
Laccase-Catalyzed 1,4-Dioxane-Mediated Synthesis of Belladine N-Oxides with Anti-Influenza A Virus Activity

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
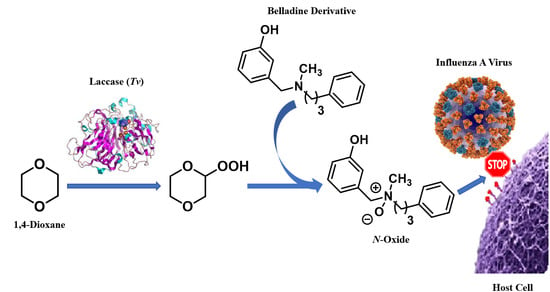
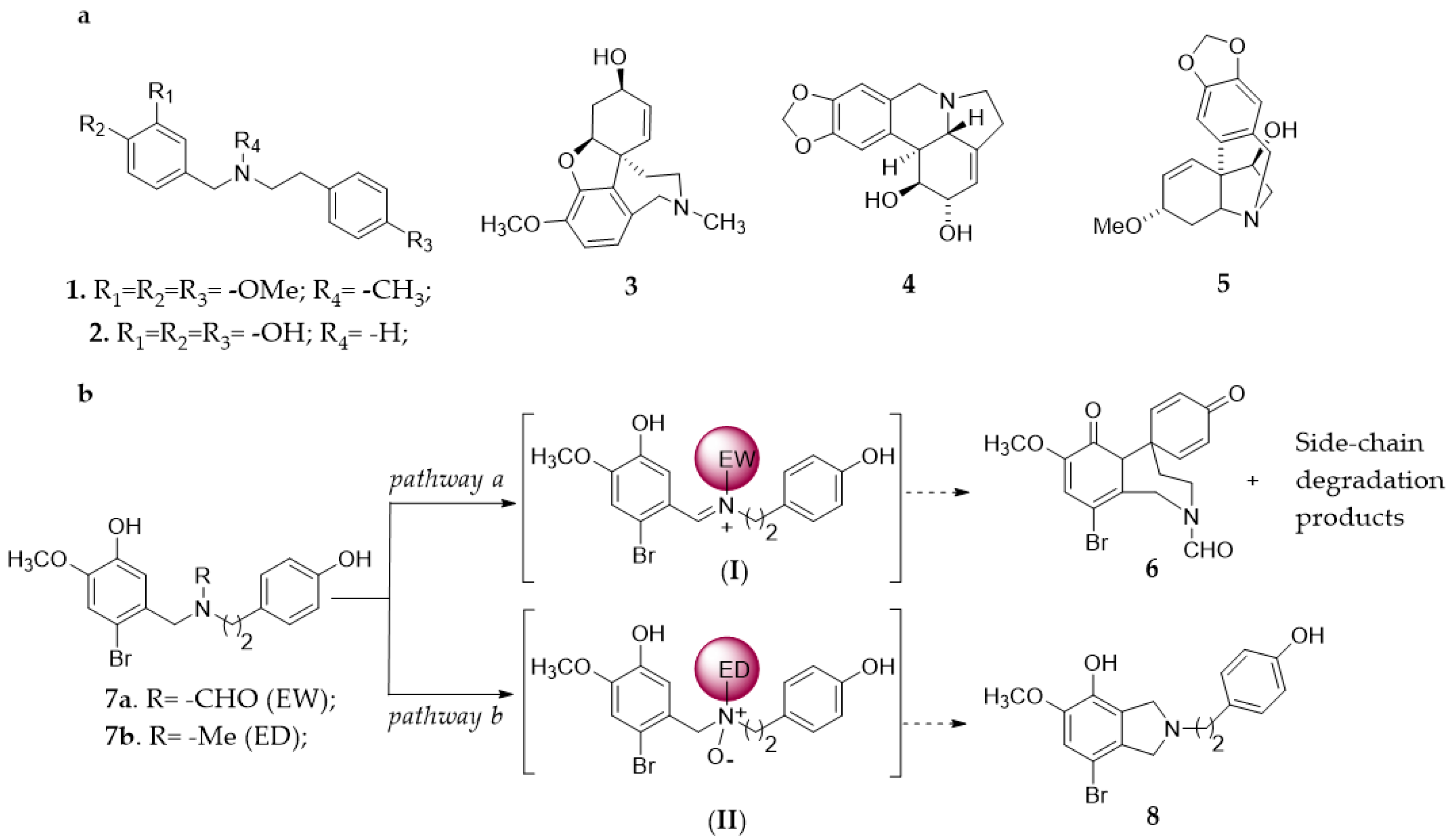
1. Introduction
2. Results and Discussion
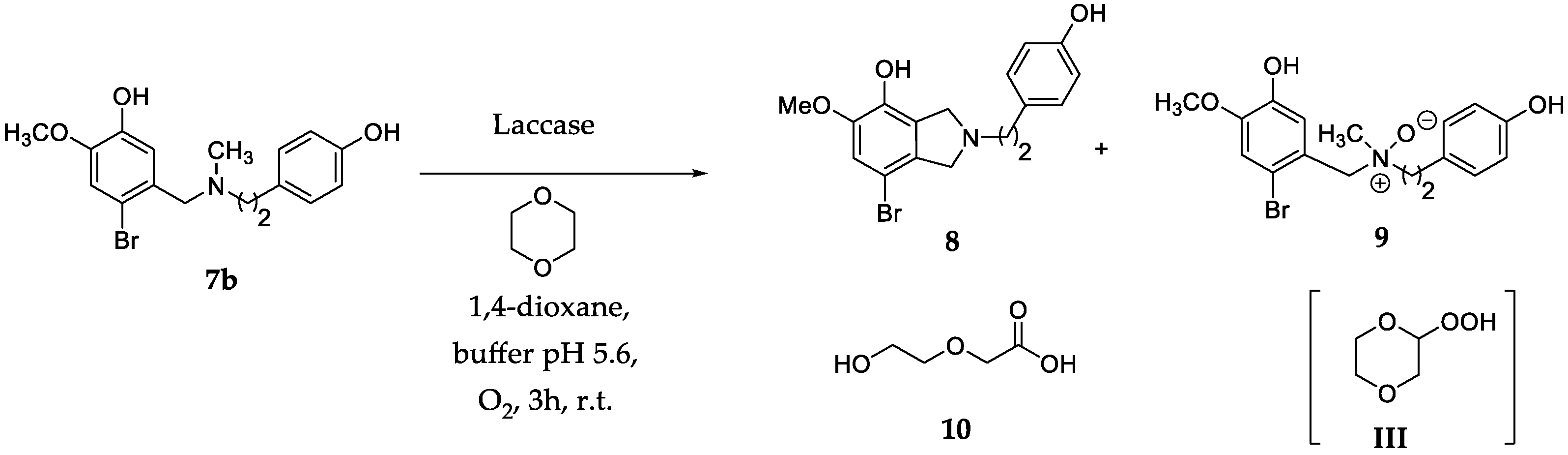
2.1. Optimization of the Reaction Conditions
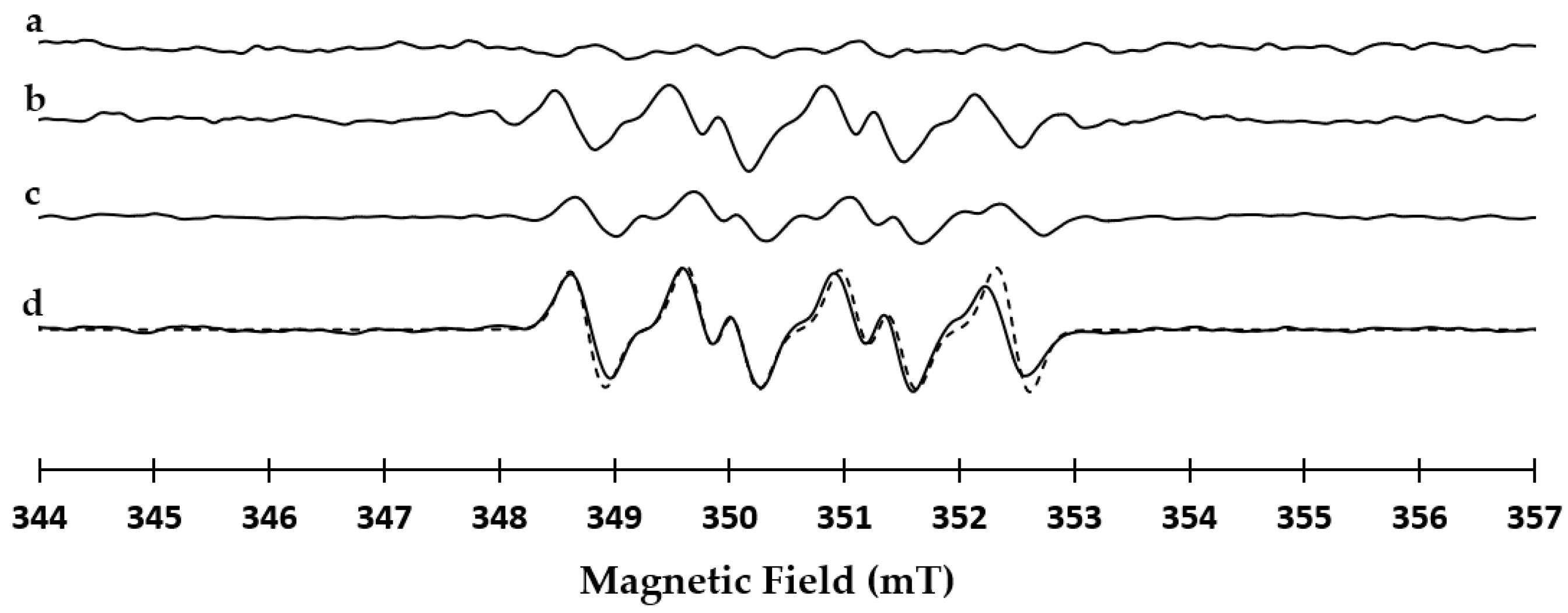
2.2. EPR Studies
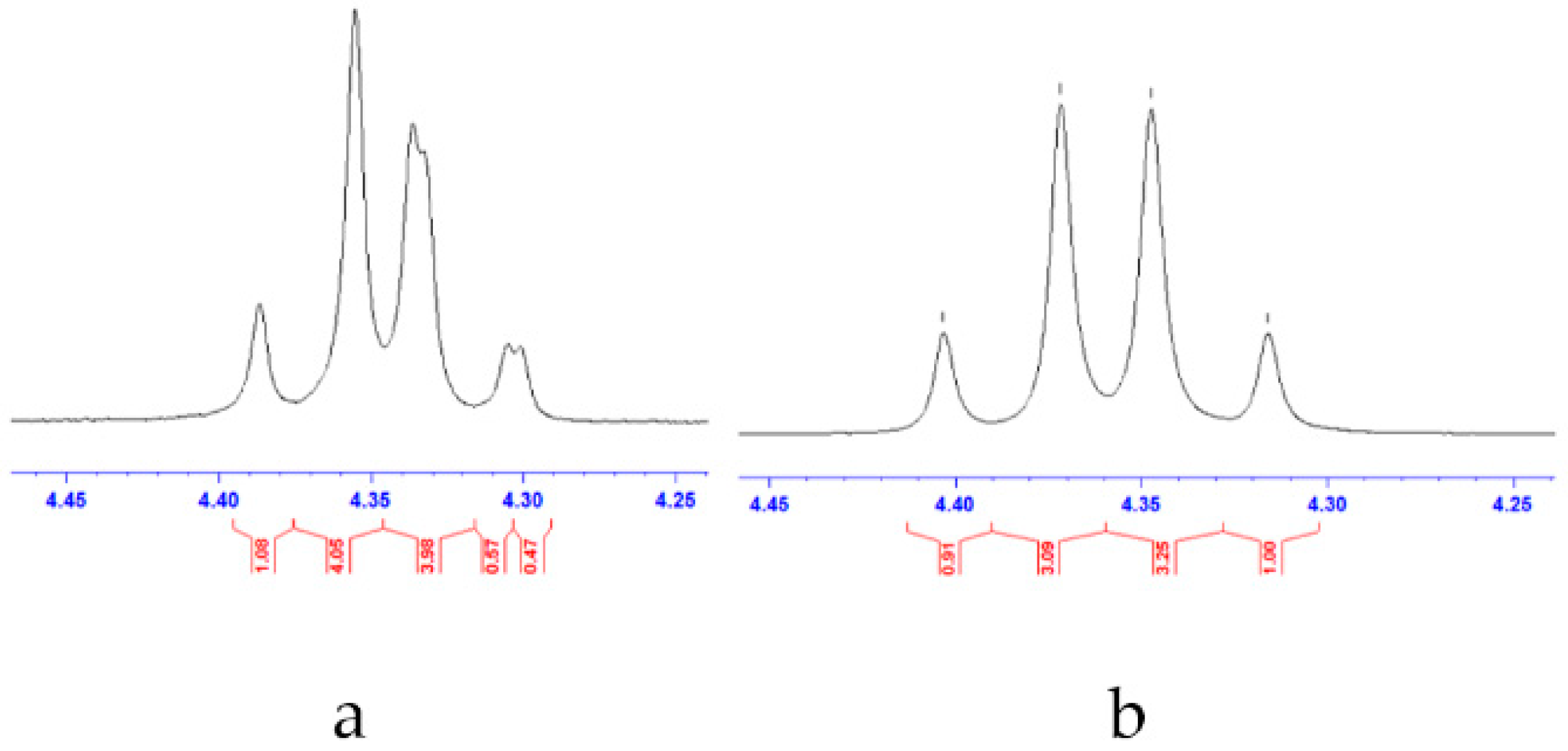
2.3. Synthesis and Characterization of Belladine N-Oxide Derivatives 12a–h
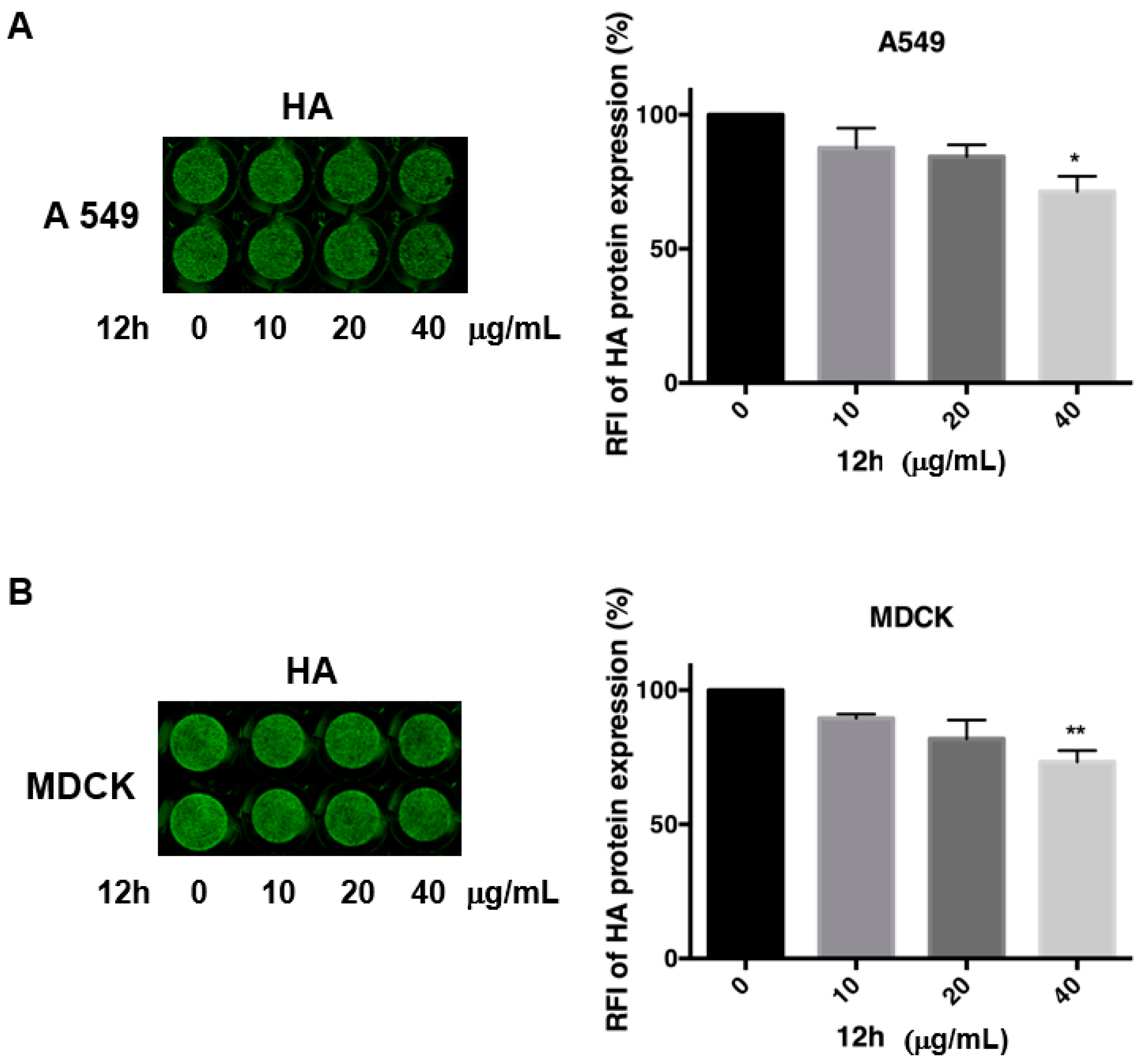
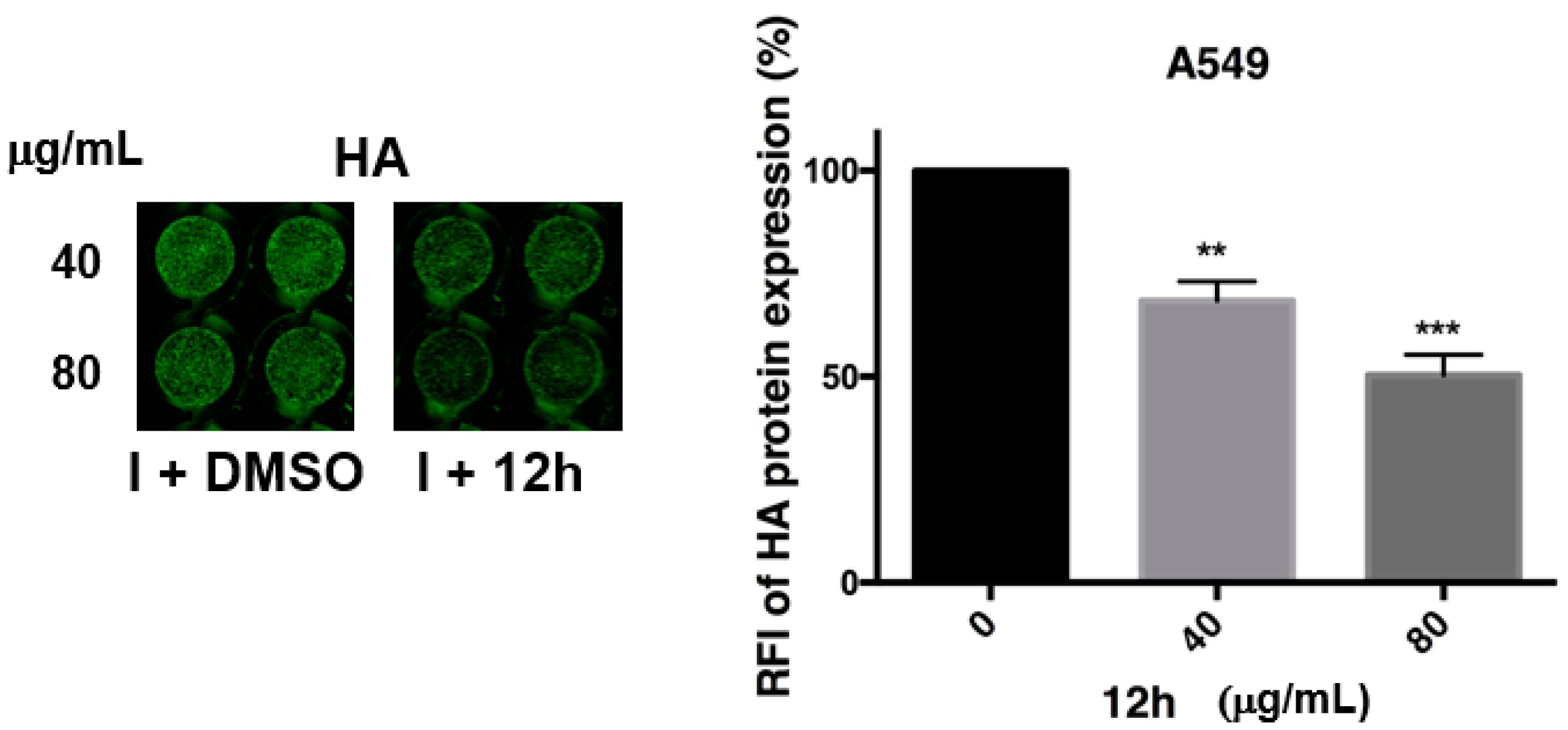
2.4. Antiviral Activity of Compound 7b and 12a–h against Influenza A Virus
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Enzyme Activity Assay
4.3. EPR Analysis
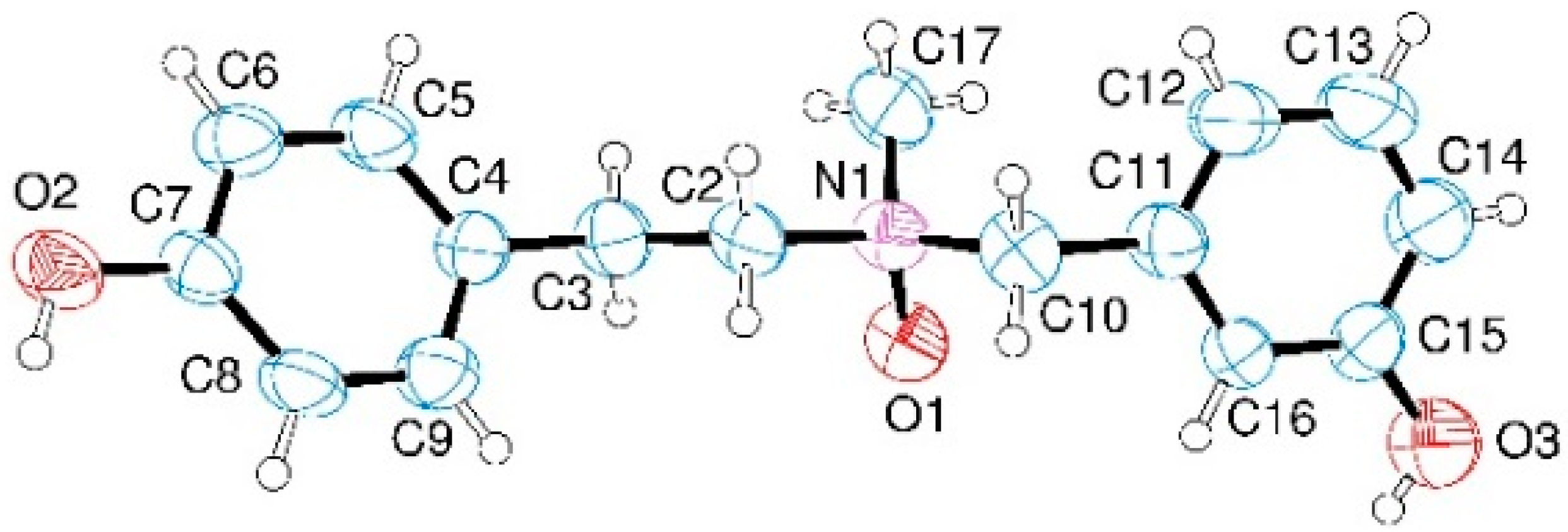
4.4. X-Ray Crystallography Data for Compound 12b
4.5. Procedure for the Synthesis of Amine N-Oxides 9 and 12a-h
4.5.1. N-(2-Bromo-5-Hydroxy-4-Methoxybenzyl)-2-(4-Hydroxyphenyl)-N-Methylethan-1-Amine Oxide (9)
4.5.2. N-(3-Hydroxy-4-Methoxybenzyl)-2-(4-Hydroxyphenyl)-N-Methylethan-1-Amine Oxide (12a)
4.5.3. N-(3-hydroxybenzyl)-2-(4-hydroxyphenyl)-N-methylethan-1-amine oxide (12b)
4.5.4. N-(4-Hydroxybenzyl)-2-(4-Hydroxyphenyl)-N-Methylethan-1-Amine Oxide (12c)
4.5.5. N-(3-Hydroxy-4-Nitrobenzyl)-2-(4-Hydroxyphenyl)-N-Methylethan-1-Amine Oxide (12d)
4.5.6. N-Benzyl-2-(4-Hydroxyphenyl)-N-Methylethan-1-Amine Oxide (12e)
4.5.7. 1-(3-hydroxyphenyl)-N,N-dimethylmethanamine oxide (12f)
4.5.8. N-(3-hydroxybenzyl)-1-(4-hydroxyphenyl)-N-methylmethanamine oxide (12g)
4.5.9. N-(3-Hydroxybenzyl)-N-Methyl-3-Phenylpropan-1-Amine Oxide (12h)
4.6. Cell Cultures
4.7. Virus Production and Infection
4.8. In Cell Western (ICW) Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, M.; Qu, C.; Gao, O.; Hu, X.; Hong, X. Biological and pharmacological activities of amaryllidaceae alkaloids. RSC Adv. 2015, 5, 16562–16574. [Google Scholar] [CrossRef]
- Hostettmann, K.; Borloz, A.; Urbain, A.; Marston, A. Natural Product Inhibitors of Acetylcholinesterase. Curr. Org. Chem. 2006, 10, 825–847. [Google Scholar] [CrossRef]
