
The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway


Abstract
1. Introduction
2. ER Stress and Unfolded Protein Response
2.1. ER Stress
2.2. Unfolded Protein Response
2.2.1. IRE1 Pathway
2.2.2. PERK Pathway
2.2.3. ATF6 Pathway
3. Ubiquitin-Proteasome System
4. Role of E3 Ubiquitin Ligase in the Regulation of ER Stress
4.1. Regulation of the Core Components of ER Stress
4.1.1. MITOL Regulates IRE1α
4.1.2. CHIP Regulates IRE1
4.1.3. HRD1 Regulates IRE1α
4.1.4. Parkin Regulates CHOP
4.2. E3 Ubiquitin Ligases which Positively Regulate the ER Stress
4.2.1. RNF183 Increases ER Stress-Induced Apoptosis by Ubiquitinating Bcl-xL
4.2.2. RNF186 Regulates ER Stress-Mediated Apoptosis by Interacting with Bnip1
4.2.3. BAR Interacts with BI-1 to Eliminate the Inhibitory Effect of BI-1 on IRE1α
4.3. E3 Ubiquitin Ligases Which Antagonize ER Stress
4.3.1. GP78 Plays Dual Roles in ER Stress Response
4.3.2. POSH
5. Deubiquitinase and ER Stress
5.1. Deubiquitinases Which Are Involved in ERAD
5.1.1. USP13
5.1.2. USP19
5.1.3. YOD1
5.2. UPR-Related Deubiquitinases
5.2.1. USP14
5.2.2. BAP1
5.2.3. Other DUBs
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [PubMed]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Wang, Y.; Le, W.-D. Autophagy and Ubiquitin-Proteasome System. In Autophagy: Biology and Diseases: Basic Science; Qin, Z.-H., Ed.; Springer: Singapore, 2019; pp. 527–550. [Google Scholar]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Bio. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.; Huang, E.Y.; Olzmann, J.A. Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu. Rev. Nutr. 2016, 36, 511–542. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Reid, D.W.; Chen, Q.; Tay, A.S.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic Reticulum Stress in Malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Cagnetta, R.; Wong, H.H.; Frese, C.K.; Mallucci, G.R.; Krijgsveld, J.; Holt, C.E. Noncanonical Modulation of the eIF2 Pathway Controls an Increase in Local Translation during Neural Wiring. Mol. Cell 2019, 73, 474–489.e5. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, M.; Li, Y.; Li, C.; Tang, S.; Qu, X.; Feng, N.; Wu, Y. The PERK-EIF2alpha-ATF4 signaling branch regulates osteoblast differentiation and proliferation by PTH. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E590–E604. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hossain, M.M.; Read, D.E.; Hetz, C.; Samali, A.; Gupta, S. PERK regulated miR-424(322)-503 cluster fine-tunes activation of IRE1 and ATF6 during Unfolded Protein Response. Sci. Rep. 2015, 5, 18304. [Google Scholar] [CrossRef]
- Leestemaker, Y.; Ovaa, H. Tools to investigate the ubiquitin proteasome system. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef]
- Passmore, L.A.; Barford, D. Getting into position: The catalytic mechanisms of protein ubiquitylation. Biochem. J. 2004, 379, 513–525. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37 Pt 5, 937–953. [Google Scholar] [CrossRef]
