
The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases

 , , , , ,
, , , , ,
Abstract
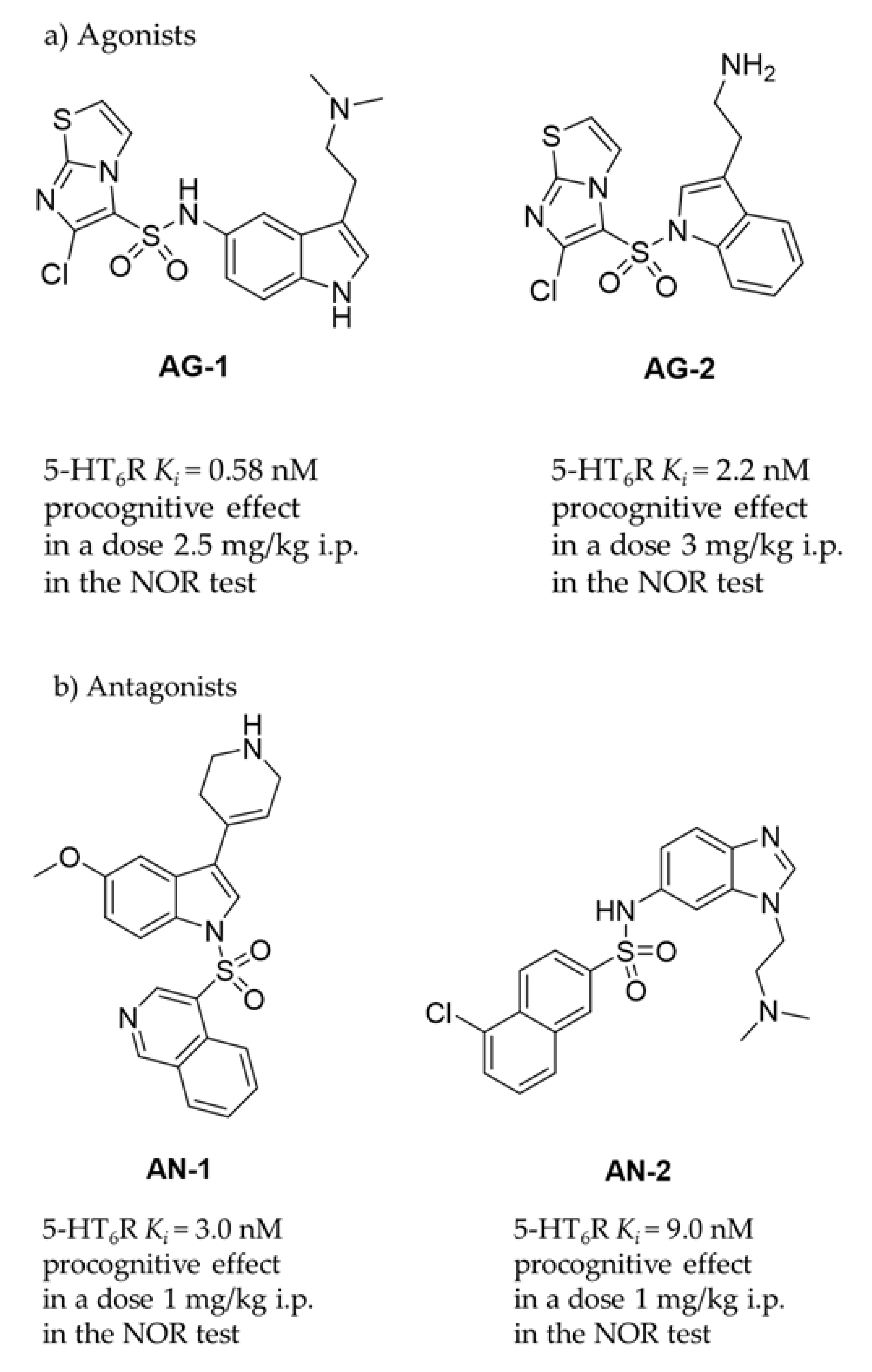
