
Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
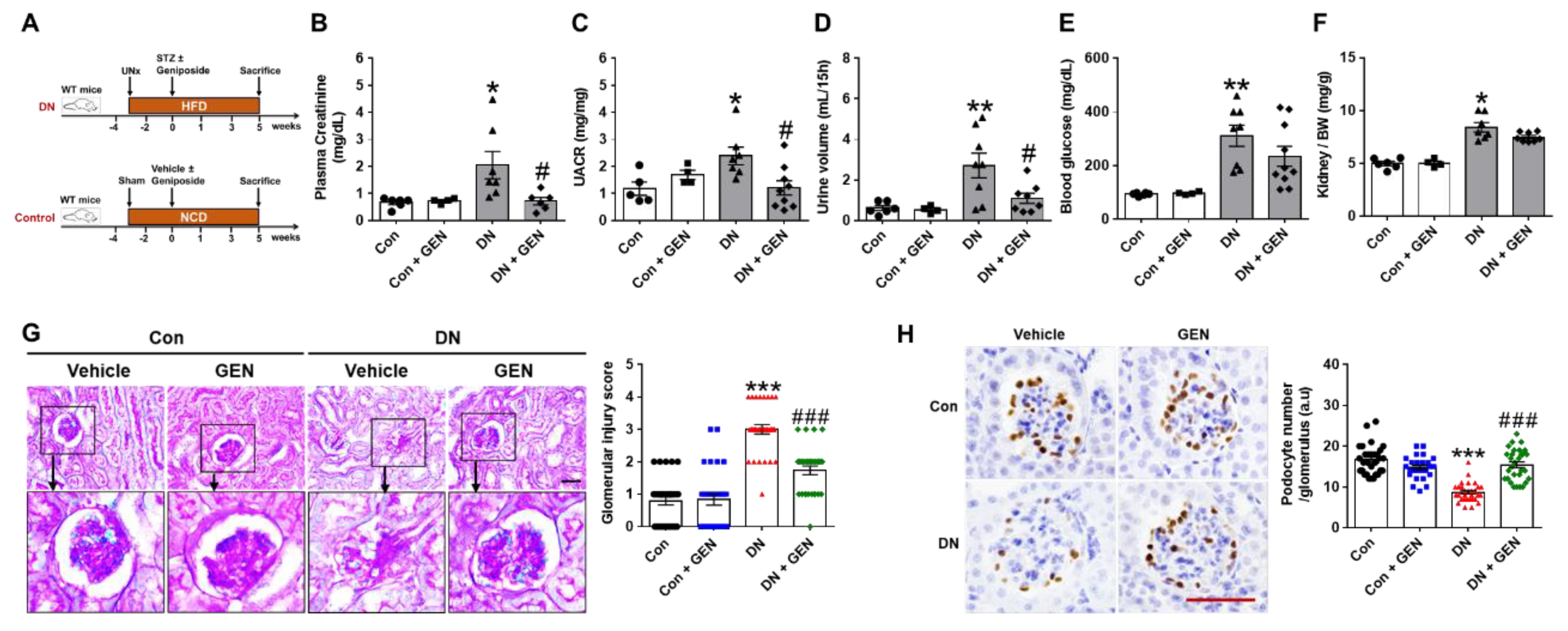
2.1. Geniposide Improved Glomerular Filtration Function and Structural Lesions in DN
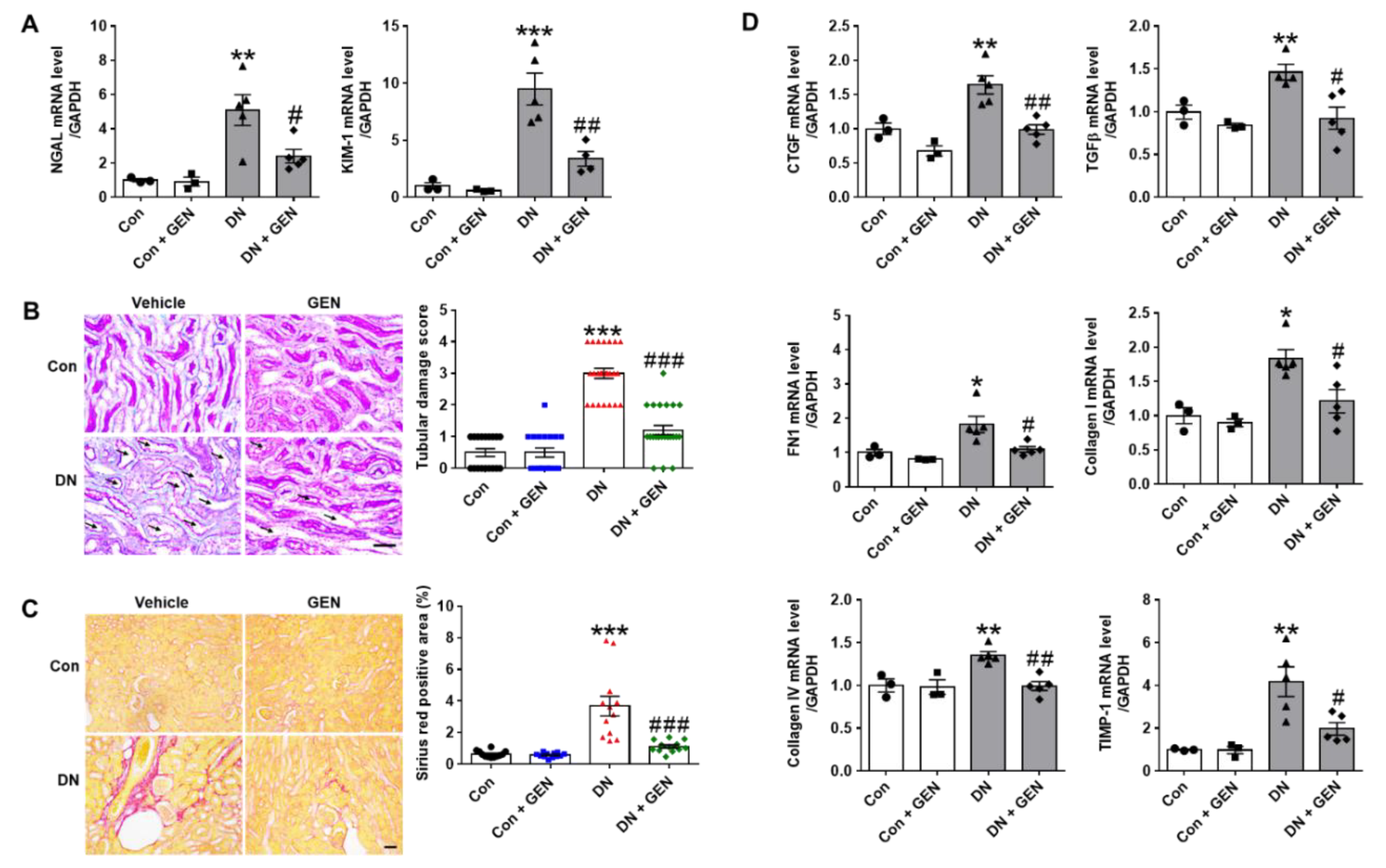
2.2. Geniposide Reduced Renal Tubular Injury and Interstitial Fibrosis in DN
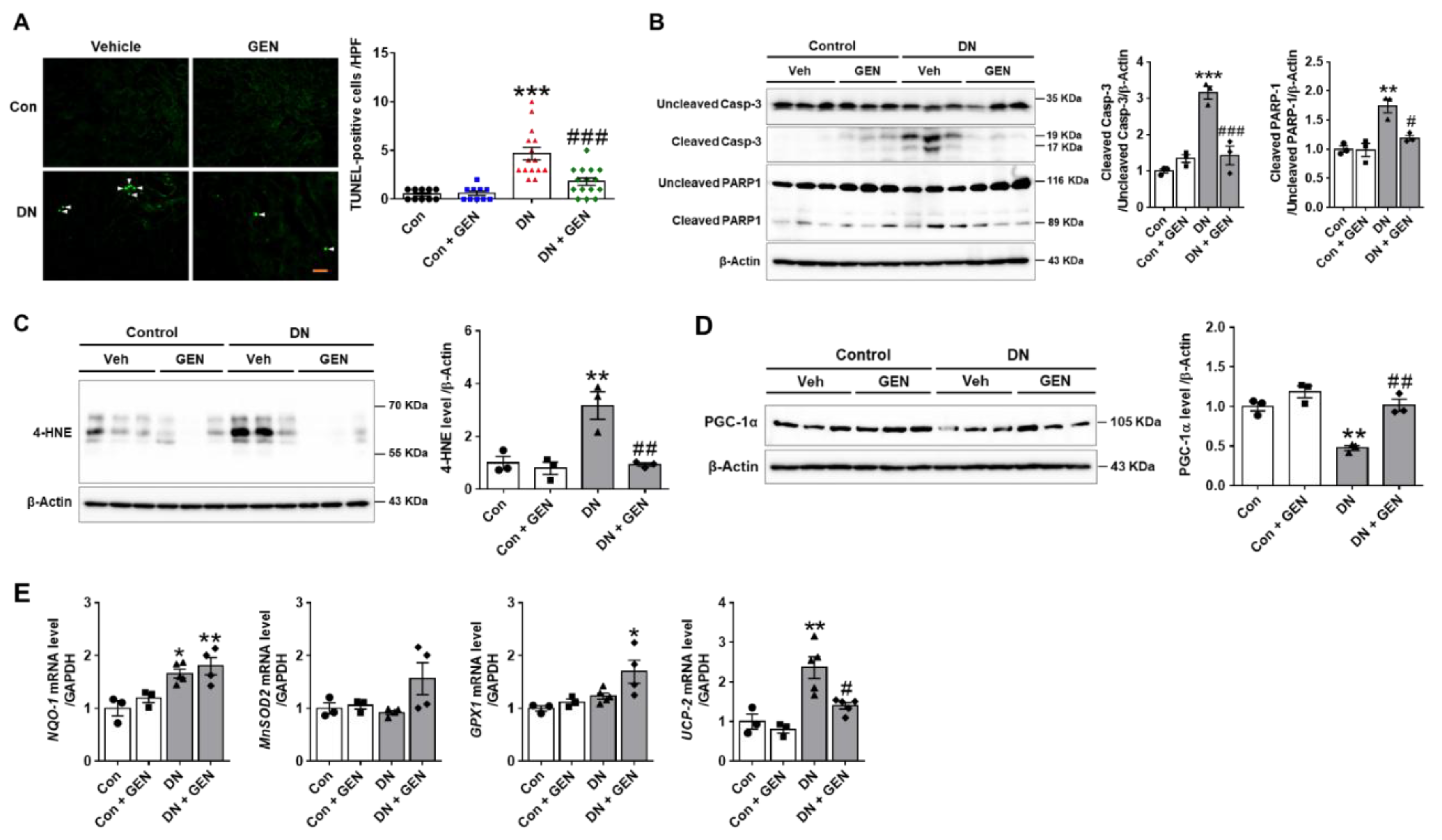
2.3. Geniposide Decreased Renal Apoptosis and Oxidative Stress in DN
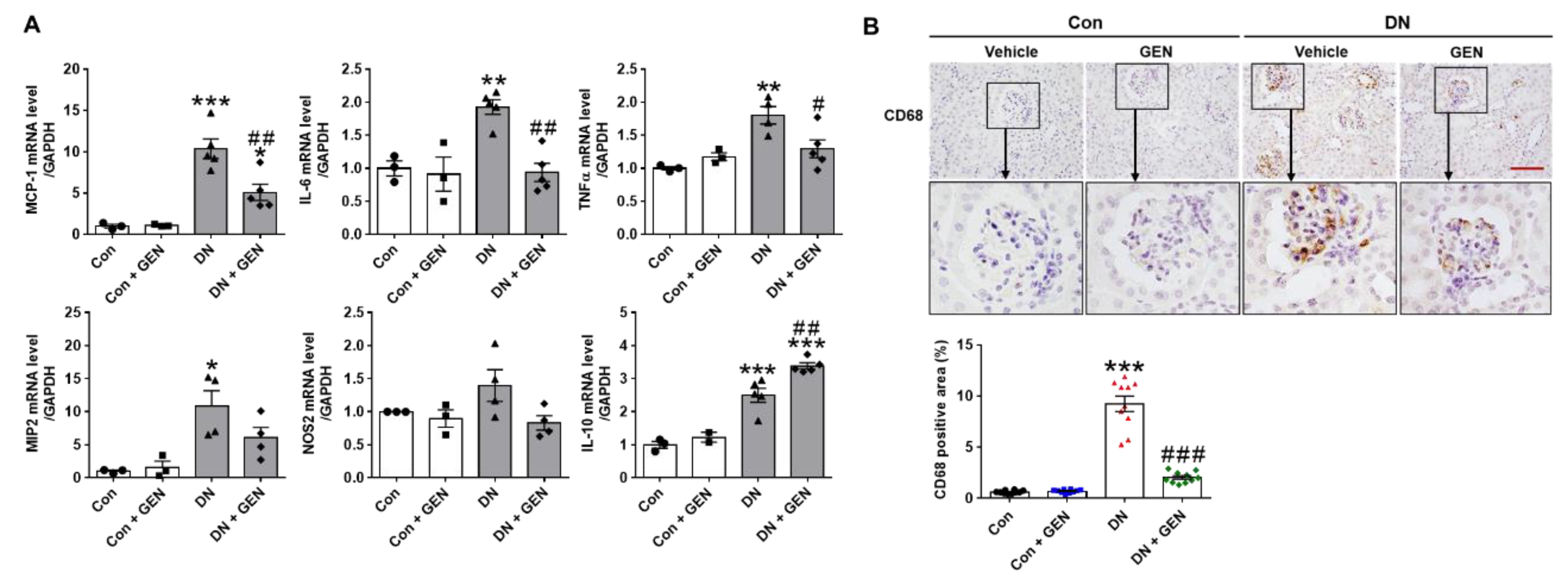
2.4. Geniposide Suppressed Macrophage Infiltration and Renal Inflammation in DN
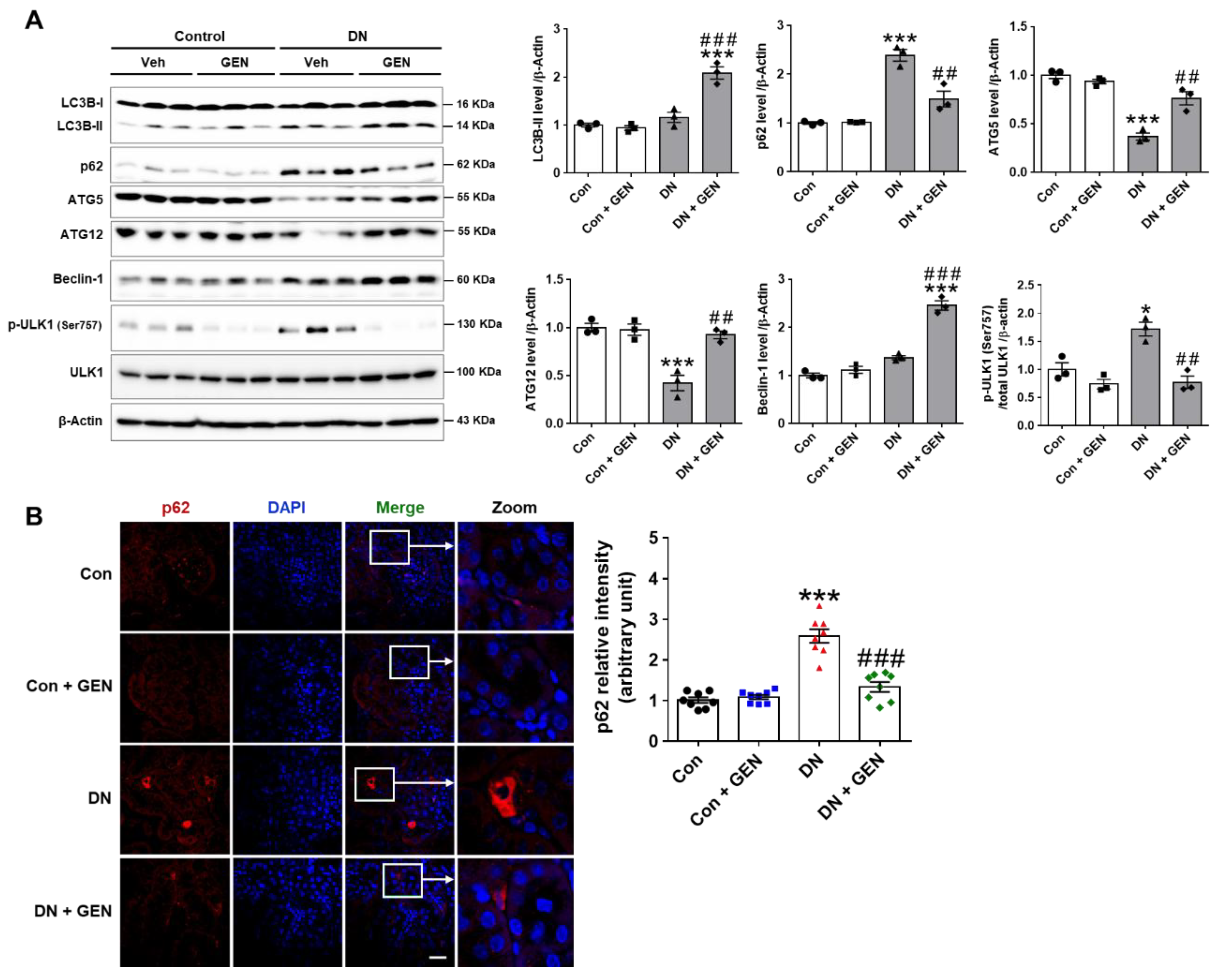
2.5. Geniposide Increased Autophagy Response in DN
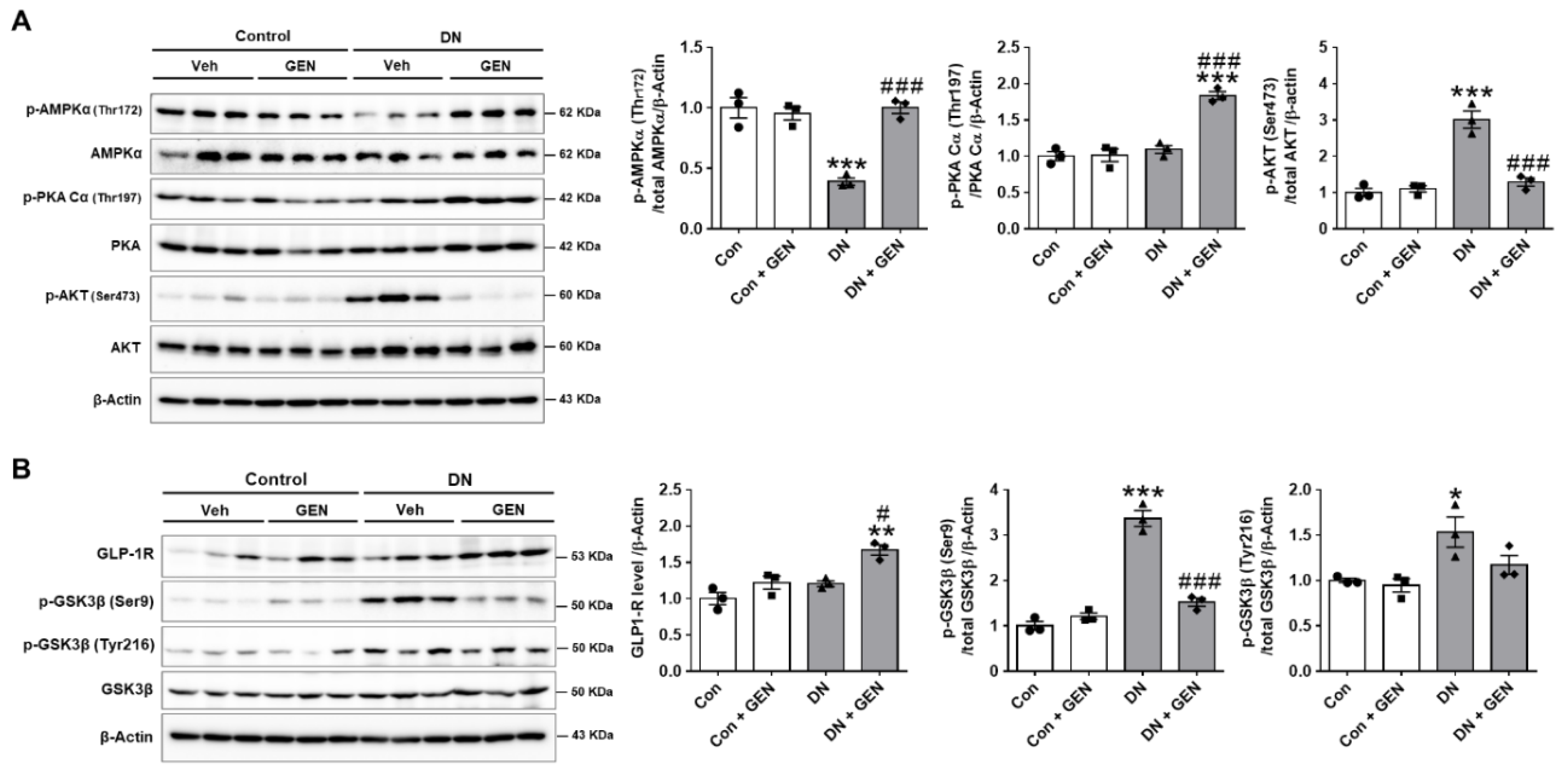
2.6. Geniposide Protected the Kidney through AMPK Activation and AKT Inhibition in DN
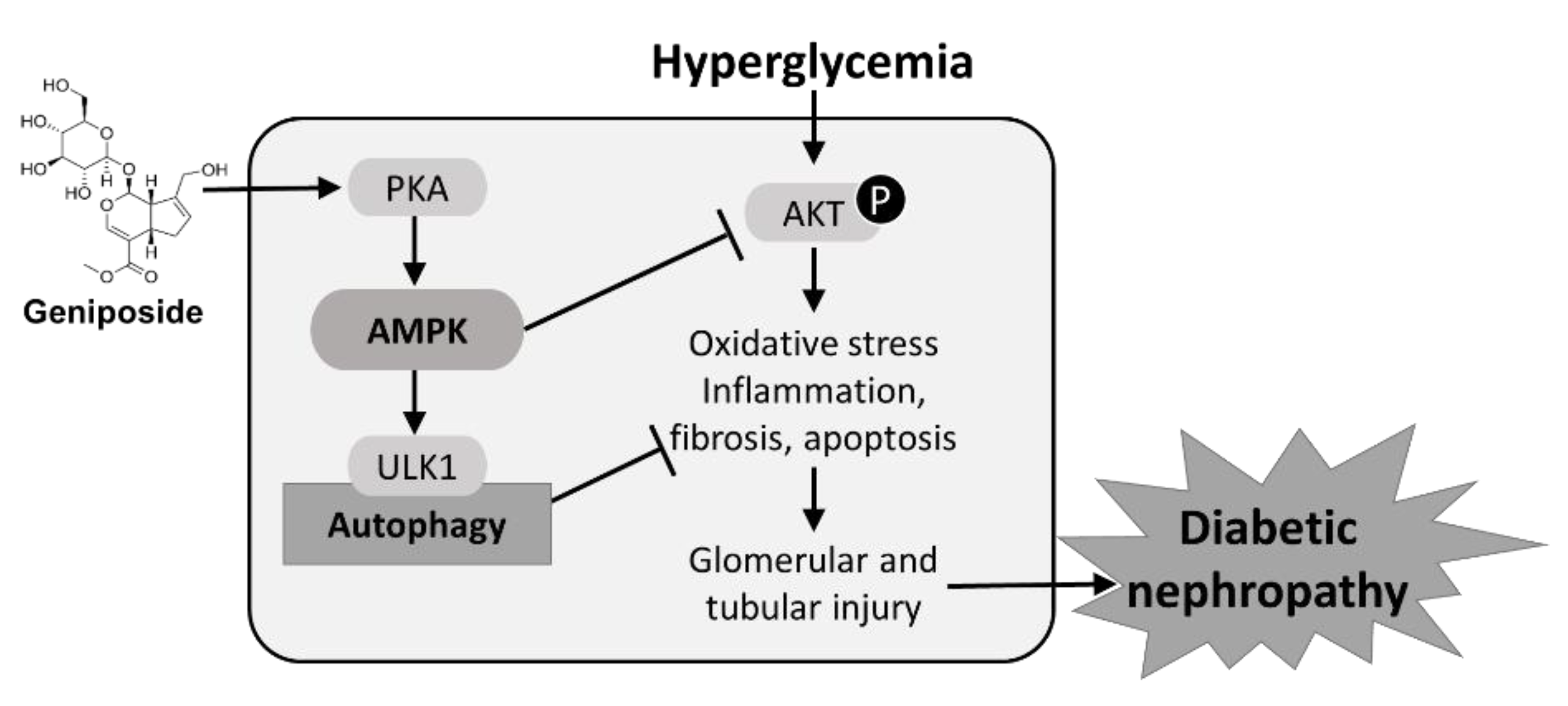
2.7. Protective Mechanism of Geniposide against DN by Improving Autophagy and Inhibiting Oxidative Stress through AMPK Activation and AKT Inhibition
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. DN Animal Model and Treatment
4.3. Biochemical Assays
4.4. Periodic Acid–Schiff (PAS) and Picro-Sirius Red Staining
4.5. Kidney Histological Examination
4.6. Immunohistochemistry (IHC) Analysis
4.7. Immunofluorescence (IF) Staining
4.8. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
4.9. Western Blot Analysis
4.10. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jefferson, J.A.; Shankland, S.J.; Pichler, R.H. Proteinuria in diabetic kidney disease: A mechanistic viewpoint. Kidney Int. 2008, 74, 22–36. [Google Scholar] [CrossRef]
- Liang, G.; Song, L.; Chen, Z.; Qian, Y.; Xie, J.; Zhao, L.; Lin, Q.; Zhu, G.; Tan, Y.; Li, X.; et al. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 2018, 93, 95–109. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. New therapeutic agents in diabetic nephropathy. Korean J. Intern. Med. 2017, 32, 11–25. [Google Scholar] [CrossRef]
- Su, J.; Li, S.J.; Chen, Z.H.; Zeng, C.H.; Zhou, H.; Li, L.S.; Liu, Z.H. Evaluation of podocyte lesion in patients with diabetic nephropathy: Wilms’ tumor-1 protein used as a podocyte marker. Diabetes Res. Clin. Pract. 2010, 87, 167–175. [Google Scholar] [CrossRef]