- Schreiber, A.; Stengel, F.; Zhang, Z.G.; Enchev, R.I.; Kong, E.H.; Morris, E.P.; Robinson, C.V.; da Fonseca, P.C.A.; Barford, D. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature 2011, 470, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of Cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Zilfou, J.T.; Lowe, S.W. Tumor Suppressive Functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Stringer, D.K.; Piper, R.C. A single ubiquitin is sufficient for cargo protein entry into MVBs in the absence of ESCRT ubiquitination. J. Cell Biol. 2011, 192, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef]
- Schmidt, M.H.H.; Dikic, I. The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 907–918. [Google Scholar] [CrossRef]
- Kuang, E.; Qi, J.; Ronai, Z.E. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 2013, 38, 453–460. [Google Scholar] [CrossRef]
- Jacomin, A.C.; Taillebourg, E.; Fauvarque, M.O. Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities? Cells 2018, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.L.; Rose, I.A. The Mechanism of Ubiquitin Activating Enzyme—A Kinetic and Equilibrium-Analysis. J. Biol. Chem. 1982, 257, 329–337. [Google Scholar] [CrossRef]
- Haas, A.L.; Warms, J.V.B.; Hershko, A.; Rose, I.A. Ubiquitin-Activating Enzyme—Mechanism and Role in Protein-Ubiquitin Conjugation. J. Biol. Chem. 1982, 257, 2543–2548. [Google Scholar] [CrossRef]
- Ciechanover, A.; Ben-Saadon, R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004, 14, 103–106. [Google Scholar] [CrossRef]
- Breitschopf, K. A novel site for ubiquitination: The N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998, 17, 5964–5973. [Google Scholar] [CrossRef]
- Reinstein, E.; Scheffner, M.; Oren, M.; Ciechanover, A.; Schwartz, A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: Targeting via ubiquitination of the N-terminal residue. Oncogene 2000, 19, 5944. [Google Scholar] [CrossRef]
- Aviel, S.; Winberg, G.; Massucci, M.; Ciechanover, A. Degradation of the Epstein-Barr Virus Latent Membrane Protein 1 (LMP1) by the Ubiquitin-Proteasome Pathway: Targeting via Ubiquitination of the N-Terminal Residue. J. Biol. Chem. 2000, 275, 23491–23499. [Google Scholar] [CrossRef]
- Fajerman, I.; Schwartz, A.L.; Ciechanover, A. Degradation of the Id2 developmental regulator: Targeting via N-terminal ubiquitination. Biochem. Biophys. Res. Commun. 2004, 314, 505–512. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Promising new therapeutic targets for regulation of inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol. Lett. 2018, 202, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N. The Role of the Transmembrane RING Finger Proteins in Cellular and Organelle Function. Membranes 2011, 1, 354–393. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248. [Google Scholar] [CrossRef]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-Translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Venne, A.S.; Kollipara, L.; Zahedi, R.P. The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 2014, 14, 513–524. [Google Scholar] [CrossRef]