:1. Introduction
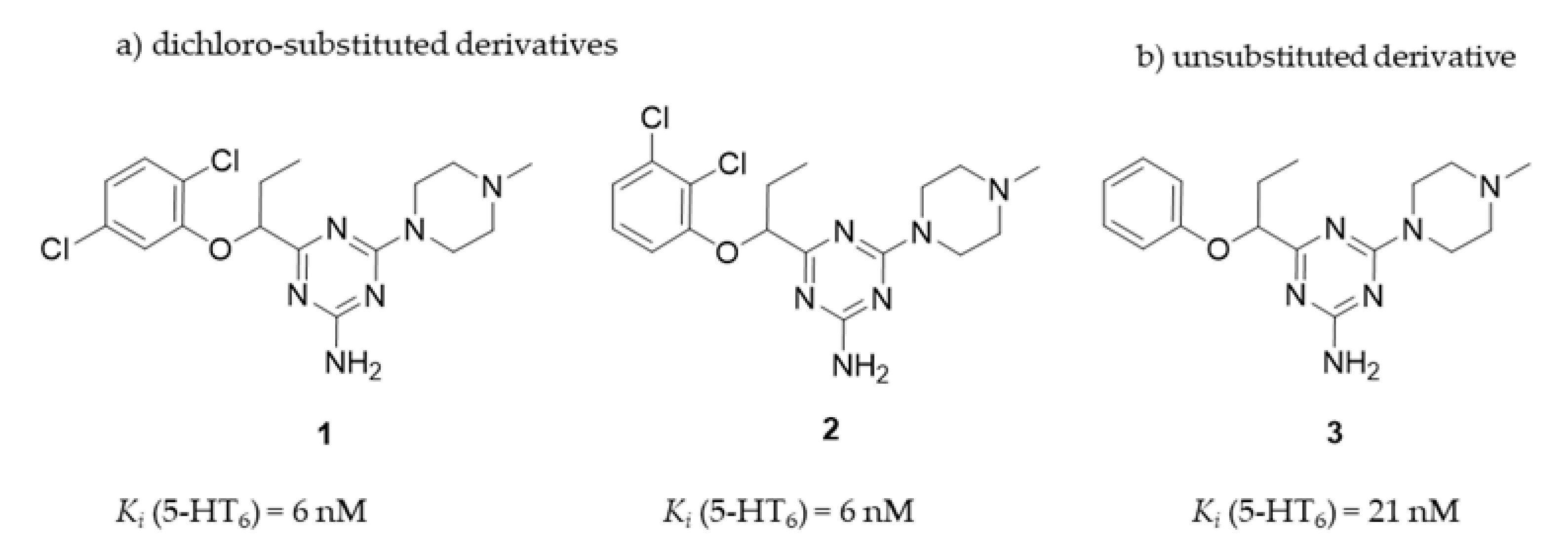
2. Results
2.1. Pharmacology In Vitro
