
Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML


 ,
,  , , ,
, , , 
Abstract
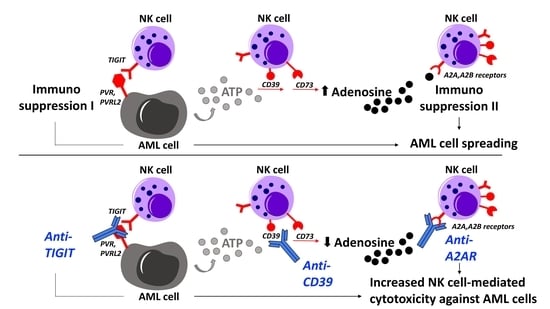
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Bone Marrow-Derived NK Cells of AML Patients Show a Shift towards CD56brightCD16− and CD56dimCD16− Cells and Are Associated with a Reduced CD56dimCD16+ Population
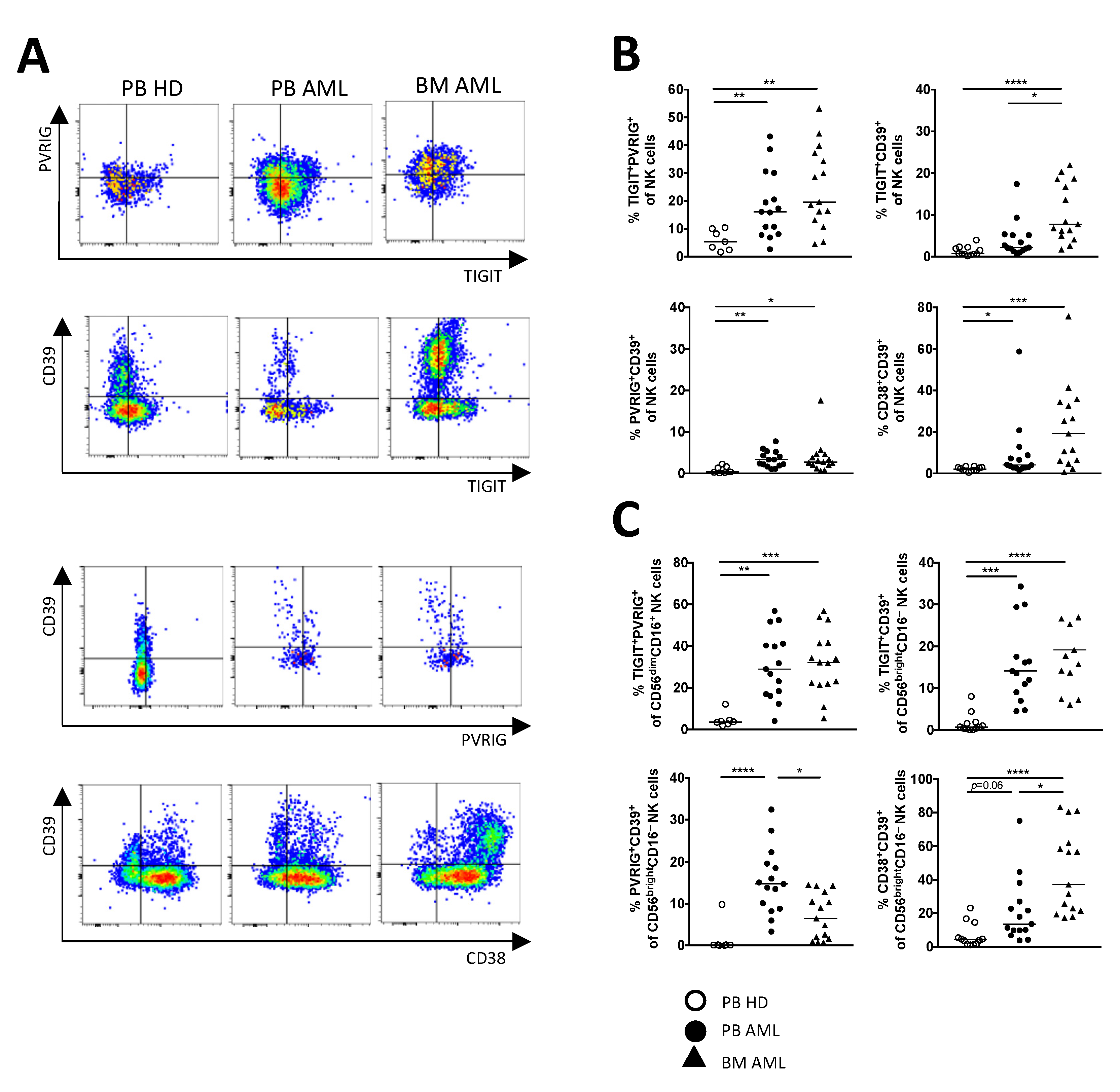
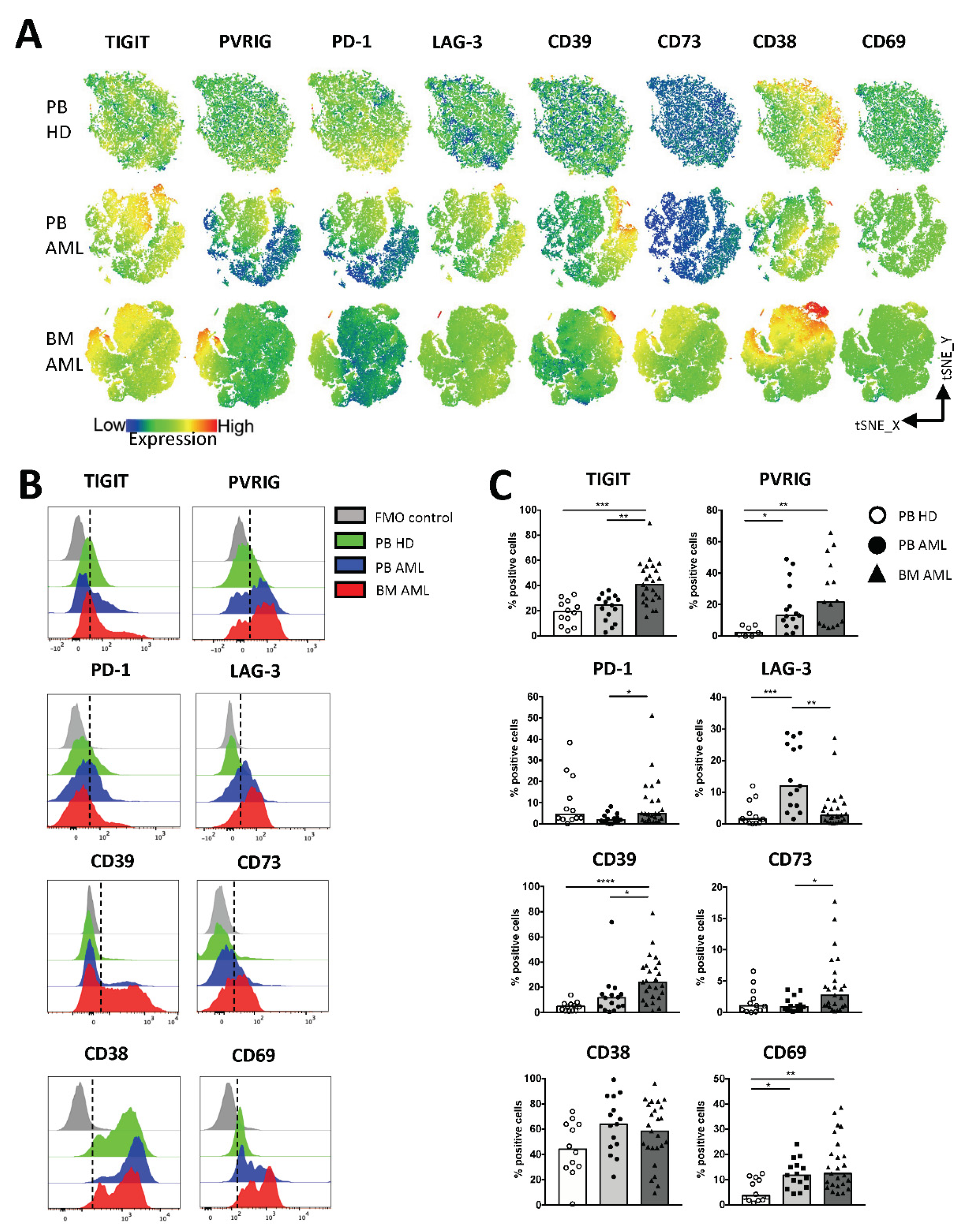
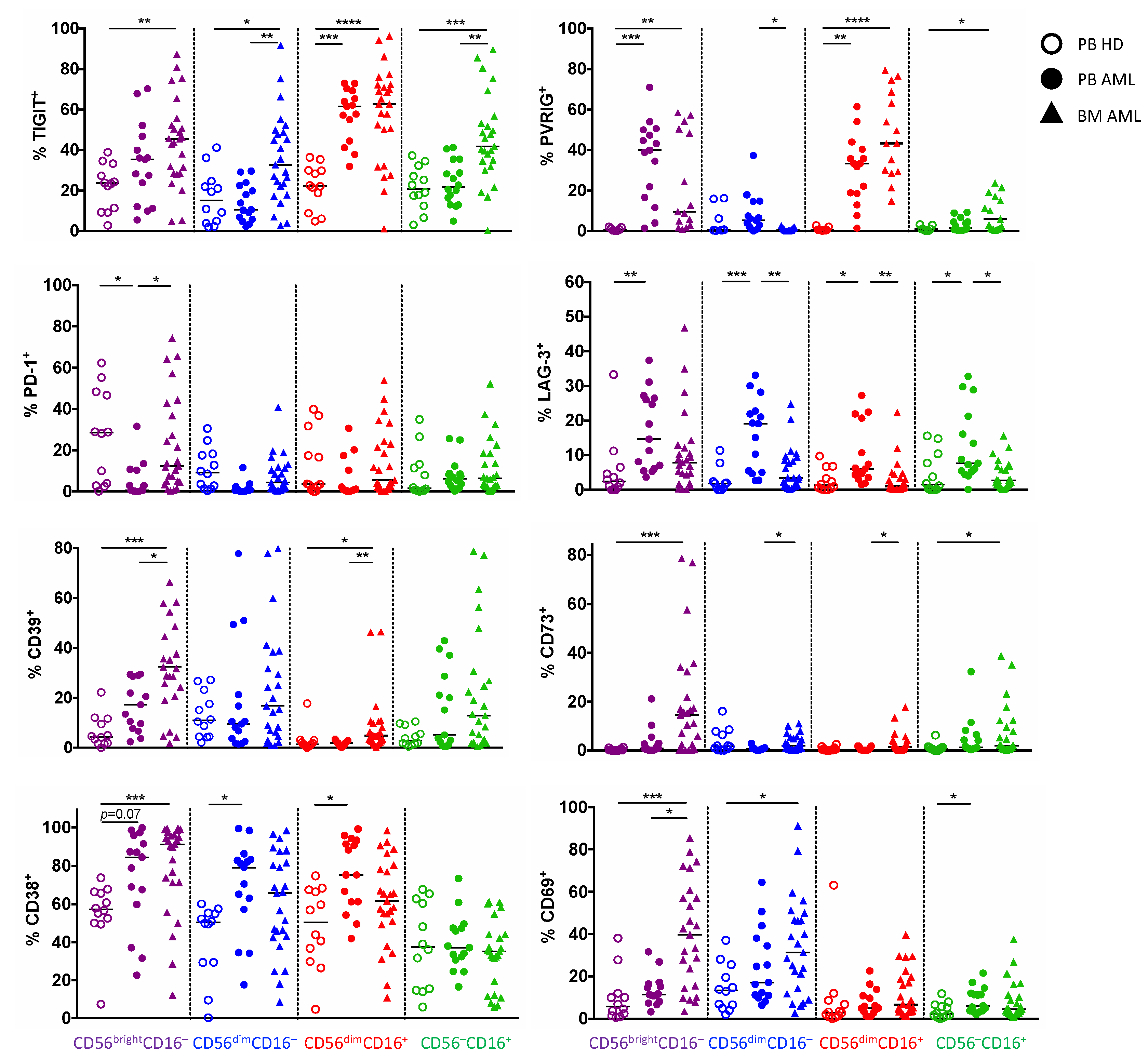
2.2. NK Cells of Patients with AML Express TIGIT, PVRIG, CD39, and CD69
2.3. TIGIT, PVRIG, CD39, and CD38 Expression Is Related to the CD56brightCD16− and the CD56dimCD16+ NK Cell Population in AML
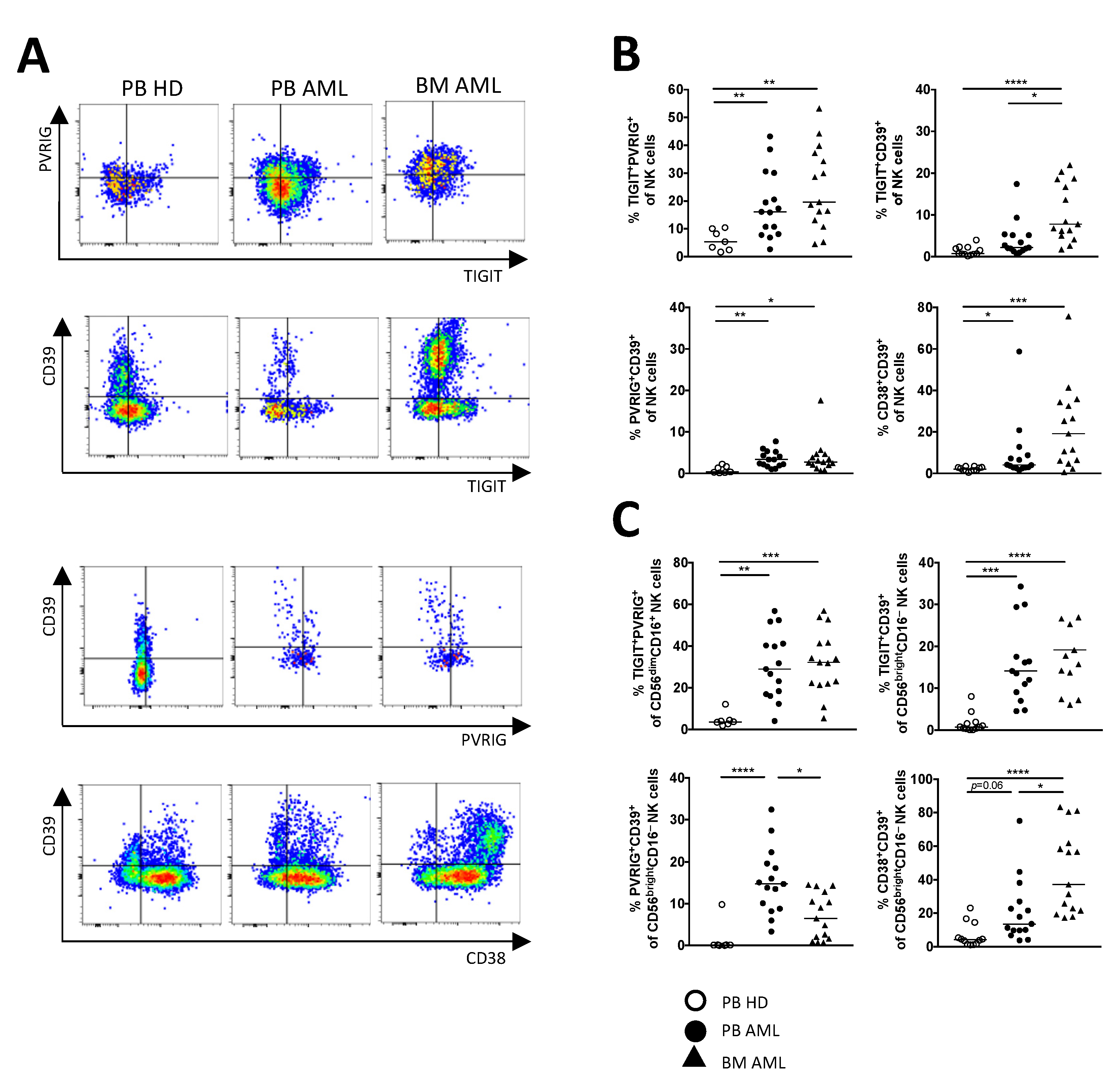
2.4. PVRIG and CD39 Are Co-Expressed with TIGIT on CD56dimCD16+ and CD56brightCD16− NK Cells in AML
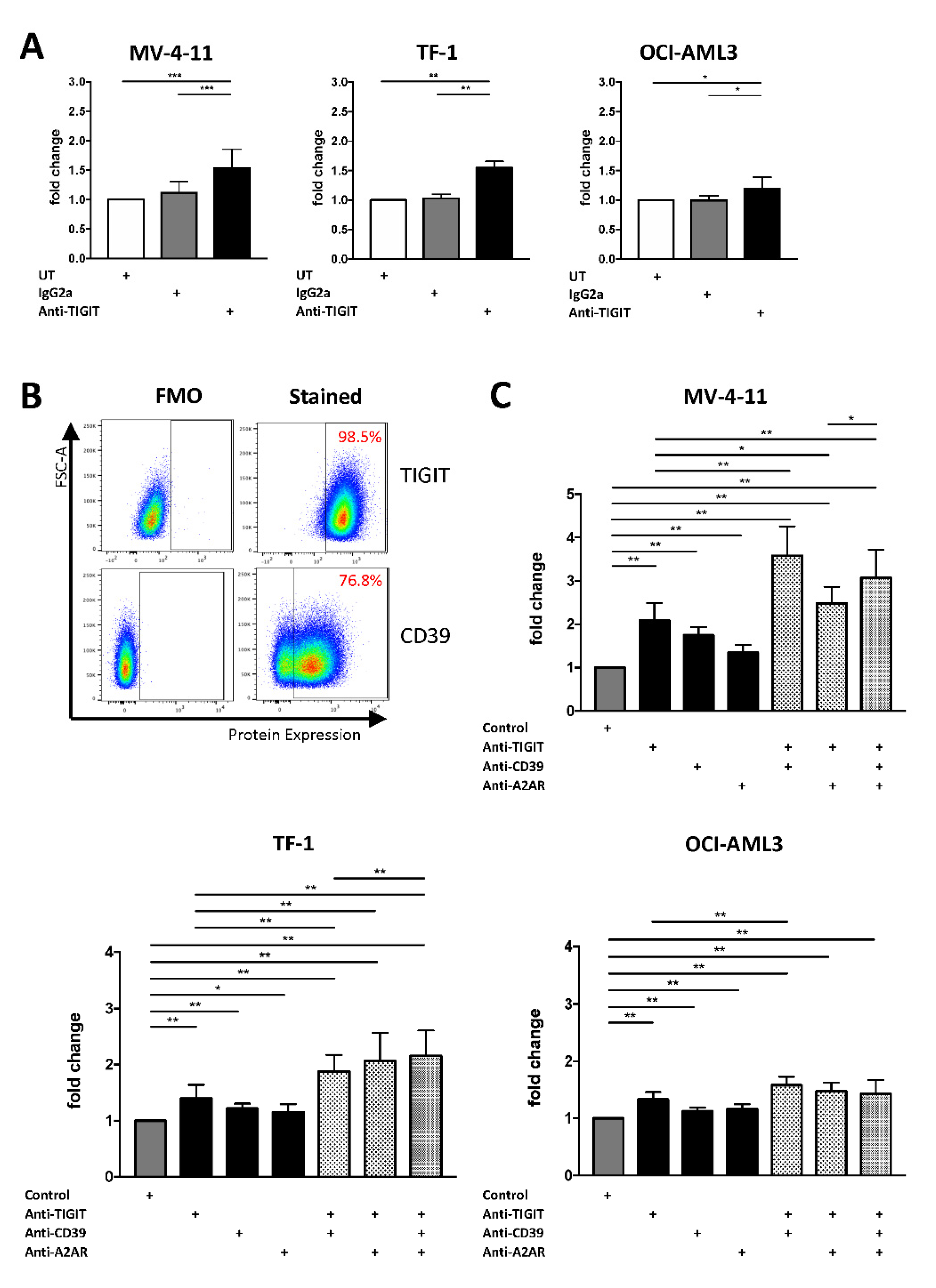
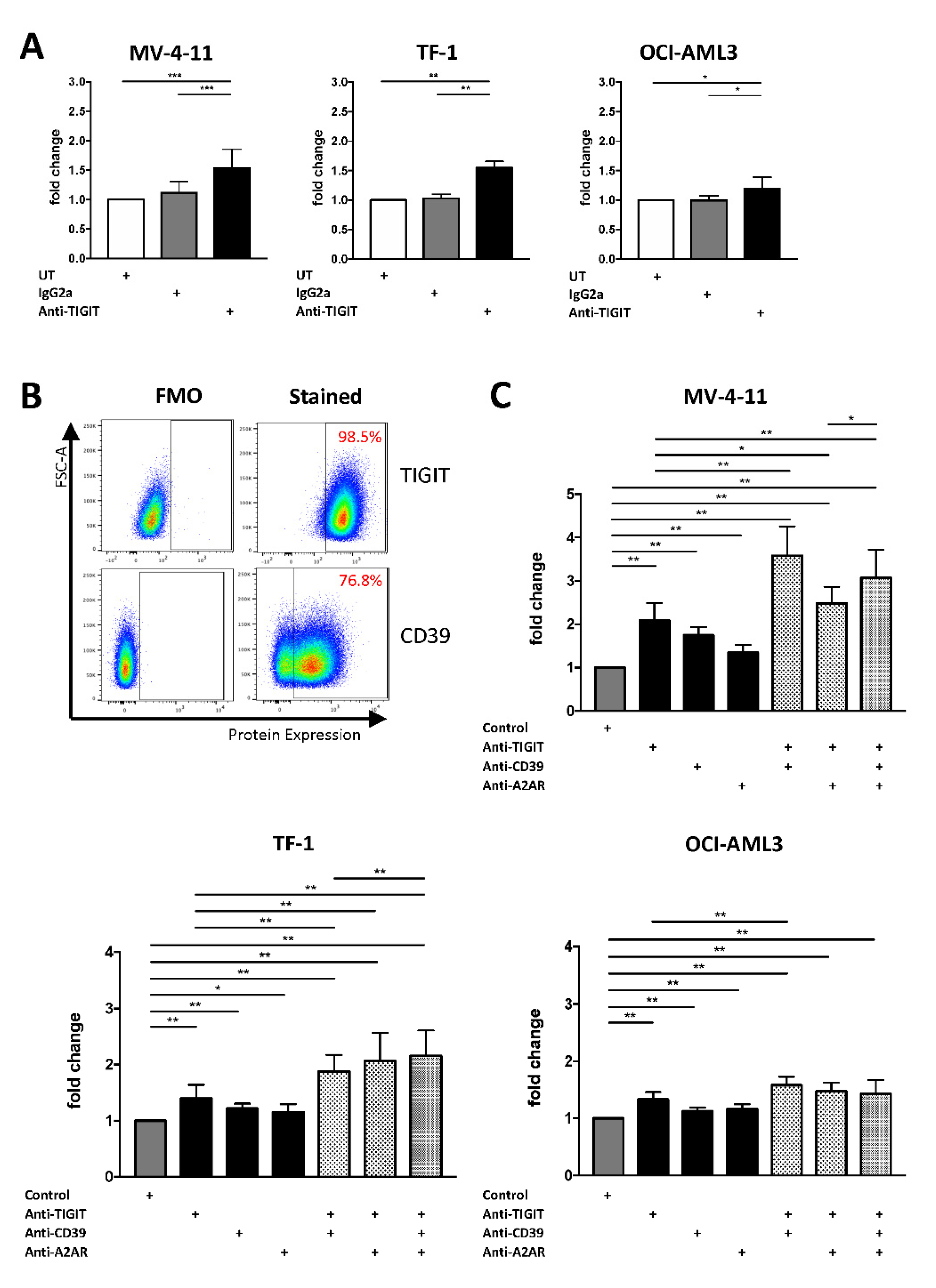
2.5. Single or Combined Checkpoint Blockade Increases NK-92 Cell-Mediated Cytotoxicity In Vitro
3. Discussion
4. Materials and Methods
4.1. Clinical Cohorts
4.2. Multiparameter Flow Cytometry and Surface Staining
4.3. NK Cell-Mediated Cytotoxicity Assay
4.4. Cell Lines
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal Transduct. Target. Ther. 2020, 5, 250. [Google Scholar] [CrossRef]
- Rosenberg, J.; Huang, J. CD8+ T cells and NK cells: Parallel and complementary soldiers of immunotherapy. Curr. Opin. Chem. Eng. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Kwon, H.-J.; Kim, N.; Kim, H.S. Molecular checkpoints controlling natural killer cell activation and their modulation for cancer immunotherapy. Exp. Mol. Med. 2017, 49, 1–11. [Google Scholar] [CrossRef]