- Patil, D.N.; Patil, S.A.; Sistla, S.; Jadhav, J.P. Comparative biophysical characterization: A screening tool for acetylcholinesterase inhibitors. PLoS ONE 2019, 14, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Maříková, J.; Hulcová, D.; Janoušek, J.; Šafratová, M.; Nováková, L.; Kučera, T.; Hrabinová, M.; Kuneš, J.; Korábečný, J.; et al. Amaryllidaceae Alkaloids of Belladine-Type from Narcissus pseudonarcissus cv. Carlton as New Selective Inhibitors of Butyrylcholinesterase. Biomolecules 2020, 10, 800. [Google Scholar] [CrossRef]
- Carmona-Viglianco, F.; Zaragoza-Puchol, D.; Parravicini, O.; Garro, A.; Enriz, R.D.; Feresin, G.E.; Kurina-Sanz, M.; Orden, A.A. Synthesis, biological evaluation and molecular modeling studies of substituted N-benzyl-2-phenylethanamines as cholinesterase inhibitors. New J. Chem. 2020, 44, 9466–9476. [Google Scholar] [CrossRef]
- Shawky, E. In-silico profiling of the biological activities of Amaryllidaceae alkaloids. J. Pharm. Pharmacol. 2017, 69, 1592–1605. [Google Scholar] [CrossRef]
- Solomon, E.I.; Baldwin, M.J.; Lowery, M.D. Electronic structures of active sites in copper proteins: Contributions to reactivity. Chem. Rev. 1992, 92, 521–542. [Google Scholar] [CrossRef]
- Zippilli, C.; Botta, L.; Bizzarri, B.M.; Baratto, M.C.; Pogni, R.; Saladino, R. Biomimetic synthesis of galantamine: Via laccase/TEMPO mediated oxidative coupling. RSC Adv. 2020, 10, 10897–10903. [Google Scholar] [CrossRef]
- Galletti, P.; Funiciello, F.; Soldati, R.; Giacomini, D. Selective Oxidation of Amines to Aldehydes or Imines using Laccase-Mediated Bio-Oxidation. Adv. Synth. Catal. 2015, 357, 1840–1848. [Google Scholar] [CrossRef]
- Polonovski, M.; Polonovski, M. Sur les aminoxydes des alcaloïdes. III. Action des anhydrides et chlorures d’acides organiques. Préparation des bases nor. Bull. Soc. Chim. Fr. 1927, 41, 1190–1208. [Google Scholar]
- Grierson, D. The Polonovski Reaction. In Organic Reactions; American Cancer Society: Atlanta, GA, USA, 2004; pp. 85–295. ISBN 9780471264187. [Google Scholar]