2.1.1. Radioligand Binding Assay
2.1.2. Functional Assays
2.2. ADMET Assays In Vitro
2.2.1. Permeability
2.2.2. Caco-2 Permeability Assay
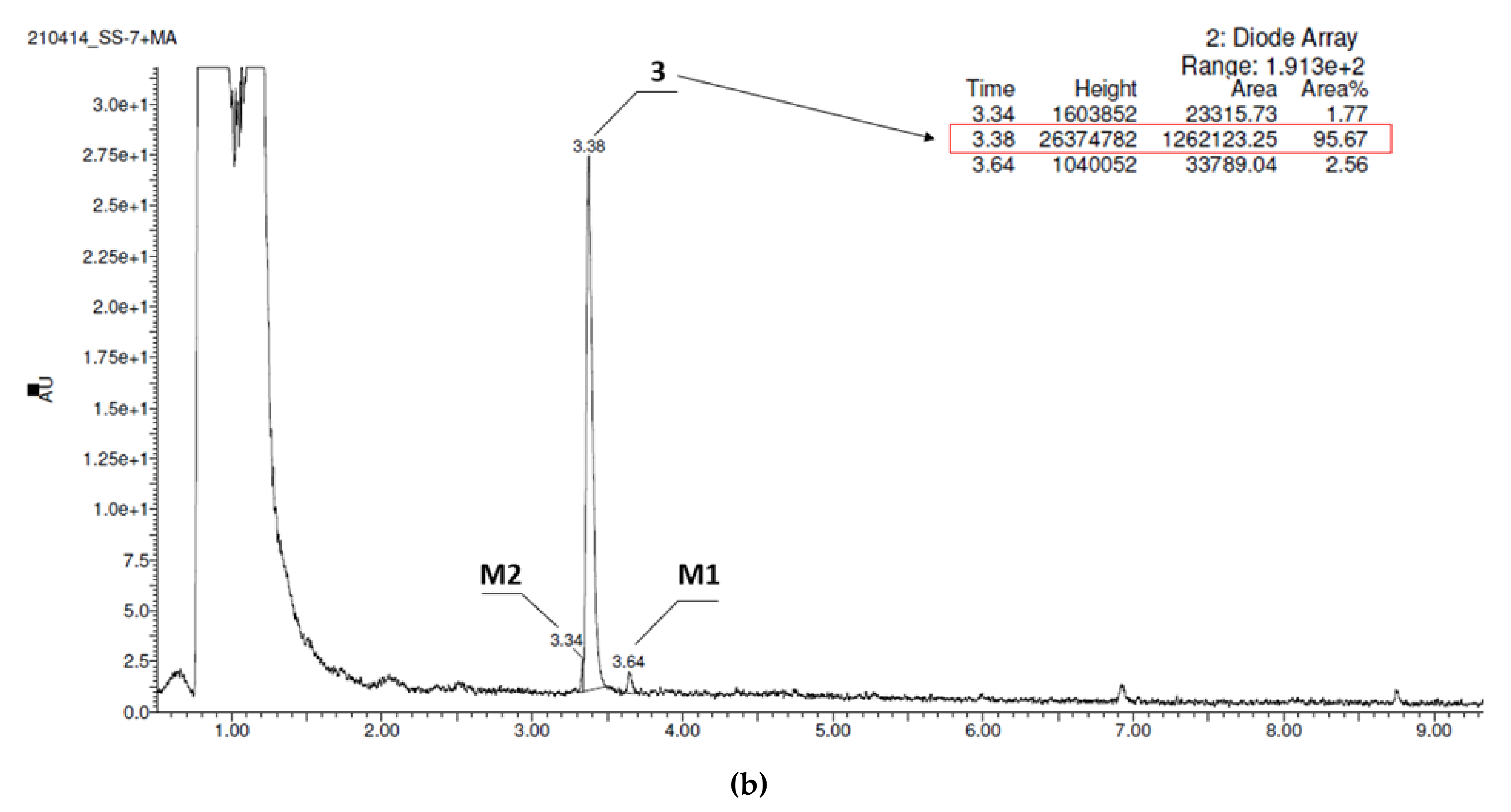
2.2.3. Metabolic Stability In Vitro
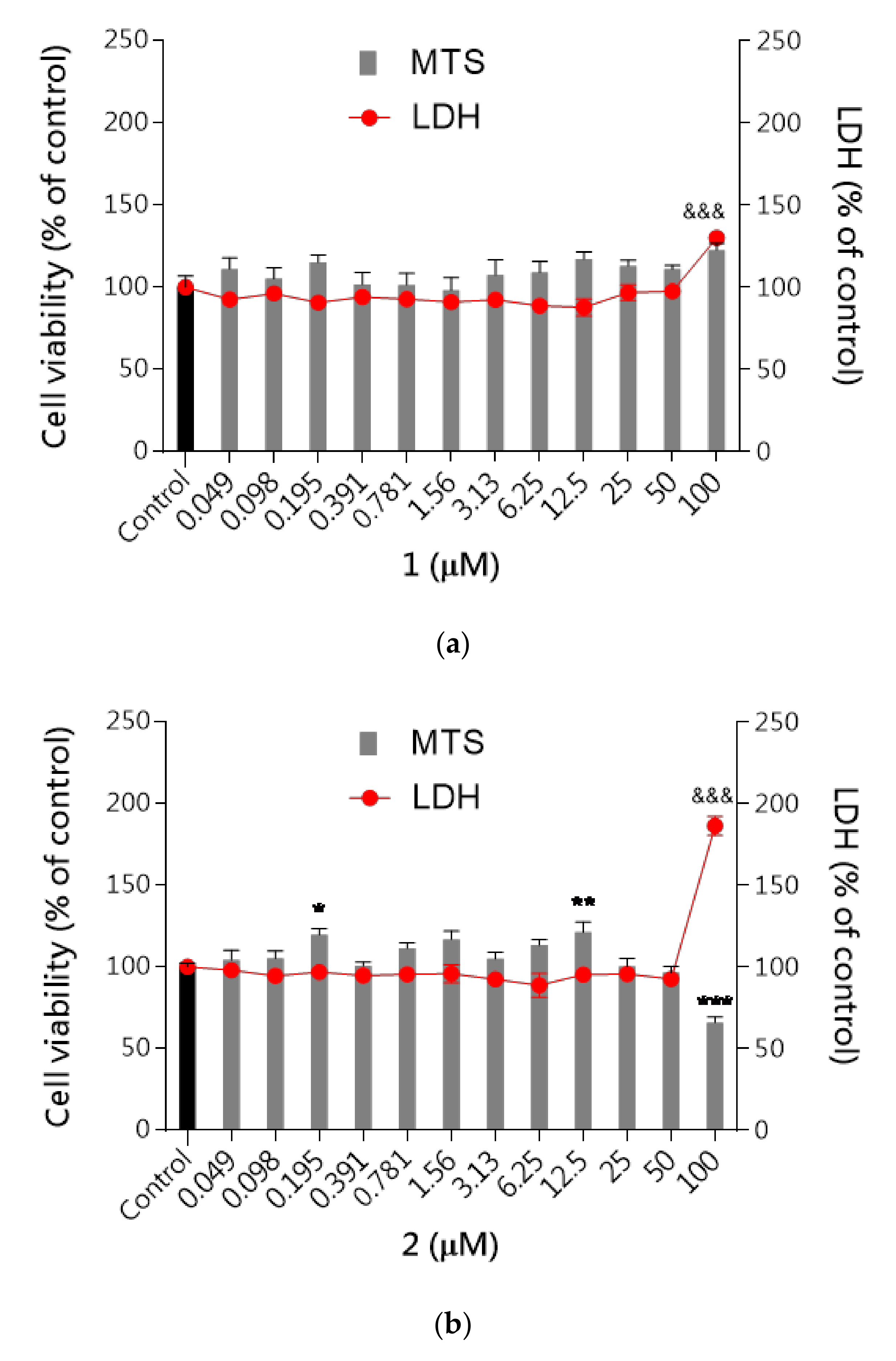