- Collins, A.J.; Foley, R.N.; Chavers, B.; Gilbertson, D.; Herzog, C.; Ishani, A.; Johansen, K.; Kasiske, B.L.; Kutner, N.; Liu, J.; et al. US Renal Data System 2013 Annual Data Report. Am. J. Kidney Dis. 2014, 63, A7. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Koya, D.; Uzu, T.; Maegawa, H. Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy. Biomed. Res. Int. 2014, 315494. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Koya, D. Autophagy: A Novel Therapeutic Target for Diabetic Nephropathy. Diabetes Metab. J. 2015, 39, 451–460. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kume, S.; Kitada, M.; Kanasaki, K.; Uzu, T.; Maegawa, H.; Koya, D. Autophagy as a therapeutic target in diabetic nephropathy. Exp. Diabetes Res. 2012, 628978. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef]
- Takiyama, Y.; Harumi, T.; Watanabe, J.; Fujita, Y.; Honjo, J.; Shimizu, N.; Makino, Y.; Haneda, M. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: A possible role of HIF-1α expression and oxygen metabolism. Diabetes 2011, 60, 981–992. [Google Scholar] [CrossRef]
- Xu, J.; Liu, L.Q.; Xu, L.L.; Xing, Y.; Ye, S. Metformin alleviates renal injury in diabetic rats by inducing Sirt1/FoxO1 autophagic signal axis. Clin. Exp. Pharmacol. Physiol. 2020, 47, 599–608. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Chowdhury, R.; Trudel, L.J.; Tee, A.R.; Slack, R.S.; Walker, C.L.; Wogan, G.N. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. USA 2013, 110, E2950–E2957. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [PubMed]
- Miyasita, S. A historical study of Chinese drugs for the treatment of Jaundice. Am. J. Chin. Med. 1976, 4, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.J.; Song, Y.S.; Kim, H.J.; Lee, Y.H.; Hong, S.M.; Kim, S.J.; Kim, B.C.; Jin, C.; Lim, C.J.; Park, E.H. Antiinflammatory effects of genipin, an active principle of gardenia. Eur. J. Pharmacol. 2004, 495, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.Y.; Lee, S.M. Protective Effects of Geniposide and Genipin against Hepatic Ischemia/Reperfusion Injury in Mice. Biomol. Ther. 2013, 21, 132–137. [Google Scholar] [CrossRef]
- Gao, C.; Liu, Y.; Jiang, Y.; Ding, J.; Li, L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3β pathway in streptozotocin-induced Alzheimer rat model. Brain Pathol. 2014, 24, 261–269. [Google Scholar] [CrossRef]