- Zhan, G.; Zhou, J.; Liu, R.; Liu, T.; Guo, G.; Wang, J.; Xiang, M.; Xue, Y.; Luo, Z.; Zhang, Y.; et al. Galanthamine, Plicamine, and Secoplicamine Alkaloids from Zephyranthes candida and Their Anti-acetylcholinesterase and Anti-inflammatory Activities. J. Nat. Prod. 2016, 79, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Katoch, D.; Kumar, D.; Padwad, Y.S.; Singh, B.; Sharma, U. Pseudolycorine N-oxide, a new N-oxide from Narcissus tazetta. Nat. Prod. Res. 2020, 34, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, P.; Sun, N.; Lu, Y.-J.; Wong, W.-L.; Fang, Z.; Zhang, K. The Diversity of Heterocyclic N-oxides Molecules: Highlights on their Potential in Organic Synthesis, Catalysis and Drug Applications. Curr. Org. Chem. 2019, 23, 616–627. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Z.; Wang, Y. Construction of N-Heterocycles through Cyclization of Tertiary Amines. Chem. A Eur. J. 2019, 25, 2423–2441. [Google Scholar] [CrossRef]
- Yanai, K.; Togo, H. Novel preparation of N-arylmethyl-N-arylmethyleneamine N-oxides from benzylic bromides with zinc and isobutyl nitrite. Tetrahedron 2019, 75, 3523–3529. [Google Scholar] [CrossRef]
- Liu, K.-J.; Deng, J.-H.; Yang, J.; Gong, S.-F.; Lin, Y.-W.; He, J.-Y.; Cao, Z.; He, W.-M. Selective oxidation of (hetero)sulfides with molecular oxygen under clean conditions. Green Chem. 2020, 22, 433–438. [Google Scholar] [CrossRef]
- Pibiri, I.; Buscemi, S.; Palumbo Piccionello, A.; Pace, A. Photochemically Produced Singlet Oxygen: Applications and Perspectives. ChemPhotoChem 2018, 2, 535–547. [Google Scholar] [CrossRef]
- Liu, K.-J.; Duan, Z.-H.; Zeng, X.-L.; Sun, M.; Tang, Z.; Jiang, S.; Cao, Z.; He, W.-M. Clean Oxidation of (Hetero)benzylic Csp3–H Bonds with Molecular Oxygen. ACS Sustain. Chem. Eng. 2019, 7, 10293–10298. [Google Scholar] [CrossRef]
- Mate, D.M.; Alcalde, M. Laccase: A multi-purpose biocatalyst at the forefront of biotechnology. Microb. Biotechnol. 2017, 10, 1457–1467. [Google Scholar] [CrossRef]
- Agrawal, K.; Chaturvedi, V.; Verma, P. Fungal laccase discovered but yet undiscovered. Bioresour. Bioprocess. 2018, 5, 4. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, G.; Ngo, H.H.; Guo, W.; Zhang, S. Advances in thermostable laccase and its current application in lignin-first biorefinery: A review. Bioresour. Technol. 2020, 298, 122511. [Google Scholar] [CrossRef]
