T.; Madsen, L.; Kristiansen, K.; Svensson-Frej, M. Chronic Trichuris muris Infection Decreases Diversity of the Intestinal Microbiota and Concomitantly Increases the Abundance of Lactobacilli. PLoS ONE 2015, 10, e0125495. [Google Scholar] [CrossRef]
- Kreisinger, J.; Bastien, G.; Hauffe, H.C.; Marchesi, J.; Perkins, S.E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140295. [Google Scholar] [CrossRef]
- Reynolds, L.A.; Smith, K.A.; Filbey, K.J.; Harcus, Y.; Hewitson, J.P.; Redpath, S.A.; Valdez, Y.; Yebra, M.J.; Finlay, B.B.; Maizels, R.M. Commensal-pathogen interactions in the intestinal tract: Lactobacilli promote infection with, and are promoted by, helminth parasites. Gut. Microbes 2014, 5, 522–532. [Google Scholar] [CrossRef]
- Walk, S.T.; Blum, A.M.; Ewing, S.A.-S.; Weinstock, J.V.; Young, V.B. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm. Bowel. Dis. 2010, 16, 1841–1849. [Google Scholar] [CrossRef]
- Lee, S.C.; Tang, M.S.; Lim, Y.A.L.; Choy, S.H.; Kurtz, Z.D.; Cox, L.M.; Gundra, U.M.; Cho, I.; Bonneau, R.; Blaser, M.J.; et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Neglect. Trop. D 2014, 8, e2880. [Google Scholar] [CrossRef] [PubMed]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Metabolism and Interaction with Food Components. Int. J. Mol. Sci. 2020, 21, 3688. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Koga, K.; Kasuya, S.; Ohtomo, H. How effective is the agar plate method for Strongyloides stercoralis? J. Parasitol. 1992, 78, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Moghaddassani, H.; Mirhendi, H.; Hosseini, M.; Rokni, M.; Mowlavi, G.; Kia, E. Molecular Diagnosis of Strongyloides stercoralis Infection by PCR Detection of Specific DNA in Human Stool Samples. Iran. J. Parasitol. 2011, 6, 23–30. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Yokono, M.; Satoh, S.; Tanaka, A. Comparative analyses of whole-genome protein sequences from multiple organisms. Sci. Rep.-UK 2018, 8, 6800. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAMPLE_ID | STATUS | Age | Total Reads Count (N) | Assigned Reads (N) | OTUs (N) |
|---|---|---|---|---|---|
| T0 | During parasite infection | 7 | 53,476 | 36,643 | 1365 |
| T1 | 1 month after parasite infection | 7 | 16,830 | 14,598 | 917 |
| T2 | 2 months after parasite infection | 7 | 44,876 | 42,857 | 790 |
| N-05-1 | healthy control | 7–8 | 58,775 | 57,578 | 1062 |
| N-05-2 | healthy control | 7–8 | 68,144 | 61,598 | 1475 |
| N-05-3 | healthy control | 7–8 | 54,796 | 52,930 | 799 |
| N-05-4 | healthy control | 7–8 | 164,725 | 159,772 | 1044 |
| N-05-5 | healthy control | 7–8 | 84,616 | 80,331 | 645 |
| N-05-6 | healthy control | 7–8 | 293,539 | 285,209 | 978 |
| N-05-7 | healthy control | 7–8 | 29,696 | 29,454 | 377 |
| N-05-8 | healthy control | 7–8 | 40,400 | 37,790 | 875 |
| N-05-9 | healthy control | 7–8 | 76,140 | 75,780 | 463 |
| N-06-1 | healthy control | 7–8 | 60,591 | 60,217 | 397 |
| N-06-2 | healthy control | 7–8 | 46,304 | 46,014 | 455 |
| N-06-3 | healthy control | 7–8 | 400,539 | 393,198 | 1647 |
| N-06-4 | healthy control | 7–8 | 69,989 | 69,092 | 385 |
| N-06-5 | healthy control | 7–8 | 75,864 | 72,235 | 781 |
| N-06-6 | healthy control | 7–8 | 40,312 | 39,751 | 932 |
| N-06-7 | healthy control | 7–8 | 9379 | 9302 | 320 |
| N-06-8 | healthy control | 7–8 | 29,337 | 26,571 | 494 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pane, S.; Sacco, A.; Iorio, A.; Romani, L.; Putignani, L. Strongyloides stercoralis Infestation in a Child: How a Nematode Can Affect Gut Microbiota. Int. J. Mol. Sci. 2021, 22, 2131. https://doi.org/10.3390/ijms22042131
Pane S, Sacco A, Iorio A, Romani L, Putignani L. Strongyloides stercoralis Infestation in a Child: How a Nematode Can Affect Gut Microbiota. International Journal of Molecular Sciences. 2021; 22(4):2131. https://doi.org/10.3390/ijms22042131
Chicago/Turabian StylePane, Stefania, Anna Sacco, Andrea Iorio, Lorenza Romani, and Lorenza Putignani. 2021. "Strongyloides stercoralis Infestation in a Child: How a Nematode Can Affect Gut Microbiota" International Journal of Molecular Sciences 22, no. 4: 2131. https://doi.org/10.3390/ijms22042131
APA StylePane, S., Sacco, A., Iorio, A., Romani, L., & Putignani, L. (2021). Strongyloides stercoralis Infestation in a Child: How a Nematode Can Affect Gut Microbiota. International Journal of Molecular Sciences, 22(4), 2131. https://doi.org/10.3390/ijms22042131