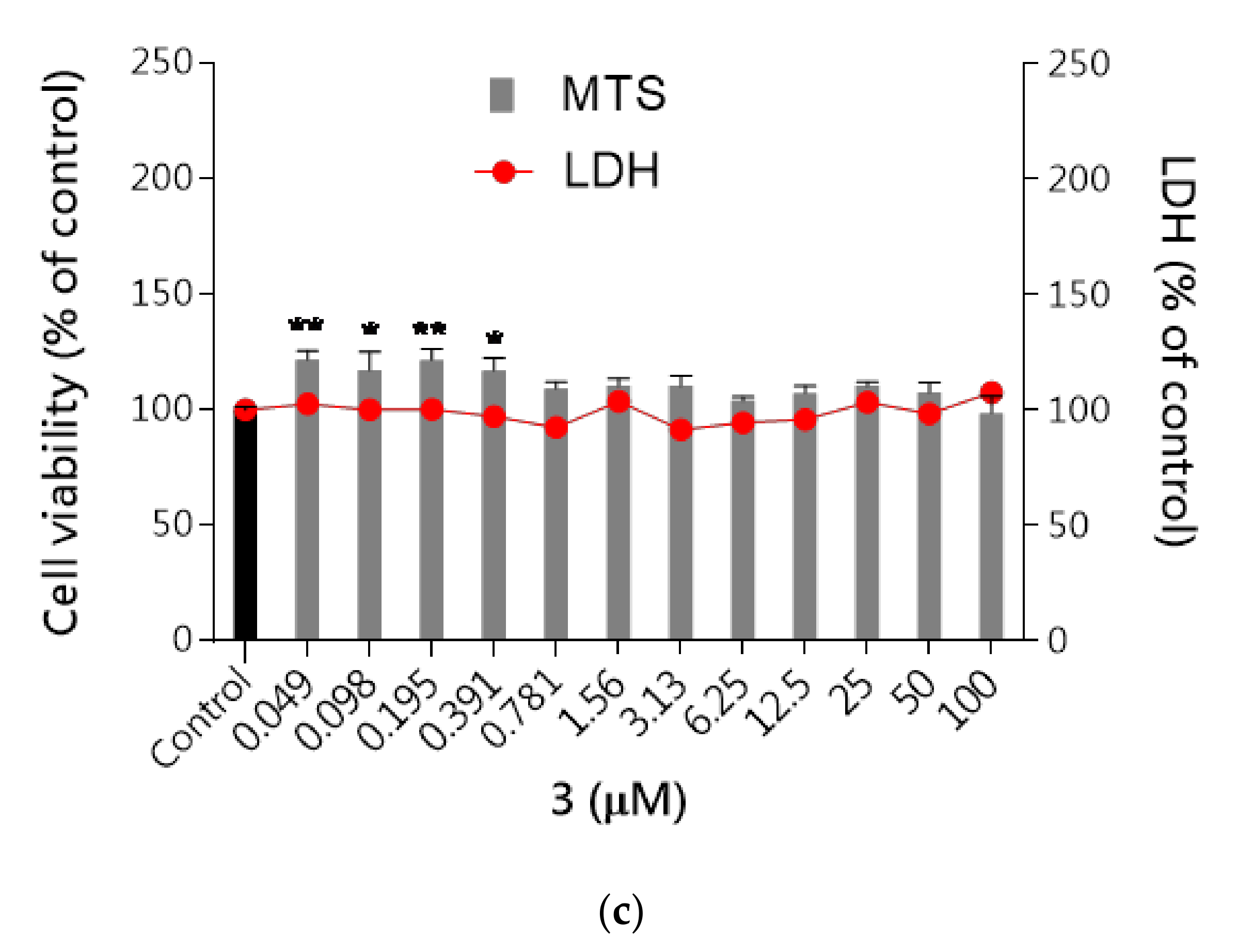
2.2.4. Safety
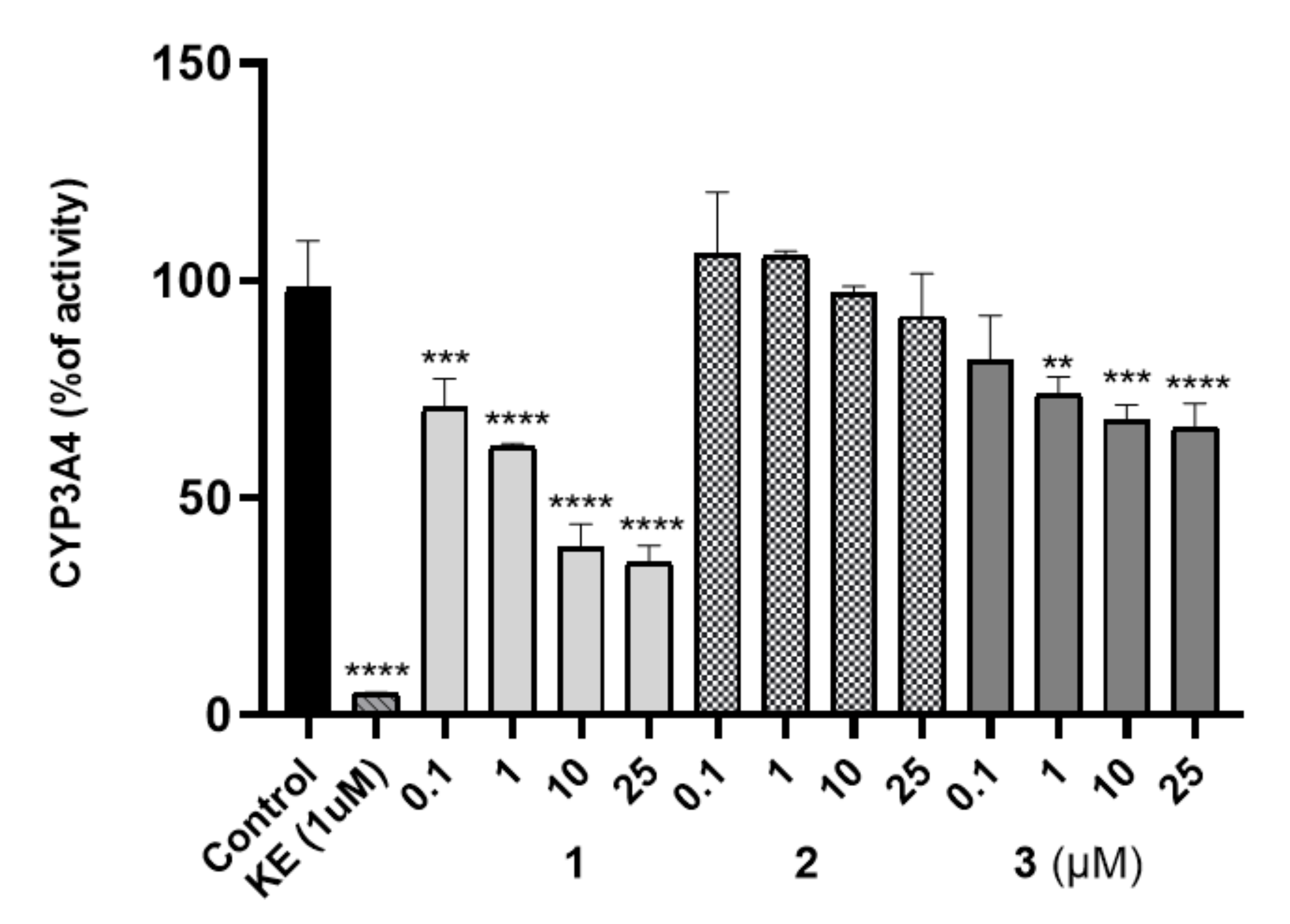
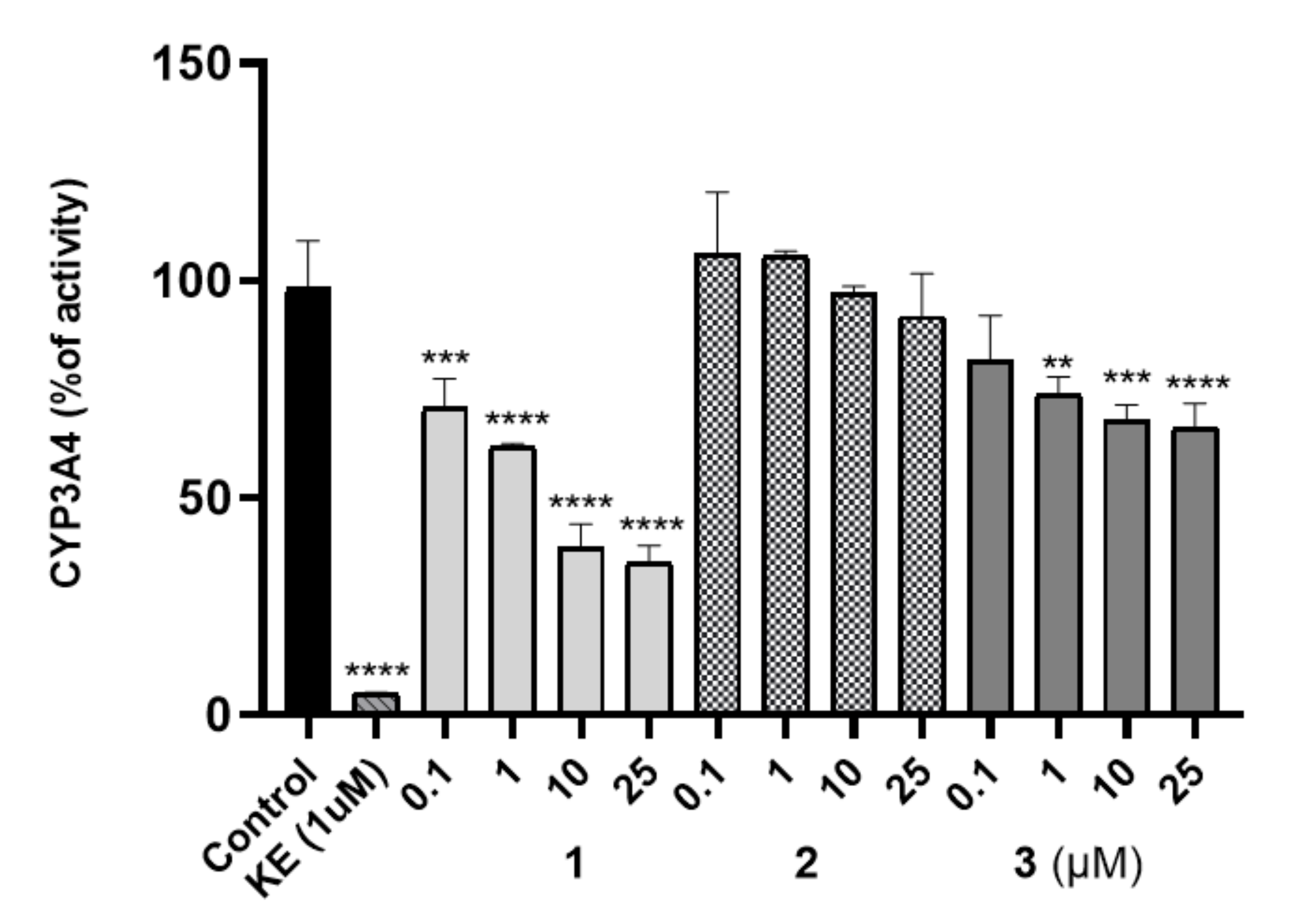