- Mandal, A.; Viswanathan, C. Natural killer cells: In health and disease. Hematol. Stem Cell Ther. 2015, 8, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Amand, M.; Iserentant, G.; Poli, A.; Sleiman, M.; Fievez, V.; Sanchez, I.P.; Sauvageot, N.; Michel, T.; Aouali, N.; Janji, B.; et al. Human CD56dimCD16dim Cells as an Individualized Natural Killer Cell Subset. Front. Immunol. 2017, 8, 699. [Google Scholar] [CrossRef]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of Distinct Natural Killer Cell Subsets in Anticancer Response. Front. Immunol. 2017, 8, 293. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2017.00293 (accessed on 21 October 2021). [CrossRef] [Green Version]
- Mavilio, D.; Lombardo, G.; Benjamin, J.; Kim, D.; Follman, D.; Marcenaro, E.; O’Shea, A.M.; Kinter, A.; Kovacs, C.; Moretta, A. Characterization of CD56-/CD16+ natural killer (NK) cells: A highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc. Natl. Acad. Sci. USA 2005, 102, 2886–2891. [Google Scholar] [CrossRef] [Green Version]
- Milush, J.M.; López-Vergès, S.A.; York, V.; Deeks, S.G.; Martin, J.N.; Hecht, F.M.; Lanier, L.L.; Nixon, D.F. CD56negCD16+NK cells are activated mature NK cells with impaired effector function during HIV-1 infection. Retrovirology 2013, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Chretien, A.-S.; Devillier, R.; Granjeaud, S.; Cordier, C.; Demerle, C.; Salem, N.; Wlosik, J.; Orlanducci, F.; Gorvel, L.; Fattori, S.; et al. High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proc. Natl. Acad. Sci. USA 2021, 118, e2020459118. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Jaeger, B.; Vivier, E. Natural Killer Cell Tolerance: Control by self or self-control? Cold Spring Harb. Perspect. Biol. 2012, 4, a007229. [Google Scholar]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 49. Available online: https://www.frontiersin.org/article/10.3389/fcimb.2020.00049 (accessed on 15 November 2021). [CrossRef] [Green Version]