- Perna, V.; Meyer, A.S.; Holck, J.; Eltis, L.D.; Eijsink, V.G.H.; Wittrup Agger, J. Laccase-Catalyzed Oxidation of Lignin Induces Production of H2O2. ACS Sustain. Chem. Eng. 2020, 8, 831–841. [Google Scholar] [CrossRef]
- Kudanga, T.; Nemadziva, B.; Le Roes-Hill, M. Laccase catalysis for the synthesis of bioactive compounds. Appl. Microbiol. Biotechnol. 2017, 101, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Janusz, G.; Pawlik, A.; Świderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkołazka, A.; Paszczyński, A. Laccase Properties, Physiological Functions, and Evolution. Int. J. Mol. Sci. 2020, 21, 966. [Google Scholar] [CrossRef] [PubMed]
- Polak, J.; Wlizło, K.; Pogni, R.; Petricci, E.; Grąz, M.; Szałapata, K.; Osińska-Jaroszuk, M.; Kapral-Piotrowska, J.; Pawlikowska-Pawlęga, B.; Jarosz-Wilkołazka, A. Structure and Bioactive Properties of Novel Textile Dyes Synthesised by Fungal Laccase. Int. J. Mol. Sci. 2020, 21, 2052. [Google Scholar] [CrossRef] [PubMed]
- Meschini, R.; D’Eliseo, D.; Filippi, S.; Bertini, L.; Bizzarri, B.M.; Botta, L.; Saladino, R.; Velotti, F. Tyrosinase-Treated Hydroxytyrosol-Enriched Olive Vegetation Waste with Increased Antioxidant Activity Promotes Autophagy and Inhibits the Inflammatory Response in Human THP-1 Monocytes. J. Agric. Food Chem. 2018, 66, 12274–12284. [Google Scholar] [CrossRef]
- Bizzarri, B.M.; Martini, A.; Serafini, F.; Aversa, D.; Piccinino, D.; Botta, L.; Berretta, N.; Guatteo, E.; Saladino, R. Tyrosinase mediated oxidative functionalization in the synthesis of DOPA-derived peptidomimetics with anti-Parkinson activity. RSC Adv. 2017, 7, 20502–20509. [Google Scholar] [CrossRef]
- Botta, G.; Bizzarri, B.M.; Garozzo, A.; Timpanaro, R.; Bisignano, B.; Amatore, D.; Palamara, A.T.; Nencioni, L.; Saladino, R. Carbon nanotubes supported tyrosinase in the synthesis of lipophilic hydroxytyrosol and dihydrocaffeoyl catechols with antiviral activity against DNA and RNA viruses. Bioorg. Med. Chem. 2015, 23, 5345–5351. [Google Scholar] [CrossRef]
- Tahmasbi, H.; Khoshayand, M.R.; Bozorgi-Koushalshahi, M.; Heidary, M.; Ghazi-Khansari, M.; Faramarzi, M.A. Biocatalytic conversion and detoxification of imipramine by the laccase-mediated system. Int. Biodeterior. Biodegrad. 2016, 108, 1–8. [Google Scholar] [CrossRef]