- Zhao, B.; Schlesiger, C.; Masucci, M.G.; Lindsten, K. The ubiquitin specific protease 4 (USP4) is a new player in the Wnt signalling pathway. J. Cell. Mol. Med. 2009, 13, 1886–1895. [Google Scholar] [CrossRef]
- Lui, T.T.; Lacroix, C.; Ahmed, S.M.; Goldenberg, S.J.; Leach, C.A.; Daulat, A.M.; Angers, S. The Ubiquitin-Specific Protease USP34 Regulates Axin Stability and Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2011, 31, 2053–2065. [Google Scholar] [CrossRef]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a Deubiquitinating Enzyme Essential for TGF beta Signaling, Controls Smad4 Monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Coulson, J.M.; Urbe, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125 Pt 2, 277–286. [Google Scholar] [CrossRef]
- Kristariyanto, Y.A.; Rehman, S.A.A.; Weidlich, S.; Knebel, A.; Kulathu, Y. A single MIU motif of MINDY-1 recognizes K48-linked polyubiquitin chains. EMBO Rep. 2017, 18, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [PubMed]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Borgermann, N.; Guo, X.H.; Typas, D.; Achuthankutty, D.; Hoffmann, S.; Shearer, R.; Sixma, T.K.; Mailand, N. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol. Cell 2018, 70, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, J.; Sun, H.; Jiang, C.; Dong, Y.; Shan, Q.; Liu, S. Ubiquitination of Inositol-requiring Enzyme 1 (IRE1) by the E3 Ligase CHIP Mediates the IRE1/TRAF2/JNK Pathway. J. Biol. Chem. 2014, 289, 30567–30577. [Google Scholar] [CrossRef]
- Doroudgar, S.; Volkers, M.; Thuerauf, D.J.; Khan, M.; Mohsin, S.; Respress, J.L.; Wang, W.; Gude, N.; Muller, O.J.; Wehrens, X.H.; et al. Hrd1 and ER-Associated Protein Degradation, ERAD, are Critical Elements of the Adaptive ER Stress Response in Cardiac Myocytes. Circ. Res. 2015, 117, 536–546. [Google Scholar] [CrossRef]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 2019, 4, e121887. [Google Scholar] [CrossRef]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Hassanzadeh, S.; Singh, K.; Menazza, S.; Nguyen, T.T.; Stevens, M.V.; Nguyen, A.; San, H.; Anderson, S.A.; Lin, Y. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci. Rep. 2017, 7, 2093. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Jia, J.; Zhang, Y.; Li, J.; Zhu, Z.; Wang, H.; Tang, J.; Hu, J. Transmembrane E3 ligase RNF183 mediates ER stress-induced apoptosis by degrading Bcl-xL. Proc. Natl. Acad. Sci. USA 2018, 115, E2762–E2771. [Google Scholar] [CrossRef]
- Wang, P.; Wu, Y.; Li, Y.; Zheng, J.; Tang, J. A novel RING finger E3 ligase RNF186 regulate ER stress-mediated apoptosis through interaction with BNip1. Cell Signal. 2013, 25, 2320–2333. [Google Scholar] [CrossRef]
- Bailly-Maitre, B.; Fondevila, C.; Kaldas, F.; Droin, N.; Luciano, F.; Ricci, J.E.; Croxton, R.; Krajewska, M.; Zapata, J.M.; Kupiec-Weglinski, J.W.; et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 2809–2814. [Google Scholar] [CrossRef]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Rong, J.; Chen, L.; Toth, J.I.; Tcherpakov, M.; Petroski, M.D.; Reed, J.C. BAR, an endoplasmic reticulum-associated E3 ubiquitin ligase, modulates BI-1 protein stability and function in ER stress. J. Biol. Chem. 2010, 286, 1453–1463. [Google Scholar] [CrossRef]