The Effect on Cytochrome P450
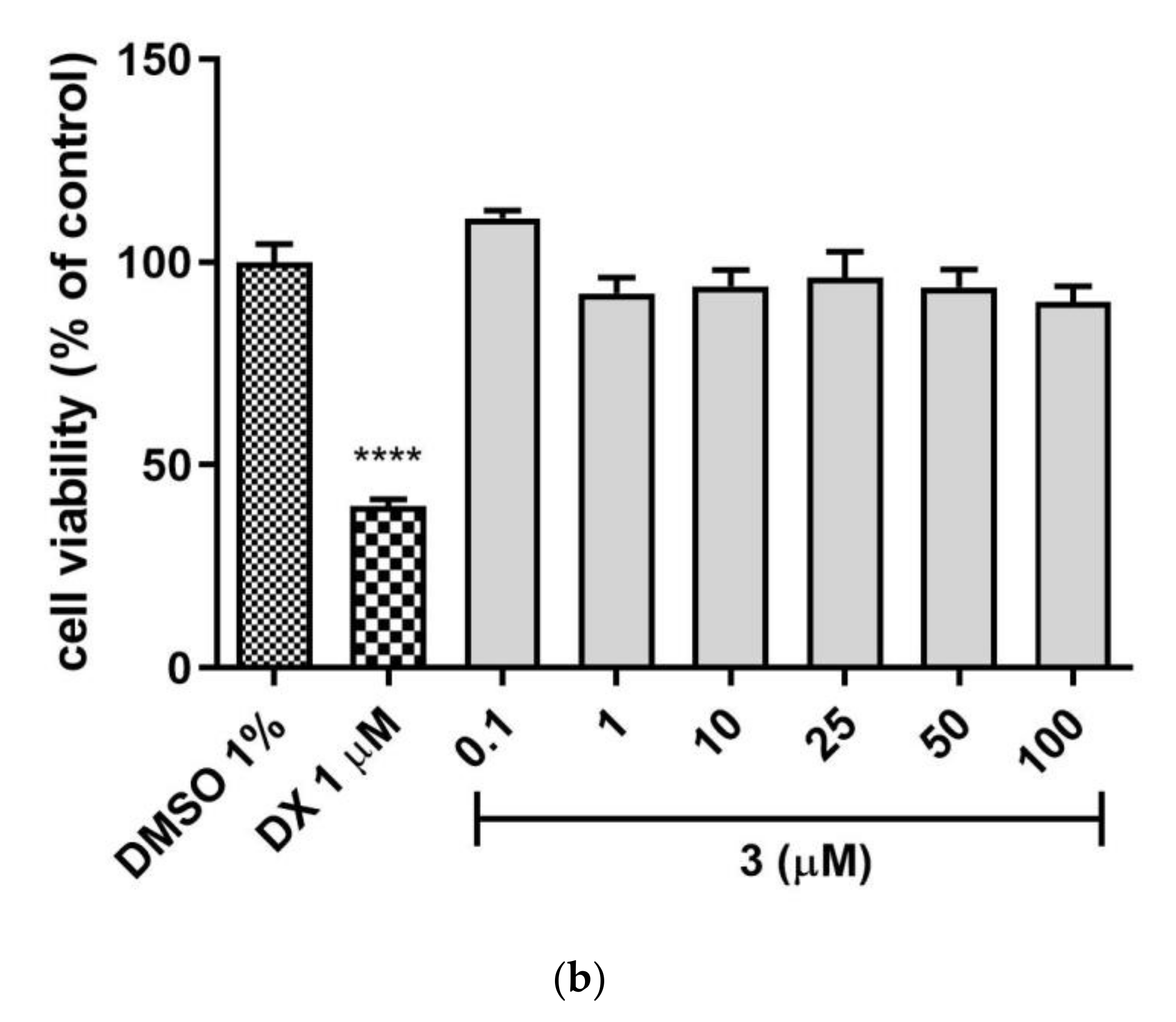
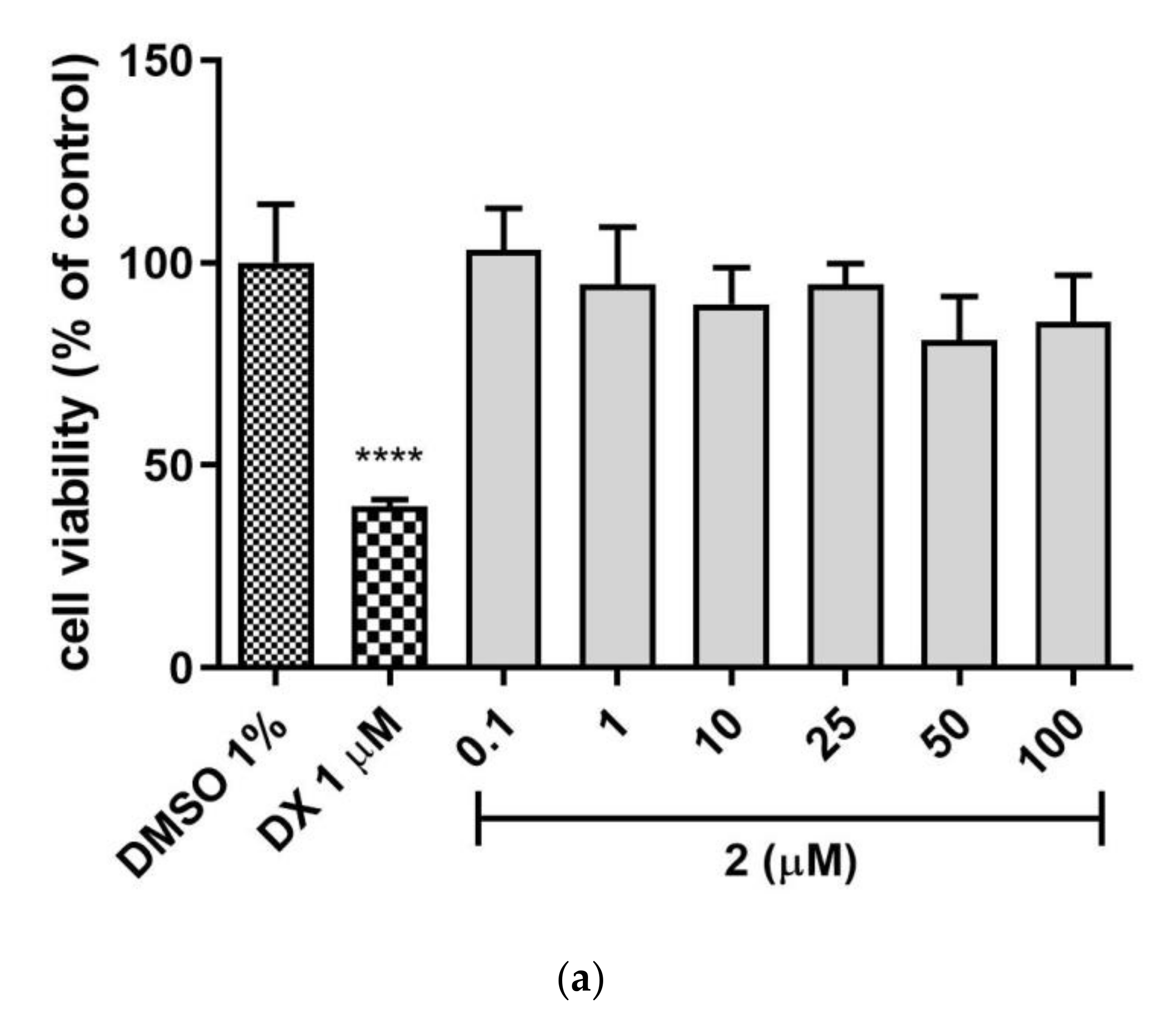