- Ferris, J.P.; Gerwe, R.D.; Gapski, G.R. Detoxication mechanisms. II. Iron-catalyzed dealkylation of trimethylamine oxide. J. Am. Chem. Soc. 1967, 89, 5270–5275. [Google Scholar] [CrossRef]
- Ferris, J.P.; Gerwe, R.D.; Gapski, G.R. Detoxication mechanisms. III. Scope and mechanism of the iron-catalyzed dealkylation of tertiary amine oxides. J. Org. Chem. 1968, 33, 3493–3498. [Google Scholar] [CrossRef]
- Aimi, N.; Yamanaka, E.; Endo, J.; Sakai, S.; Haginiwa, J. Transformation of indole alkaloids—I: Conversion of oxindole alkaloids into indole alkaloids. Tetrahedron 1973, 29, 2015–2021. [Google Scholar] [CrossRef]
- Aimi, N.; Yamanaka, E.; Endo, J.; Sakai, S.; Haginiwa, J. Conversion of oxindole alkaloids into indole alkaloids. Tetrahedron Lett. 1972, 13, 1081–1084. [Google Scholar] [CrossRef]
- Galli, C.; Gentili, P. Chemical messengers: Mediated oxidations with the enzyme laccase. J. Phys. Org. Chem. 2004, 17, 973–977. [Google Scholar] [CrossRef]
- Baiocco, P.; Barreca, A.M.; Fabbrini, M.; Galli, C.; Gentili, P. Promoting laccase activity towards non-phenolic substrates: A mechanistic investigation with some laccase–mediator systems. Org. Biomol. Chem. 2003, 1, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Milstein, O.; Nicklas, B.; Hüttermann, A. Oxidation of aromatic compounds in organic solvents with laccase from Trametes versicolor. Appl. Microbiol. Biotechnol. 1989, 31, 70–74. [Google Scholar] [CrossRef]
- Wu, M.-H.; Lin, M.-C.; Lee, C.-C.; Yu, S.-M.; Wang, A.H.-J.; Ho, T.-H.D. Enhancement of laccase activity by pre-incubation with organic solvents. Sci. Rep. 2019, 9, 9754. [Google Scholar] [CrossRef]
- Gerchikov, A.Y.; Akhatova, G.R.; Sharipova, G.M.; Mustafin, A.G.; Sakhibgareeva, M.V.; Spivak, S.I. Investigation of the mechanism of the inhibited oxidation of 1,4-dioxane by mathematical modeling. Kinet. Catal. 2015, 56, 300–303. [Google Scholar] [CrossRef]
- Nasibullina, R.A.; Gimadieva, A.R.; Yakupova, L.R.; Safiullin, R.L. Free-radical chain oxidation of 1,4-dioxane inhibited by 2-thio-6-aminouracil. Kinet. Catal. 2016, 57, 154–158. [Google Scholar] [CrossRef]