- Chen, Z.; Ballar, P.; Fu, Y.; Luo, J.; Du, S.; Fang, S. The E3 ubiquitin ligase gp78 protects against ER stress in zebrafish liver. J. Genet. Genom. 2014, 41, 357–368. [Google Scholar] [CrossRef]
- Yan, L.; Liu, W.X.; Zhang, H.H.; Liu, C.; Shang, Y.L.; Ye, Y.H.; Zhang, X.D.; Li, W. Ube2g2-gp78-mediated HERP polyubiquitylation is involved in ER stress recovery. J. Cell Sci. 2014, 127, 1417–1427. [Google Scholar] [CrossRef]
- Tuvia, S.; Taglicht, D.; Erez, O.; Alroy, I.; Alchanati, I.; Bicoviski, V.; Dori-Bachash, M.; Ben-Avraham, D.; Reiss, Y. The ubiquitin E3 ligase POSH regulates calcium homeostasis through spatial control of Herp. J. Cell Biol. 2007, 177, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Shun, N.; Takumi, T.; Shigeru, Y. Mitochondrial quality control by mitochondrial ubiquitin ligase MITOL/MARCH5. Nihon Yakurigaku Zasshi Folia Pharmacol. Jpn. 2017, 149, 254. [Google Scholar]
- Nagashima, S.; Tokuyama, T.; Yonashiro, R.; Inatome, R.; Yanagi, S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J. Biochem. 2014, 155, 273–279. [Google Scholar] [CrossRef]
- Park, Y.Y.; Lee, S.; Karbowski, M.; Neutzner, A.; Youle, R.J.; Cho, H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J. Cell Sci. 2010, 123 Pt 4, 619–626. [Google Scholar] [CrossRef]
- Park, Y.Y.; Nguyen, O.T.; Kang, H.; Cho, H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Li, Q.; Huang, S.; Lu, W.; Cheng, F.; Gao, P.; Wang, C.; Miao, L.; Mei, Y.; Wu, M. Mitochondrial E3 ligase March5 maintains stemness of mouse ES cells via suppression of ERK signalling. Nat. Commun. 2015, 6, 7112. [Google Scholar] [CrossRef]
- Wang, J.; Aung, L.H.; Prabhakar, B.S.; Li, P. The mitochondrial ubiquitin ligase plays an anti-apoptotic role in cardiomyocytes by regulating mitochondrial fission. J. Cell. Mol. Med. 2016, 20, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Jang, K.W.; Lee, K.H.; Kim, S.H.; Jin, T.; Choi, E.Y.; Jeon, H.J.; Kim, E.; Han, Y.S.; Chung, J.H. Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-κB inactivation to regulate breast cancer cell invasion. J. Cell. Biochem. 2011, 112, 3612–3620. [Google Scholar] [CrossRef]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, D. Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 2020, 109, 102423. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Ishiguro, M.; Niinuma, Y.; Uesugi, M.; Nomura, Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. 2002, 532, 147–152. [Google Scholar] [CrossRef]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Rudensky, A. The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol. Rev. 2006, 212, 86–98. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis. 2014, 13, 69–71. [Google Scholar] [CrossRef]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef]