- Meng, F.; Li, L.; Lu, F.; Yue, J.; Liu, Z.; Zhang, W.; Fu, R. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front. Oncol. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Q.; Yang, J.; Li, X.; Xian, L.; Li, W.; Lin, T.; Cheng, J.; Lin, Q.; Xu, X.; et al. Increased TIGIT expressing NK cells with dysfunctional phenotype in AML patients correlated with poor prognosis. Cancer Immunol. Immunother. 2021, 21, 2978. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2011, 90, 109–115. [Google Scholar] [CrossRef]
- Stamm, H.; Klingler, F.; Grossjohann, E.-M.; Muschhammer, J.; Vettorazzi, E.; Heuser, M.; Mock, U.; Thol, F.; Vohwinkel, G.; Latuske, E.; et al. Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene 2018, 37, 5269–5280. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29855615 (accessed on 15 November 2021). [CrossRef] [Green Version]
- Quatrini, L.; Mariotti, F.R.; Munari, E.; Tumino, N.; Vacca, P.; Moretta, L. The immune checkpoint PD-1 in natural killer cells: Expression, function and targeting in tumour immunotherapy. Cancers 2020, 12, 3285. [Google Scholar] [CrossRef]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. PD-1-positive Natural Killer Cells have a weaker antitumor function than that of PD-1-negative Natural Killer Cells in Lung Cancer. Int. J. Med. Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Ahl, P.J.; Bijin, V.A.; Kaliaperumal, N.; Lim, S.G.; Wang, C.I.; Fairhurst, A.M.; Connolly, J.E. LAG3 is a central regulator of NK cell cytokine production. bioRxiv 2020. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef]