- Weinstein, A.B.; Stahl, S.S. Palladium catalyzed aryl C–H amination with O2via in situ formation of peroxide-based oxidant(s) from dioxane. Catal. Sci. Technol. 2014, 4, 4301–4307. [Google Scholar] [CrossRef]
- Clément, J.L.; Ferré, N.; Siri, D.; Karoui, H.; Rockenbauer, A.; Tordo, P. Assignment of the EPR spectrum of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) superoxide spin adduct. J. Org. Chem. 2005, 70, 1198–1203. [Google Scholar] [CrossRef]
- Tori, K.; Yoshimura, Y.; Kainosho, M.; Ajisaka, K. Evidence for the presence of contact term contribution to lanthanide induced shifts in 1H and 13C NMR spectra of pyridine N-oxides. Tetrahedron Lett. 1973, 14, 1573–1576. [Google Scholar] [CrossRef]
- Shaw, M.L.; Palese, P. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1647–1689. [Google Scholar]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza Neuraminidase Inhibitors Possessing a Novel Hydrophobic Interaction in the Enzyme Active Site: Design, Synthesis, and Structural Analysis of Carbocyclic Sialic Acid Analogues with Potent Anti-Influenza Activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef]
- Bizzarri, B.M.; Fanelli, A.; Piccinino, D.; De Angelis, M.; Dolfa, C.; Palamara, A.T.; Nencioni, L.; Zippilli, C.; Crucianelli, M.; Saladino, R. Synthesis of Stilbene and Chalcone Inhibitors of Influenza A Virus by SBA-15 Supported Hoveyda-Grubbs Metathesis. Catalysts 2019, 9, 983. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The Amphibian Antimicrobial Peptide Temporin B Inhibits In Vitro Herpes Simplex Virus 1 Infection. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Madej, A.; Koszelewski, D.; Paprocki, D.; Brodzka, A.; Ostaszewski, R. The amine as carbonyl precursor in the chemoenzymatic synthesis of Passerini adducts in aqueous medium. Catal. Commun. 2020, 145, 106118. [Google Scholar] [CrossRef]
- Correia Cordeiro, R.S.; Ríos-Lombardía, N.; Morís, F.; Kourist, R.; González-Sabín, J. One-Pot Transformation of Ketoximes into Optically Active Alcohols and Amines by Sequential Action of Laccases and Ketoreductases or ω-Transaminases. ChemCatChem 2019, 11, 1272–1277. [Google Scholar] [CrossRef]