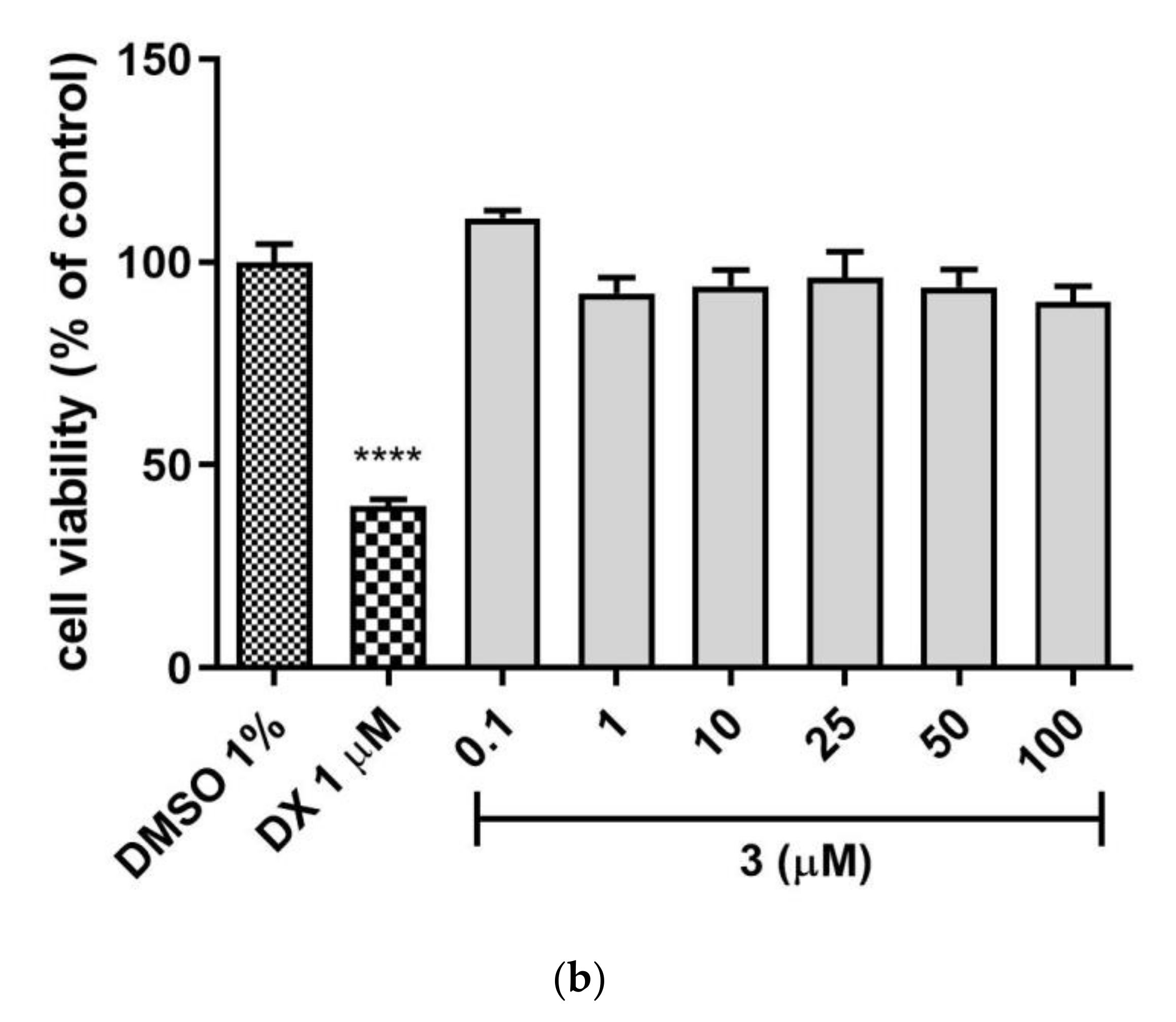
Hepatotoxicity
Mutagenicity
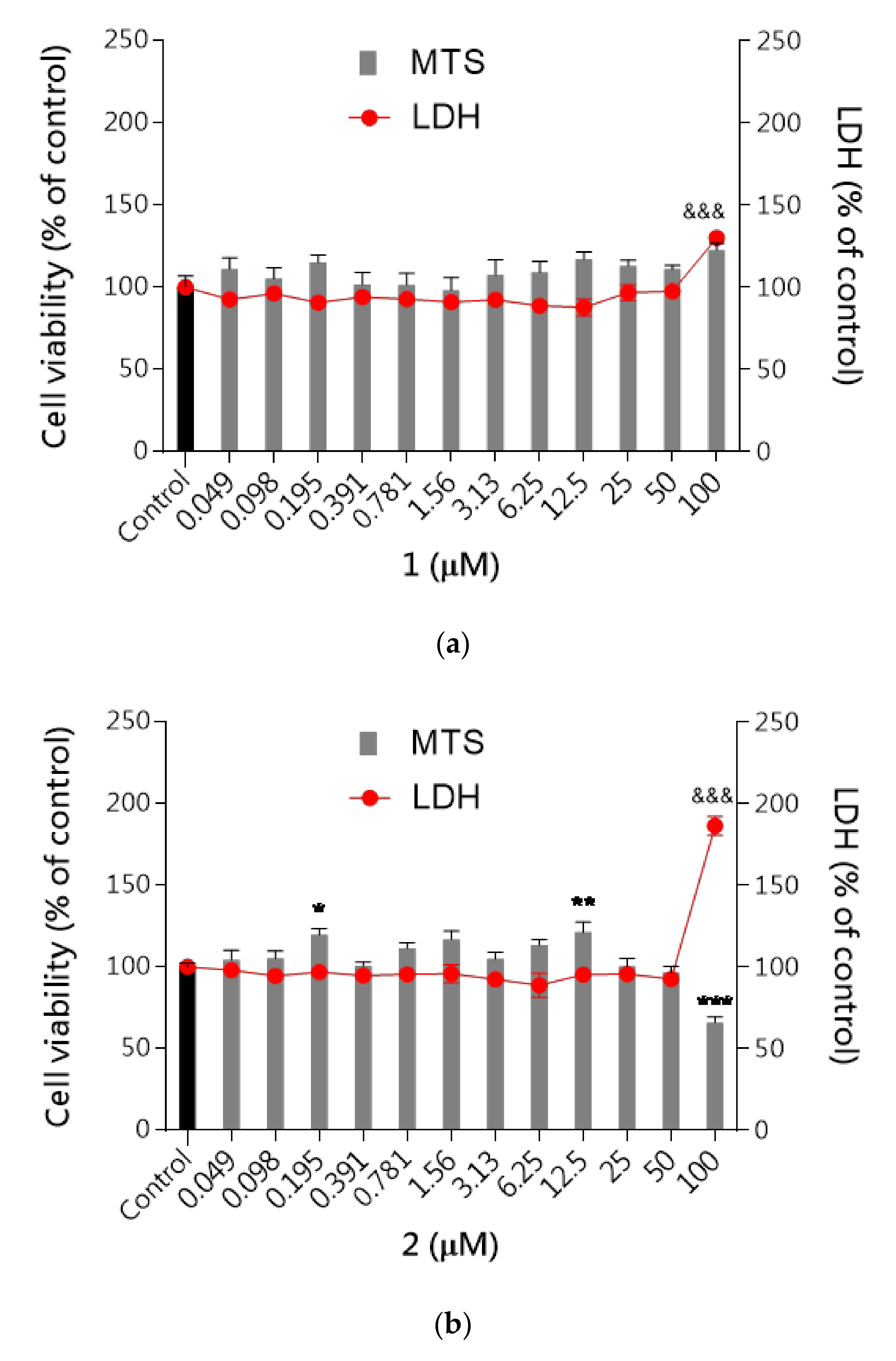