- Ohta, A. A metabolic immune checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2019.01187 (accessed on 21 October 2021). [CrossRef] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Lanman, N.M.A.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Du, Y.; Wei, Y. Therapeutic Potential of Natural Killer Cells in Gastric Cancer. Front. Immunol. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, Y.; Lian, J.; Yang, H.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF-α-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J. Transl. Med. 2019, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Charap, A.J.; Enokida, T.; Brody, R.; Sfakianos, J.; Miles, B.; Bhardwaj, N.; Horowitz, A. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e001523. Available online: http://jitc.bmj.com/content/8/2/e001523.abstract (accessed on 21 October 2021). [CrossRef]
- Lee, H.; Da Silva, I.; Palendira, U.; Scolyer, R.; Long, G.; Wilmott, J. Targeting NK Cells to Enhance Melanoma Response to Immunotherapies. Cancers 2021, 13, 1363. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2019.01205 (accessed on 21 October 2021). [CrossRef] [PubMed]
- Carlsten, M.; Järås, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy—More Than a Pipe Dream. Front. Immunol. 2021, 11, 3466. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2020.618427 (accessed on 21 October 2021). [CrossRef] [PubMed]
- Xu, J.; Niu, T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 167. [Google Scholar] [CrossRef] [PubMed]
- Kaweme, N.M.; Zhou, F. Optimizing NK Cell-Based Immunotherapy in Myeloid Leukemia: Abrogating an Immunosuppressive Microenvironment. Front. Immunol. 2021, 12, 2348. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2021.683381 (accessed on 21 October 2021). [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- JJudge, S.J.; Dunai, C.; Aguilar, E.G.; Vick, S.C.; Sturgill, I.R.; Khuat, L.T.; Stoffel, K.M.; Van Dyke, J.; Longo, D.L.; Darrow, M.A.; et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 2020, 130, 3051–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J. Clin. Investig. 