- de Aguiar, V.M.; Esquinelato Silva, R.; Leão, R.A.C.; de Souza, R.O.M.A.; Gonçalves, R.S.B.; de Mariz e Miranda, L.S. Studies on the laccases catalyzed oxidation of norbelladine like acetamides. Mol. Catal. 2020, 485, 110788. [Google Scholar] [CrossRef]
- Feng, Y.; Shen, M.; Wang, Z.; Liu, G. Transformation of atenolol by a laccase-mediator system: Efficiencies, effect of water constituents, and transformation pathways. Ecotoxicol. Environ. Saf. 2019, 183, 109555. [Google Scholar] [CrossRef]
- Wellington, K.W.; Govindjee, V.P.; Steenkamp, P. A laccase-catalysed synthesis of triaminated cyclohexa-2,4-dienones from catechol. J. Catal. 2018, 368, 306–314. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Neri, V.; Farina, A.; Crestini, C.; Nencioni, L.; Palamara, A.T. A Novel and Efficient Synthesis of Tocopheryl Quinones by Homogeneous and Heterogeneous Methyltrioxorhenium/Hydrogen Peroxide Catalytic Systems. Adv. Synth. Catal. 2008, 350, 321–331. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | pH | Solvent | Solvent/Buffer Ratio | Conversion (%) | Product(s) | Yield 5 (%) |
|---|---|---|---|---|---|---|
| 1 | 4.5 | 1,4-dioxane | 1:4 | 81 | 8 | 76 2 |
| 2 | 4.5 | 1,4-dioxane | 1:4 | 45 | 8(9) | 34(7) 3 |
| 3 | 5.6 | 1,4-dioxane | 1:4 | 50 | 8(9) | 19(28) 3 |
| 4 | 5.6 | 1,4-dioxane | 10:1 | 60 | 8(9){10} | 5(47){5} 3 |
| 5 | 5.6 | 1,4-dioxane | 10:1 | 71 | 8(9){10} | 2(56){8} 3, 4 |
| 6 | 5.6 | THF | 10:1 | 16 | (9) | (2) 3 |
| 7 | 5.6 | CH3CN | 10:1 | 5 | 8 | 2 3 |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Compd | X | R1 | R2 | R3 | R4 | Product | Yield [%] 2 | 3 |
| 1 | 7b | -(CH2)2- | -OH | -OCH3 | -Br | -OH | 9 | 56 | +12.8 |
| 2 | 11a | -(CH2)2- | -OH | -OCH3 | -H | -OH | 12a | 55 | +17.4 |
| 3 | 11b | -(CH2)2- | -OH | -H | -H | -OH | 12b | 53 | +11.5 |
| 4 | 11c | -(CH2)2- | -H | -OH | -H | -OH | 12c | 52 | +21.5 |
| 5 | 11d | -(CH2)2- | -OH | -NO2 | -H | -OH | 12d | 70 | +6.4 |
| 6 | 11e | -(CH2)2- | -H | -H | -H | -OH | 12e | 71 | +18.6 |
| 7 | 11f | -CH3 | -OH | -H | -H | none | 12f | 67 | - |
| 8 | 11g | -(CH2)- | -OH | -H | -H | -OH | 12g | 55 | +8.9 |
| 9 | 11h | -(CH2)3- | -OH | -H | -H | -H | 12h | 78 | −7.1 |
| Entry | Compd | A549 | MDCK | ||||
|---|---|---|---|---|---|---|---|