- Rosen, K.M.; Moussa, C.E.H.; Lee, H.K.; Kumar, P.; Kitada, T.; Qin, G.J.; Fu, Q.H.; Querfurth, H.W. Parkin Reverses Intracellular beta-Amyloid Accumulation and Its Negative Effects on Proteasome Function. J. Neurosci. Res. 2010, 88, 167–178. [Google Scholar] [CrossRef]
- Hebron, M.; Chen, W.; Miessau, M.J.; Lonskaya, I.; Moussa, C.E. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem. 2014, 129, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Muller-Rischart, A.K.; Pilsl, A.; Beaudette, P.; Patra, M.; Hadian, K.; Funke, M.; Peis, R.; Deinlein, A.; Schweimer, C.; Kuhn, P.H.; et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell 2013, 49, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Tan, X.; Wu, J.; Pan, Z.; Yi, M.; Shi, W.; Liu, R.; Yao, C.; Wang, G.; Lin, J.; et al. RNF183 promotes proliferation and metastasis of colorectal cancer cells via activation of NF-kappaB-IL-8 axis. Cell Death Dis. 2017, 8, e2994. [Google Scholar] [CrossRef]
- Park, H.A.; Broman, K.; Stumpf, A.; Kazyak, S.; Jonas, E.A. Nutritional Regulators of Bcl-xL in the Brain. Molecules 2018, 23, 3019. [Google Scholar] [CrossRef]
- Yasuda, M.; Theodorakis, P.; Subramanian, T.; Chinnadurai, G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 1998, 273, 12415–12421. [Google Scholar] [CrossRef]
- Nakajima, K.; Hirose, H.; Taniguchi, M.; Kurashina, H.; Arasaki, K.; Nagahama, M.; Tani, K.; Yamamoto, A.; Tagaya, M. Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J. 2004, 23, 3216–3226. [Google Scholar] [CrossRef]
- Ryu, S.W.; Choi, K.; Yoon, J.; Kim, S.; Choi, C. Endoplasmic reticulum-specific BH3-only protein BNIP1 induces mitochondrial fragmentation in a Bcl-2- and Drp1-dependent manner. J. Cell. Physiol. 2012, 227, 3027–3035. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, Q.; Wang, L.; Ji, Y.; Zhang, L.; Xie, L.; Zhang, H. RNF186 impairs insulin sensitivity by inducing ER stress in mouse primary hepatocytes. Cell. Signal. 2018, 52, 155–162. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, Q.L.; Krajewski, S.; Krajewska, M.; Xie, Z.H.; Fuess, S.; Kitada, S.; Pawlowski, K.; Godzik, A.; Reed, J.C. BAR: An apoptosis regulator at the intersection of caspases and Bcl-2 family proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 2597–2602. [Google Scholar] [CrossRef]
- Roth, W.; Kermer, P.; Krajewska, M.; Welsh, K.; Davis, S.; Krajewski, S.; Reed, J.C. Bifunctional apoptosis inhibitor (BAR) protects neurons from diverse cell death pathways. Cell Death Differ. 2003, 10, 1178–1187. [Google Scholar] [CrossRef]
- Nabi, I.R.; Raz, A. Cell shape modulation alters glycosylation of a metastatic melanoma cell-surface antigen. Int. J. Cancer 1987, 40, 396–402. [Google Scholar] [CrossRef]
- Madore, H.P.; England, J.M. Rabies virus protein synthesis in infected BHK-21 cells. J. Virol. 1977, 22, 102–112. [Google Scholar] [CrossRef]
- Nabi, I.R.; Watanabe, H.; Silletti, S.; Raz, A. Tumor cell autocrine motility factor receptor. EXS 1991, 59, 163–177. [Google Scholar]
- Fang, S.Y.; Ferrone, M.; Yang, C.H.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef]