- Koo, H.J.; Lee, S.; Shin, K.H.; Kim, B.C.; Lim, C.J.; Park, E.H. Geniposide, an anti-angiogenic compound from the fruits of Gardenia jasminoides. Planta Med. 2004, 70, 467–469. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, X.; Jin, G.; Shi, Z.; Sun, W.; Chen, F. Geniposide reduces development of streptozotocin-induced diabetic nephropathy via regulating nuclear factor-kappa B signaling pathways. Fundam. Clin. Pharmacol. 2017, 31, 54–63. [Google Scholar] [CrossRef]
- Wu, S.Y.; Wang, G.F.; Liu, Z.Q.; Rao, J.J.; Lu, L.; Xu, W.; Wu, S.G.; Zhang, J.J. Effect of geniposide, a hypoglycemic glucoside, on hepatic regulating enzymes in diabetic mice induced by a high-fat diet and streptozotocin. Acta Pharmacol. Sin. 2009, 30, 202–208. [Google Scholar] [CrossRef]
- Yao, D.D.; Yang, L.; Wang, Y.; Liu, C.; Wei, Y.J.; Jia, X.B.; Yin, W.; Shu, L. Geniposide promotes beta-cell regeneration and survival through regulating beta-catenin/TCF7L2 pathway. Cell Death Dis. 2015, 6, e1746. [Google Scholar] [CrossRef]
- Li, F.; Chen, Y.; Li, Y.; Huang, M.; Zhao, W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-kappaB pathway. Eur. J. Pharmacol. 2020, 886, 173449. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodriguez, E.; Fernandez-Prado, R.; Martin-Cleary, C.; Pizarro-Sanchez, M.S.; Sanchez-Nino, M.D.; Sanz, A.B.; Fernandez-Fernandez, B.; Ortiz, A. Kidney Injury Marker 1 and Neutrophil Gelatinase-Associated Lipocalin in Chronic Kidney Disease. Nephron 2017, 136, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Sabbisetti, V.S.; Waikar, S.S.; Antoine, D.J.; Smiles, A.; Wang, C.; Ravisankar, A.; Ito, K.; Sharma, S.; Ramadesikan, S.; Lee, M.; et al. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J. Am. Soc. Nephrol. 2014, 25, 2177–2186. [Google Scholar] [CrossRef]
- Dusabimana, T.; Kim, S.R.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. P2Y2R contributes to the development of diabetic nephropathy by inhibiting autophagy response. Mol. Metab. 2020, 42, 101089. [Google Scholar] [CrossRef] [PubMed]
- Hinden, L.; Udi, S.; Drori, A.; Gammal, A.; Nemirovski, A.; Hadar, R.; Baraghithy, S.; Permyakova, A.; Geron, M.; Cohen, M.; et al. Modulation of Renal GLUT2 by the Cannabinoid-1 Receptor: Implications for the Treatment of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Burlaka, I.; Nilsson, L.M.; Scott, L.; Holtback, U.; Eklof, A.C.; Fogo, A.B.; Brismar, H.; Aperia, A. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int. 2016, 90, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Gandolfo, M.T.; Ferrario, F.; Rastaldi, M.P.; Villaggio, B.; Gianiorio, F.; Giannoni, M.; Rimoldi, L.; Lauria, F.; Miji, M.; et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007, 72, 1262–1272. [Google Scholar] [CrossRef]
- Wang, X.; Meng, L.; Zhao, L.; Wang, Z.; Liu, H.; Liu, G.; Guan, G. Resveratrol ameliorates hyperglycemia-induced renal tubular oxidative stress damage via modulating the SIRT1/FOXO3a pathway. Diabetes Res. Clin. Pract. 2017, 126, 172–181. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocrinol. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Li, W.J.; Wang, C.M. The role of uncoupling proteins in diabetes mellitus. J. Diabetes Res. 2013, 2013, 585897. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Gong, Q.; Qi, X.M.; Wang, K.; Wu, Y.G. Paeoniflorin Ameliorates Macrophage Infiltration and Activation by Inhibiting the TLR4 Signaling Pathway in Diabetic Nephropathy. Front. Pharmacol. 2019, 10, 566. [Google Scholar] [CrossRef]
- Soetikno, V.; Sari, F.R.; Veeraveedu, P.T.; Thandavarayan, R.A.; Harima, M.; Sukumaran, V.; Lakshmanan, A.P.; Suzuki, K.; Kawachi, H.; Watanabe, K. Curcumin ameliorates macrophage infiltration by inhibiting NF-kB activation and proinflammatory cytokines in streptozotocin induced-diabetic nephropathy. Nutr. Metab. 2011, 8, 35. [Google Scholar] [CrossRef]
- Seok, S.J.; Lee, E.S.; Kim, G.T.; Hyun, M.; Lee, J.H.; Chen, S.; Choi, R.; Kim, H.M.; Lee, E.Y.; Chung, C.H. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol. Dial. Transplant. 2013, 28, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Dusabimana, T.; Kim, S.R.; Je, J.; Jeong, K.; Kang, M.C.; Cho, K.M.; Kim, H.J.; Park, S.W. Supplementation of Abelmoschus manihot Ameliorates Diabetic Nephropathy and Hepatic Steatosis by Activating Autophagy in Mice. Nutrients 2018, 10, 1703. [Google Scholar] [CrossRef]