| IC50 | CC50 | SI | IC50 | CC50 | SI | ||
| 1 | 9 | 376.88 ± 11.4 | 210.65 ± 8.1 | 0.56 | 86.84 ± 5.1 | 205.90 ± 7.9 | 2.37 |
| 2 | 12a | 78.76 ± 4.3 | 200 ± 7.1 | 2.5 | 137.23 ± 5.9 | 137.48 ± 5.8 | 1.00 |
| 3 | 12b | 83.23 ± 5.1 | 149.31 ± 6.2 | 1.79 | 138.28 ± 6.1 | 129.18 ± 6.0 | 0.93 |
| 4 | 12c | 80.20 ± 4.9 | 83.79 ± 5.0 | 1.04 | 84.73 ± 4.8 | 113.86 ± 5.2 | 1.34 |
| 5 | 12d | 1243.97 ± 30.2 | 112.69 ± 6.1 | 0.09 | 70.15 ± 4.0 | 100.51 ± 4.9 | 1.43 |
| 6 | 12e | ND 2 | ND 2 | ND 2 | 133.32 ± 5.8 | 108.61 ± 5.1 | 0.81 |
| 7 | 12f | ND 2 | ND 2 | ND 2 | ND 2 | ND 2 | ND 2 |
| 8 | 12g | ND 2 | ND 2 | ND 2 | ND 2 | ND 2 | ND 2 |
| 9 | 12h | 70.40 ± 3.1 | 287.28 ± 8.3 | 4.0 | 73.20 ± 3.3 | 220.74 ± 8.9 | 3.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zippilli, C.; Botta, L.; Bizzarri, B.M.; Nencioni, L.; De Angelis, M.; Protto, V.; Giorgi, G.; Baratto, M.C.; Pogni, R.; Saladino, R. Laccase-Catalyzed 1,4-Dioxane-Mediated Synthesis of Belladine N-Oxides with Anti-Influenza A Virus Activity. Int. J. Mol. Sci. 2021, 22, 1337. https://doi.org/10.3390/ijms22031337
Zippilli C, Botta L, Bizzarri BM, Nencioni L, De Angelis M, Protto V, Giorgi G, Baratto MC, Pogni R, Saladino R. Laccase-Catalyzed 1,4-Dioxane-Mediated Synthesis of Belladine N-Oxides with Anti-Influenza A Virus Activity. International Journal of Molecular Sciences. 2021; 22(3):1337. https://doi.org/10.3390/ijms22031337
Chicago/Turabian StyleZippilli, Claudio, Lorenzo Botta, Bruno Mattia Bizzarri, Lucia Nencioni, Marta De Angelis, Virginia Protto, Gianluca Giorgi, Maria Camilla Baratto, Rebecca Pogni, and Raffaele Saladino. 2021. "Laccase-Catalyzed 1,4-Dioxane-Mediated Synthesis of Belladine N-Oxides with Anti-Influenza A Virus Activity" International Journal of Molecular Sciences 22, no. 3: 1337. https://doi.org/10.3390/ijms22031337
APA StyleZippilli, C., Botta, L., Bizzarri, B. M., Nencioni, L., De Angelis, M., Protto, V., Giorgi, G., Baratto, M. C., Pogni, R., & Saladino, R. (2021). Laccase-Catalyzed 1,4-Dioxane-Mediated Synthesis of Belladine N-Oxides with Anti-Influenza A Virus Activity. International Journal of Molecular Sciences, 22(3), 1337. https://doi.org/10.3390/ijms22031337