- Shen, Y.X.; Ballar, P.; Apostolou, A.; Doong, H.; Fang, S.Y. ER stress differentially regulates the stabilities of ERAD ubiquitin ligases and their substrates. Biochem. Bioph. Res. Commun. 2007, 352, 919–924. [Google Scholar] [CrossRef]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a New cis-Acting Element Responsible for the ATF6-dependent Mammalian Unfolded Protein Response. J. Biol. Chem. 2001, 276, 9199. [Google Scholar] [CrossRef]
- Nogalska, A.; Engel, W.K.; Mcferrin, J.; Kokame, K.; Komano, H.; Askanas, V. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J. Neurochem. 2010, 96, 1491–1499. [Google Scholar] [CrossRef]
- Jarosch, E.; Taxis, C.; Volkwein, C.; Bordallo, J.; Finley, D.; Wolf, D.H.; Sommer, T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 2002, 4, 134–139. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Ichinoda, F.; Yamaguchi, A.; Tamatani, T.; Taniguchi, M.; Koyama, Y.; Katayama, T.; Tohyama, M.; Stern, D.M.; Ozawa, K.; et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells 2004, 9, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Nagata, K.; Lamarche, N.; Hall, A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J. 1998, 17, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Belal, C.; Ameli, N.J.; Kommos, A.E.; Bezalel, S.; Al’Khafaji, A.M.; Mughal, M.R.; Mattson, M.P.; Kyriazis, G.A.; Tyrberg, B.; Chan, S.L. The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant α-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum. Mol. Genet. 2011, 21, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Fu, W.; Zhang, P.; Cheng, A.; Lee, J.; Kokame, K.; Mattson, M.P. Herp Stabilizes Neuronal Ca2+ Homeostasis and Mitochondrial Function during Endoplasmic Reticulum Stress. J. Biol. Chem. 2004, 279, 28733. [Google Scholar] [CrossRef]
- Liu, Y.; Soetandyo, N.; Lee, J.-G.; Liu, L.; Xu, Y.; Clemons, W.M.; Ye, Y. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. eLife 2014, 3, e01369. [Google Scholar] [CrossRef]
- Hassink, G.C.; Zhao, B.; Sompallae, R.; Altun, M.; Gastaldello, S.; Zinin, N.V.; Masucci, M.G.; Lindsten, K. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009, 10, 755–761. [Google Scholar] [CrossRef]
- Harada, K.; Kato, M.; Nakamura, N. USP19-Mediated Deubiquitination Facilitates the Stabilization of HRD1 Ubiquitin Ligase. Int. J. Mol. Sci. 2016, 17, 1829. [Google Scholar] [CrossRef]
- Schuberth, C.; Buchberger, A. UBX domain proteins: Major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. 2008, 65, 2360–2371. [Google Scholar] [CrossRef]
- Ernst, R.; Mueller, B.; Ploegh, H.L.; Schlieker, C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell 2009, 36, 28–38. [Google Scholar] [CrossRef]