2020, 130, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Dogra, P.; Rancan, C.; Ma, W.; Toth, M.; Senda, T.; Carpenter, D.J.; Kubota, M.; Matsumoto, R.; Thapa, P.; Szabo, P.A.; et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell 2020, 180, 749–763.e13. [Google Scholar] [CrossRef] [Green Version]
- Molgora, M.; Cortez, V.; Colonna, M. Killing the Invaders: NK Cell Impact in Tumors and Anti-Tumor Therapy. Cancers 2021, 13, 595. [Google Scholar] [CrossRef]
- Salomé, B.; Gomez-Cadena, A.; Loyon, R.; Suffiotti, M.; Salvestrini, V.; Wyss, T.; Vanoni, G.; Ruan, D.F.; Rossi, M.; Tozzo, A.; et al. CD56 as a marker of an ILC1-like population with NK cell properties that is functionally impaired in AML. Blood Adv. 2019, 3, 3674–3687. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Bañas, H.; Casas-Avilés, I.; Durán, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: Novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Sanchez-Correa, B.; Guerrero, B.; Bergua, J.M.; Arcos, M.J.; Bañas, H.; Casas-Avilés, I.; et al. Characterization of the DNAM-1, TIGIT and TACTILE Axis on Circulating NK, NKT-Like and T Cell Subsets in Patients with Acute Myeloid Leukemia. Cancers 2020, 12, 2171. [Google Scholar] [CrossRef]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer–mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef]
- Maas, R.J.; Evert, J.S.H.-V.; Van der Meer, J.M.; Mekers, V.; Rezaeifard, S.; Korman, A.J.; de Jonge, P.K.; Cany, J.; Woestenenk, R.; Schaap, N.P.; et al. TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM-1/TIGIT/CD96 checkpoint molecules in ovarian cancer. Oncoimmunology 2020, 9, 1843247. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Cao, G.; Zheng, X.; Sun, C.; Wei, H.; Tian, Z.; Xiao, W.; Sun, R.; Sun, H. Blockade of checkpoint receptor PVRIG unleashes anti-tumor immunity of NK cells in murine and human solid tumors. J. Hematol. Oncol. 2021, 14, 100. [Google Scholar] [CrossRef]
- Li, J.; Whelan, S.; Kotturi, M.F.; Meyran, D.; D’Souza, C.; Hansen, K.; Liang, S.; Hunter, J.; Trapani, J.A.; Neeson, P.J. PVRIG is a novel NK cell immune checkpoint receptor in acute myeloid leukemia. Haematologica 2020, 25, 8547. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef]
- Dierks, P.; Wroblewski, R.; Eberhard, J.M.; Martrus, G.; Degen, O.; Hertling, S.; Schmiedel, S.; Lunemann, S.; Hüfner, A.; Lohse, A.W.; et al. Brief Report: Increased Frequency of CD39+ CD56bright Natural Killer Cells in HIV-1 Infection Correlates With Immune Activation and Disease Progression. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 74, 467–472. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.; Tang, B.; Zhao, Q.; Wang, D.; Liu, Y.; Guo, M.; Zhao, S.; Qi, Y.; Zhang, Y.; et al. IL-6-induced CD39 expression on tumor-infiltrating NK cells predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Immunol. Immunother. 2020, 69, 2371–2380. [Google Scholar] [CrossRef]
- Zhang, H.; Vijayan, D.; Li, X.-Y.; Robson, S.C.; Geetha, N.; Teng, M.W.L.; Smyth, M.J. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology 2019, 8, e1593809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, X.-Y.; Aguilera, A.R.; Xiao, C.; Jacoberger-Foissac, C.; Nowlan, B.; Robson, S.C.; Beers, C.; Moesta, A.K.; Geetha, N.; et al. Control of Metastases via Myeloid CD39 and NK Cell Effector Function. Cancer Immunol. Res. 2020, 8, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Sconocchia, G.; Titus, J.A.; Mazzoni, A.; Visintin, A.; Pericle, F.; Hicks, S.W.; Malavasi, F.; Segal, D.M. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood 1999, 94, 3864–3871. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56 bright CD16 − NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4 + T Cell Proliferation. J Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo–Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [Green Version]
- Mahaweni, N.M.; Bos, G.M.J.; Mitsiades, C.S.; Tilanus, M.G.J.; Wieten, L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol. Immunother. 2018, 67, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.; Ye, M.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.; Sachsenmeier, K.; Clarke, J.D.; et al. Small molecule AZD4635 inhibitor of A2AR signaling rescues immune cell function including CD103+ dendritic cells enhancing anti-tumor immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30500073 (accessed on 15 November 2021). [CrossRef] [Green Version]
- Dixon, K.O.; Schorer, M.; Nevin, J.; Etminan, Y.; Amoozgar, Z.; Kondo, T.; Kurtulus, S.; Kassam, N.; Sobel, R.A.; Fukumura, D.; et al. Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J. Immunol. 2018, 200, 3000–3007. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- KKoch-Nolte, F.; Reyelt, J.; Schößow, B.; Schwarz, N.; Scheuplein, F.; Rothenburg, S.; Haag, F.; Alzogaray, V.; Cauerhff, A.; Goldbaum, F.A. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2in vivo. FASEB J. 2007, 21, 3490–3498. [Google Scholar] [CrossRef]
- Eden, T.; Menzel, S.; Wesolowski, J.; Bergmann, P.; Nissen, M.; Dubberke, G.; Seyfried, F.; Albrecht, B.; Haag, F.; Koch-Nolte, F. A cDNA Immunization Strategy to Generate Nanobodies against Membrane Proteins in Native Conformation. Front. Immunol. 2018, 8, 1989. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2017.01989 (accessed on 21 October 2021). [CrossRef] [PubMed] [Green Version]