- Yao, F.; Zhang, M.; Chen, L. 5′-Monophosphate-activated protein kinase (AMPK) improves autophagic activity in diabetes and diabetic complications. Acta Pharm. Sin. B 2016, 6, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Udi, S.; Hinden, L.; Earley, B.; Drori, A.; Reuveni, N.; Hadar, R.; Cinar, R.; Nemirovski, A.; Tam, J. Proximal Tubular Cannabinoid-1 Receptor Regulates Obesity-Induced CKD. J. Am. Soc. Nephrol. 2017, 28, 3518–3532. [Google Scholar] [CrossRef]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim. Biophys. Acta 2014, 1846, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, P.; Chen, B.; Ge, Y.; Gong, A.Y.; Flickinger, B.; Malhotra, D.K.; Wang, L.J.; Dworkin, L.D.; Liu, Z.; et al. Glycogen synthase kinase 3beta hyperactivity in urinary exfoliated cells predicts progression of diabetic kidney disease. Kidney Int. 2020, 97, 175–192. [Google Scholar] [CrossRef]
- Mariappan, M.M.; Prasad, S.; D’Silva, K.; Cedillo, E.; Sataranatarajan, K.; Barnes, J.L.; Choudhury, G.G.; Kasinath, B.S. Activation of glycogen synthase kinase 3β ameliorates diabetes-induced kidney injury. J. Biol. Chem. 2014, 289, 35363–35375. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Lee, B.H.; Ahn, S.G.; Yoon, J.H.; Oh, S.H. Serine 9 and tyrosine 216 phosphorylation of GSK-3β differentially regulates autophagy in acquired cadmium resistance. Toxicol. Sci. 2013, 135, 380–389. [Google Scholar] [CrossRef]
- Gilbert, R.E. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes 2017, 66, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Zeni, L.; Norden, A.G.W.; Cancarini, G.; Unwin, R.J. A more tubulocentric view of diabetic kidney disease. J. Nephrol. 2017, 30, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1801–1802. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef]
- He, X.; Zhang, T.; Tolosa, M.; Goru, S.K.; Chen, X.; Misra, P.S.; Robinson, L.A.; Yuen, D.A. A new, easily generated mouse model of diabetic kidney fibrosis. Sci. Rep. 2019, 9, 12549. [Google Scholar] [CrossRef]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE 2014, 9, e97985. [Google Scholar] [CrossRef]
- Aghadavod, E.; Khodadadi, S.; Baradaran, A.; Nasri, P.; Bahmani, M.; Rafieian-Kopaei, M. Role of Oxidative Stress and Inflammatory Factors in Diabetic Kidney Disease. Iran. J. Kidney Dis. 2016, 10, 337–343. [Google Scholar]
- Liu, G.; Ji, W.; Huang, J.; Liu, L.; Wang, Y. 4-HNE expression in diabetic rat kidneys and the protective effects of probucol. J. Endocrinol. Investig. 2016, 39, 865–873. [Google Scholar] [CrossRef]