- Nagai, A.; Kadowaki, H.; Maruyama, T.; Takeda, K.; Nishitoh, H.; Ichijo, H. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem. Biophys. Res. Commun. 2009, 379, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Hyrskyluoto, A.; Bruelle, C.; Lundh, S.H.; Do, H.T.; Kivinen, J.; Rappou, E.; Reijonen, S.; Waltimo, T.; Petersen, A.; Lindholm, D.; et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: Involvement of the proteasome and ER stress-activated kinase IRE1alpha. Hum. Mol. Genet. 2014, 23, 5928–5939. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Lee, H.; Zhang, Y.; Zhuang, L.; Yao, H.; Xi, Y.; Xiao, Z.D.; You, M.J.; Li, W.; Su, X.; et al. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc. Natl. Acad. Sci. USA 2017, 114, 3192–3197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, W.; Zhou, L.; Chen, Y.; Qin, S.; Zhang, L.; Liu, J.; He, Y.; Lei, Y.; Chen, H.N.; et al. Repurposing Brigatinib for the Treatment of Colorectal Cancer Based on Inhibition of ER-phagy. Theranostics 2019, 9, 4878–4892. [Google Scholar] [CrossRef] [PubMed]
- Wiles, B.; Miao, M.; Coyne, E.; Larose, L.; Cybulsky, A.V.; Wing, S.S. USP19 deubiquitinating enzyme inhibits muscle cell differentiation by suppressing unfolded-protein response signaling. Mol. Biol. Cell 2015, 26, 913–923. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar] [CrossRef]
- Rabinovich, E.; Kerem, A.; Frohlich, K.U.; Diamant, N.; Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 2002, 22, 626–634. [Google Scholar] [CrossRef]
- Ballar, P.; Shen, Y.; Yang, H.; Fang, S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2006, 281, 35359–35368. [Google Scholar] [CrossRef]
- Kaneko, M.; Nomura, Y. ER signaling in unfolded protein response. Life Sci. 2003, 74, 199–205. [Google Scholar] [CrossRef]
- Sanyal, S.; Claessen, J.H.; Ploegh, H.L. A viral deubiquitylating enzyme restores dislocation of substrates from the endoplasmic reticulum (ER) in semi-intact cells. J. Biol. Chem. 2012, 287, 23594–23603. [Google Scholar] [CrossRef] [PubMed]
- Rodighiero, C.; Tsai, B.; Rapoport, T.A.; Lencer, W.I. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002, 3, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Lencer, W.I.; Tsai, B. The intracellular voyage of cholera toxin: Going retro. Trends Biochem. Sci. 2003, 28, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Williams, J.M.; Kikkert, M.; van Voorden, S.; Wiertz, E.J.; Ye, Y.; Tsai, B. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol. Biol. Cell 2010, 21, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Williams, J.M.; Inoue, T.; Schultz, A.; Tsai, B. A deubiquitinase negatively regulates retro-translocation of nonubiquitinated substrates. Mol. Biol. Cell 2013, 24, 3545–3556. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Reijonen, S.; Putkonen, N.; Norremolle, A.; Lindholm, D.; Korhonen, L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef]
- Vidal, R.; Caballero, B.; Couve, A.; Hetz, C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr. Mol. Med. 2011, 11, 1–12. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, Z.; Hu, Y.; Lu, Y.; Li, D.; Liu, J.; Liao, S.; Hu, M.; Wang, Y.; Zhang, D.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. USA 2019, 116, 21732–21738. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Turner, N.; Yang, H. The role of oxysterol-binding protein and its related proteins in cancer. Semin. Cell Dev. Biol. 2018, 81, 149–153. [Google Scholar] [CrossRef]