- Baum, N.; Fliegert, R.; Bauche, A.; Hambach, J.; Menzel, S.; Haag, F.; Bannas, P.; Koch-Nolte, F. Daratumumab and Nanobody-Based Heavy Chain Antibodies Inhibit the ADPR Cyclase but not the NAD+ Hydrolase Activity of CD38-Expressing Multiple Myeloma Cells. Cancers 2020, 13, 76. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brauneck, F.; Seubert, E.; Wellbrock, J.; Schulze zur Wiesch, J.; Duan, Y.; Magnus, T.; Bokemeyer, C.; Koch-Nolte, F.; Menzel, S.; Fiedler, W. Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML. Int. J. Mol. Sci. 2021, 22, 12919. https://doi.org/10.3390/ijms222312919
Brauneck F, Seubert E, Wellbrock J, Schulze zur Wiesch J, Duan Y, Magnus T, Bokemeyer C, Koch-Nolte F, Menzel S, Fiedler W. Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML. International Journal of Molecular Sciences. 2021; 22(23):12919. https://doi.org/10.3390/ijms222312919
Chicago/Turabian StyleBrauneck, Franziska, Elisa Seubert, Jasmin Wellbrock, Julian Schulze zur Wiesch, Yinghui Duan, Tim Magnus, Carsten Bokemeyer, Friedrich Koch-Nolte, Stephan Menzel, and Walter Fiedler. 2021. "Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML" International Journal of Molecular Sciences 22, no. 23: 12919. https://doi.org/10.3390/ijms222312919
APA StyleBrauneck, F., Seubert, E., Wellbrock, J., Schulze zur Wiesch, J., Duan, Y., Magnus, T., Bokemeyer, C., Koch-Nolte, F., Menzel, S., & Fiedler, W. (2021). Combined Blockade of TIGIT and CD39 or A2AR Enhances NK-92 Cell-Mediated Cytotoxicity in AML. International Journal of Molecular Sciences, 22(23), 12919. https://doi.org/10.3390/ijms222312919