- Miranda-Diaz, A.G.; Pazarin-Villasenor, L.; Yanowsky-Escatell, F.G.; Andrade-Sierra, J. Oxidative Stress in Diabetic Nephropathy with Early Chronic Kidney Disease. J. Diabetes Res. 2016, 2016, 7047238. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.R.; Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 2004, 37, 937–945. [Google Scholar] [CrossRef]
- Chaudhary, P.; Sharma, R.; Sharma, A.; Vatsyayan, R.; Yadav, S.; Singhal, S.S.; Rauniyar, N.; Prokai, L.; Awasthi, S.; Awasthi, Y.C. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry 2010, 49, 6263–6275. [Google Scholar] [CrossRef] [PubMed]
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013, 1, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Tabak, O.; Gelisgen, R.; Erman, H.; Erdenen, F.; Muderrisoglu, C.; Aral, H.; Uzun, H. Oxidative lipid, protein, and DNA damage as oxidative stress markers in vascular complications of diabetes mellitus. Clin. Investig. Med. 2011, 34, E163–E171. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Targeting Redox Imbalance as an Approach for Diabetic Kidney Disease. Biomedicines 2020, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Mauer, M.; Doria, A. Uric Acid and Diabetic Nephropathy Risk. Contrib. Nephrol. 2018, 192, 103–109. [Google Scholar] [CrossRef]
- Xiong, Q.; Liu, J.; Xu, Y. Effects of Uric Acid on Diabetes Mellitus and Its Chronic Complications. Int. J. Endocrinol. 2019, 9691345. [Google Scholar] [CrossRef]
- Butler, R.; Morris, A.D.; Belch, J.J.; Hill, A.; Struthers, A.D. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 2000, 35, 746–751. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Allopurinol protects human glomerular endothelial cells from high glucose-induced reactive oxygen species generation, p53 overexpression and endothelial dysfunction. Int. Urol. Nephrol. 2018, 50, 179–186. [Google Scholar] [CrossRef]
- Rodrigues, A.F.; Roecker, R.; Junges, G.M.; de Lima, D.D.; da Cruz, J.G.; Wyse, A.T.; Dal Magro, D.D. Hypoxanthine induces oxidative stress in kidney of rats: Protective effect of vitamins E plus C and allopurinol. Cell Biochem. Funct. 2014, 32, 387–394. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, Y.; Zhang, Q.Y.; Wang, F.M.; Kong, L.D. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS ONE 2012, 7, e38285. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Huang, S.S.; Ding, D.F.; Chen, S.; Dong, C.L.; Ye, X.L.; Yuan, Y.G.; Feng, Y.M.; You, N.; Xu, J.R.; Miao, H.; et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci. Rep. 2017, 7, 45692. [Google Scholar] [CrossRef]