- Zhong, W.; Qin, S.; Zhu, B.; Pu, M.; Liu, F.; Wang, L.; Ye, G.; Yi, Q.; Yan, D. Oxysterol-binding protein-related protein 8 (ORP8) increases sensitivity of hepatocellular carcinoma cells to Fas-mediated apoptosis. J. Biol. Chem. 2015, 290, 8876–8887. [Google Scholar] [CrossRef]
- Nakanishi, K.; Dohmae, N.; Morishima, N. Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB J. 2007, 21, 2994–3003. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef]
- Sundaram, P.; Pang, Z.; Miao, M.; Yu, L.; Wing, S.S. USP19-deubiquitinating enzyme regulates levels of major myofibrillar proteins in L6 muscle cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1283–E1290. [Google Scholar] [CrossRef]



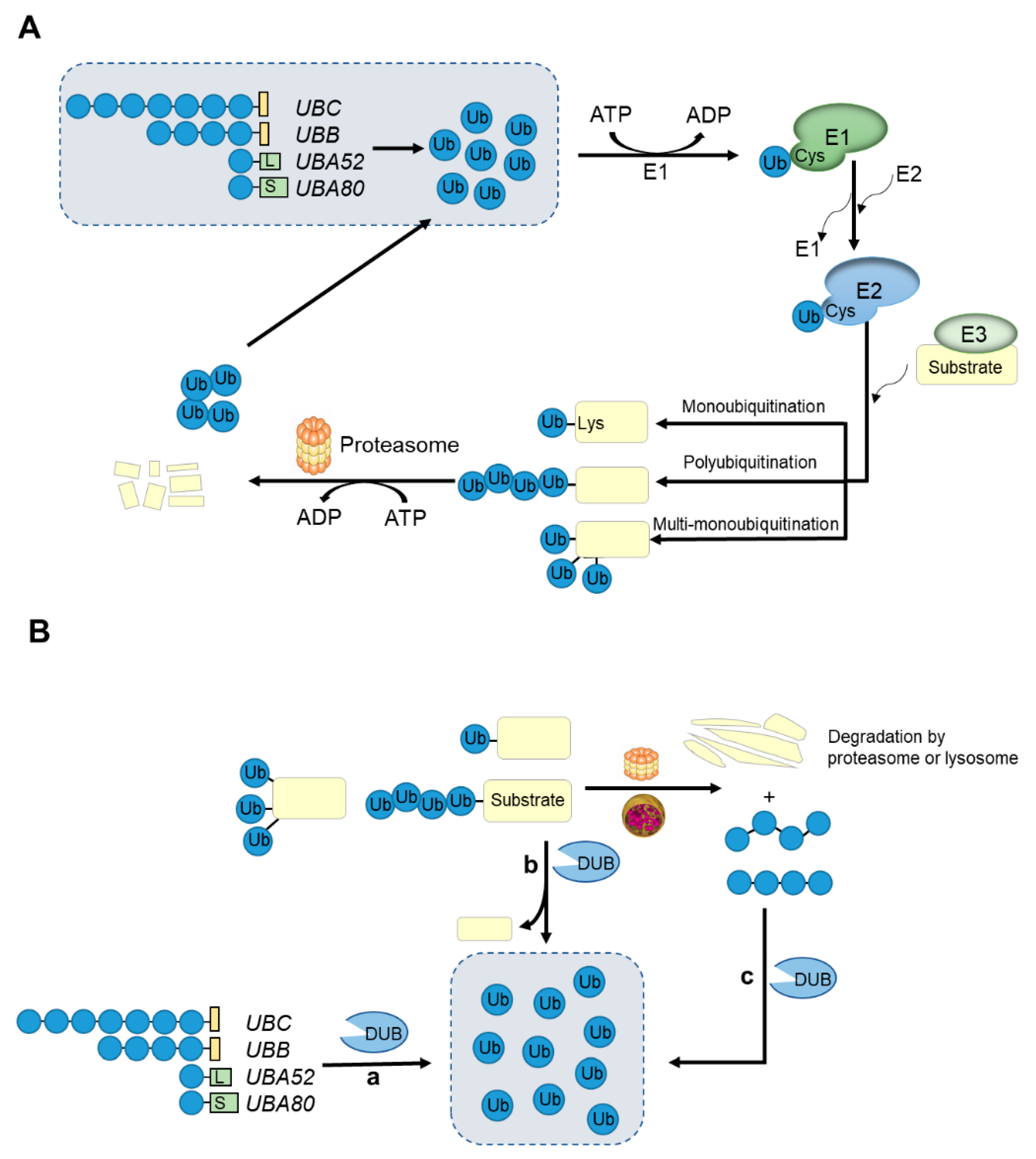{kind=link}
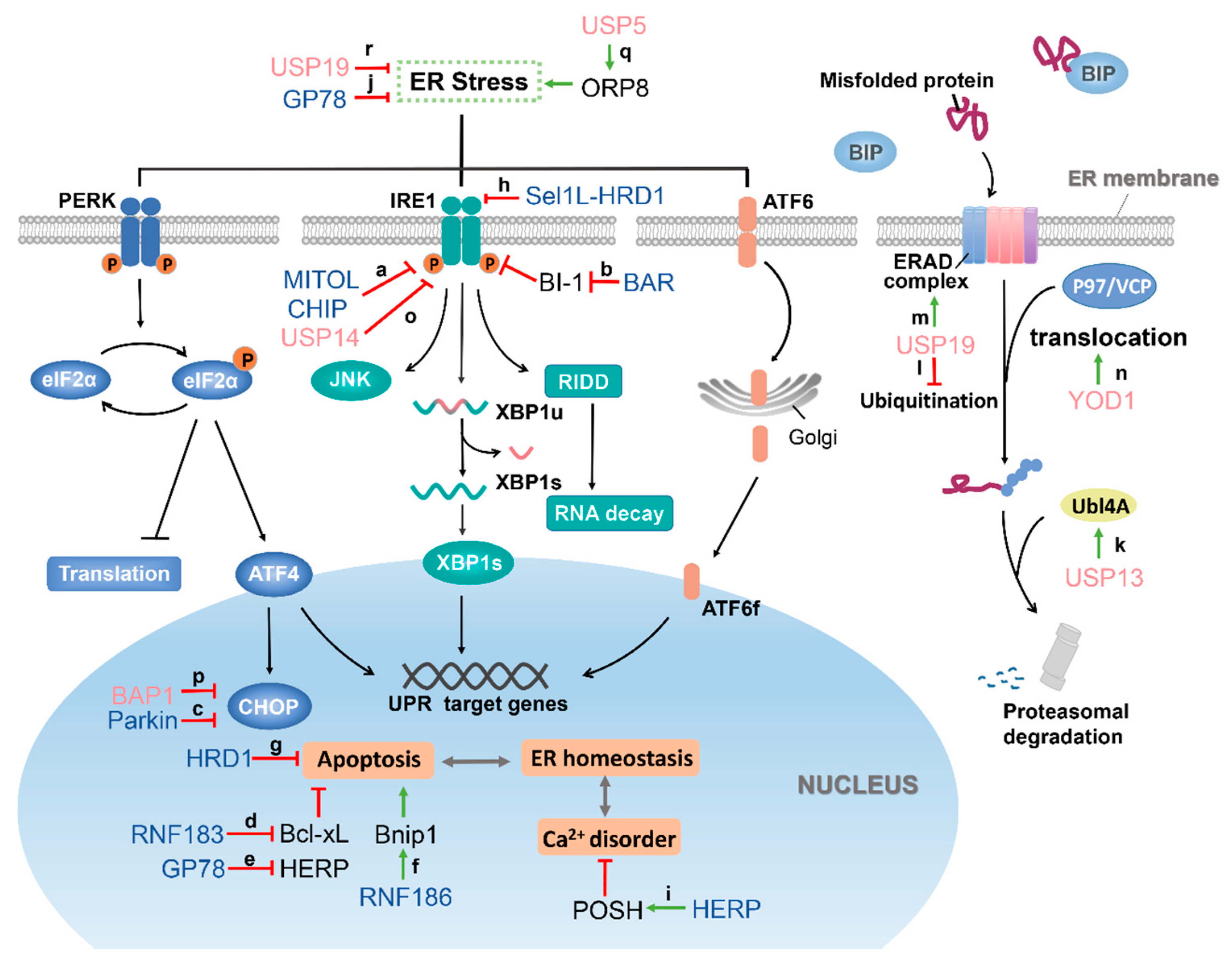{kind=link}
| E3 ligase | Target | Impact on ER Stress | References |
|---|---|---|---|
| MITOL | IRE1α | Inhibits ER stress | [78] |
| CHIP | IRE1 | Antagonizes ER stress-induced cellular senescence | [79] |
| HRD1 | IRE1α | Inhibits ER stress | [80,81,82] |
| Parkin | CHOP | Inhibits ER stress | [82,83,84] |
| RNF183 | Bcl-xL | Increases ER stress-induced apoptosis | [85] |
| RNF186 | Bnip1 | Increases ER stress-induced apoptosis | [86] |
| BAR | BI-1 | Increases ER stress | [87,88,89] |
| GP78 | ? | Protects against ER stress and regulates lipid metabolism in zebrafish liver | [90] |
| GP78 | HERP | Initiates the process of ER stress recovery | [91] |
| POSH | HERP | Maintains calcium homeostasis during ER stress | [92] |
| DUB | Target | Impact on ER stress | References |
|---|---|---|---|
| ERAD | |||
| USP13 | Ubl4A | Promotes ERAD | [137] |
| USP19 | TCRa and other ERAD substrates | Saves the substrate degradation caused by ERAD | [138] |
| HRD1 | Maintains normal ERAD process | [139] | |
| YOD1 | RI332 and other ERAD substrates | Promotes the reverse transport of misfolded proteins | [140,141] |
| UPR | |||
| USP14 | IRE1α | Inhibits ER stress-induced cell death | [142,143] |
| BAP1 | CHOP/ATF3 | Inhibits ER stress-induced apoptosis | [144] |
| USP5 | ORP8 | Increases ER stress | [145] |
| USP19 | ? | Inhibits the UPR reaction | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. https://doi.org/10.3390/ijms22041526
Qu J, Zou T, Lin Z. The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. International Journal of Molecular Sciences. 2021; 22(4):1526. https://doi.org/10.3390/ijms22041526
Chicago/Turabian StyleQu, Junyan, Tingting Zou, and Zhenghong Lin. 2021. "The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway" International Journal of Molecular Sciences 22, no. 4: 1526. https://doi.org/10.3390/ijms22041526
APA StyleQu, J., Zou, T., & Lin, Z. (2021). The Roles of the Ubiquitin–Proteasome System in the Endoplasmic Reticulum Stress Pathway. International Journal of Molecular Sciences, 22(4), 1526. https://doi.org/10.3390/ijms22041526