- Fang, Y.; Li, F.; Qi, C.; Mao, X.; Wang, F.; Zhao, Z.; Chen, J.K.; Zhang, Z.; Wu, H. Metformin effectively treats Tsc1 deletion-caused kidney pathology by upregulating AMPK phosphorylation. Cell Death Discov. 2020, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Decleves, A.E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, L.; Wu, H.; Zhou, H. Antioxidative Property and Molecular Mechanisms Underlying Geniposide-Mediated Therapeutic Effects in Diabetes Mellitus and Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7480512. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef]
- Mariappan, M.M.; Shetty, M.; Sataranatarajan, K.; Choudhury, G.G.; Kasinath, B.S. Glycogen synthase kinase 3β is a novel regulator of high glucose- and high insulin-induced extracellular matrix protein synthesis in renal proximal tubular epithelial cells. J. Biol. Chem. 2008, 283, 30566–30575. [Google Scholar] [CrossRef]
- Hurcombe, J.A.; Hartley, P.; Lay, A.C.; Ni, L.; Bedford, J.J.; Leader, J.P.; Singh, S.; Murphy, A.; Scudamore, C.L.; Marquez, E.; et al. Podocyte GSK3 is an evolutionarily conserved critical regulator of kidney function. Nat. Commun. 2019, 10, 403. [Google Scholar] [CrossRef]
- Welsh, G.I.; Hale, L.J.; Eremina, V.; Jeansson, M.; Maezawa, Y.; Lennon, R.; Pons, D.A.; Owen, R.J.; Satchell, S.C.; Miles, M.J.; et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zhou, F.; Xu, Y.; Liu, X.; Zhang, Y.; Gu, M.; Su, Z.; Zhao, D.; Zhang, L.; Jia, Y. Geniposide regulates the miR-101/MKP-1/p38 pathway and alleviates atherosclerosis inflammatory injury in ApoE(−/−) mice. Immunobiology 2019, 224, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Dai, J.; Zhang, W.B.; Yuan, Y.; Liao, H.H.; Zhang, N.; Bian, Z.Y.; Tang, Q.Z. Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor/AMPKα signalling pathway. Br. J. Pharmacol. 2016, 173, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, Y.; Shi, S.; Katoh, M.; Kitada, M.; Kanasaki, K.; Koya, D. Dipeptidyl peptidase-4 plays a pathogenic role in BSA-induced kidney injury in diabetic mice. Sci. Rep. 2019, 9, 7519. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primers (5′–3′) | Reverse Primers (5′–3′) |
|---|---|---|
| KIM-1 | GAGAGTGACAGTGGTCTGTATTG | CCTTGTAGTTGTGGGTCTTCTT |
| NGAL | CACCACGGACTACAACCAGTTCGC | TCAGTTGTCAATGCATTGGTCGGTG |
| CTGF | CAGCTGGGAGAACTGTGTACGG | CACACTCCGATCTTGCGGTTGG |
| TGFβ | CGAAGCGGACTACTATGCTAAA | TCCCGAATGTCTGACGTATTG |
| FN1 | TACGGAGAGACAGGAGGAAATA | CATACAGGGTGATGGTGTAGTC |
| COL I | AGACCTGTGTGTTCCCTACT | GAATCCATCGGTCATGCTCTC |
| COL IV | CTGCTCTGCGTGGAGTATTT | AGGATGAAGGAGGCTAACAAAG |
| TIMP-1 | CCAGTCATGGAAAGCCTCTGTGGA | CTTTGCTGAGCAGGGCTCAGAGTA |
| MCP-1 | ACCTTTGAATGTGAAGTTGA | CTACAGAAGTGCTTGAGGTG |
| IL-6 | CCAATTCATCTTGAAATCAC | GGAATGTCCACAAACTGATA |
| TNFα | CATATACCTGGGAGGAGTCT | GAGCAATGACTCCAAAGTAG |
| MIP2 | AGAGGGTGAGTTGGGAACTA | GCCATCCGACTGCATCTATT |
| NOS2 | GGAATCTTGGAGCGAGTTGT | CCTCTTGTCTTTGACCCAGTAG |
| IL-10 | GGGAAGACAATAACTGCAC | TGAAAGAAAGTCTTCACCTG |
| NQO1 | ATGACATCACAGGTGAGCTGAAGG | CTCAAACCAGCCTTTCAGAATGGC |
| MnSOD2 | CCACCGAGGAGAAGTACCACGAG | CTCCTTATTGAAGCCAAGCCAGCC |
| GPX1 | GAGAAGTGCGAAGTGAATGGTGAG | CACACCGGAGACCAAATGATGTAC |
| UCP2 | GCG TTC TGG GTA CCA TCC TAA C | GCG ACC AGC CCA TTG TAG AG |
| GAPDH | GTGGCAAAGTGGAGATTGTTG | TTGACTGTGCCGTTGAATTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusabimana, T.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation. Int. J. Mol. Sci. 2021, 22, 1651. https://doi.org/10.3390/ijms22041651
Dusabimana T, Park EJ, Je J, Jeong K, Yun SP, Kim HJ, Kim H, Park SW. Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation. International Journal of Molecular Sciences. 2021; 22(4):1651. https://doi.org/10.3390/ijms22041651
Chicago/Turabian StyleDusabimana, Theodomir, Eun Jung Park, Jihyun Je, Kyuho Jeong, Seung Pil Yun, Hye Jung Kim, Hwajin Kim, and Sang Won Park. 2021. "Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation" International Journal of Molecular Sciences 22, no. 4: 1651. https://doi.org/10.3390/ijms22041651
APA StyleDusabimana, T., Park, E. J., Je, J., Jeong, K., Yun, S. P., Kim, H. J., Kim, H., & Park, S. W. (2021). Geniposide Improves Diabetic Nephropathy by Enhancing ULK1-Mediated Autophagy and Reducing Oxidative Stress through AMPK Activation. International Journal of Molecular Sciences, 22(4), 1651. https://doi.org/10.3390/ijms22041651