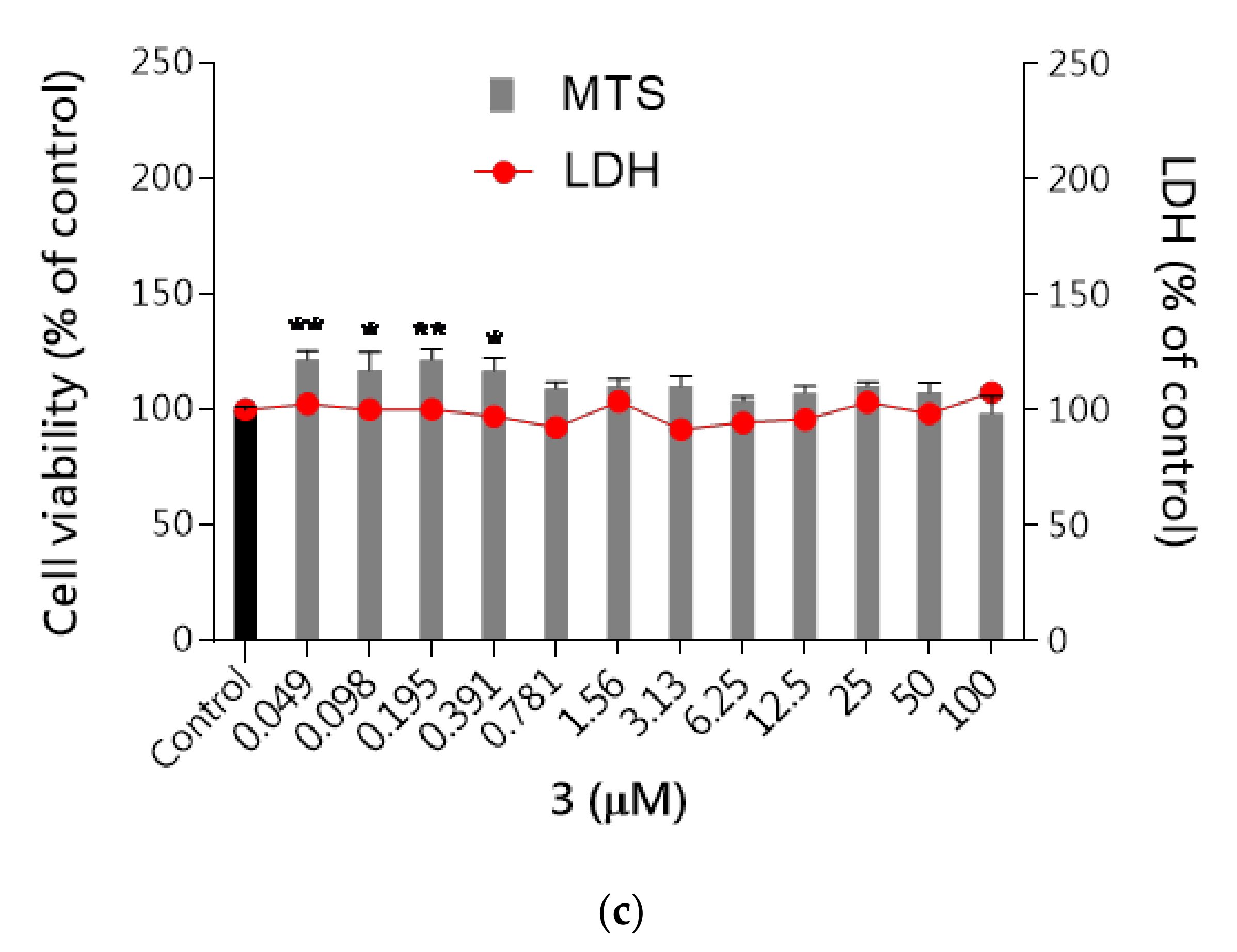
Neurotoxicity
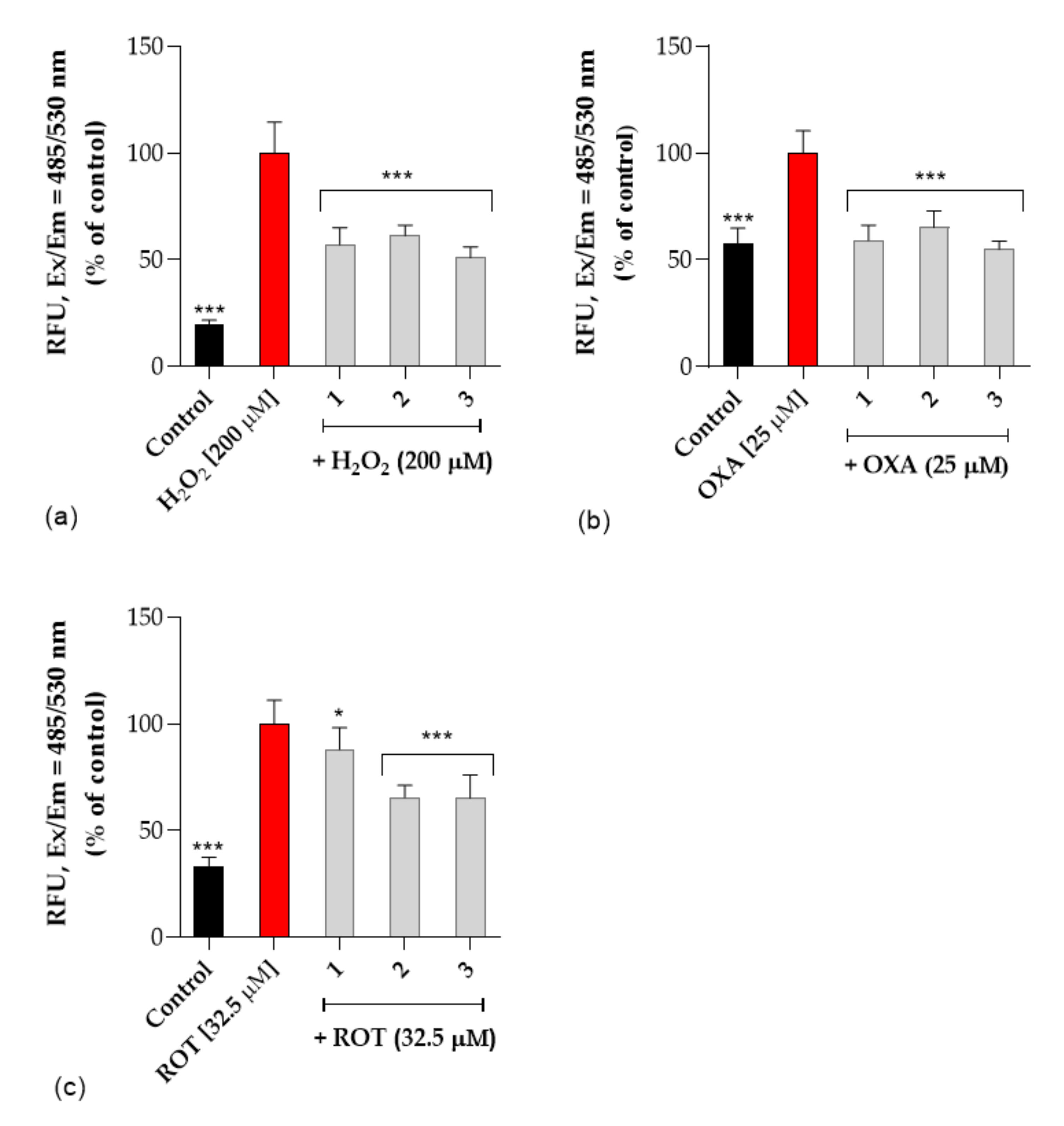
Neuroprotective Activity
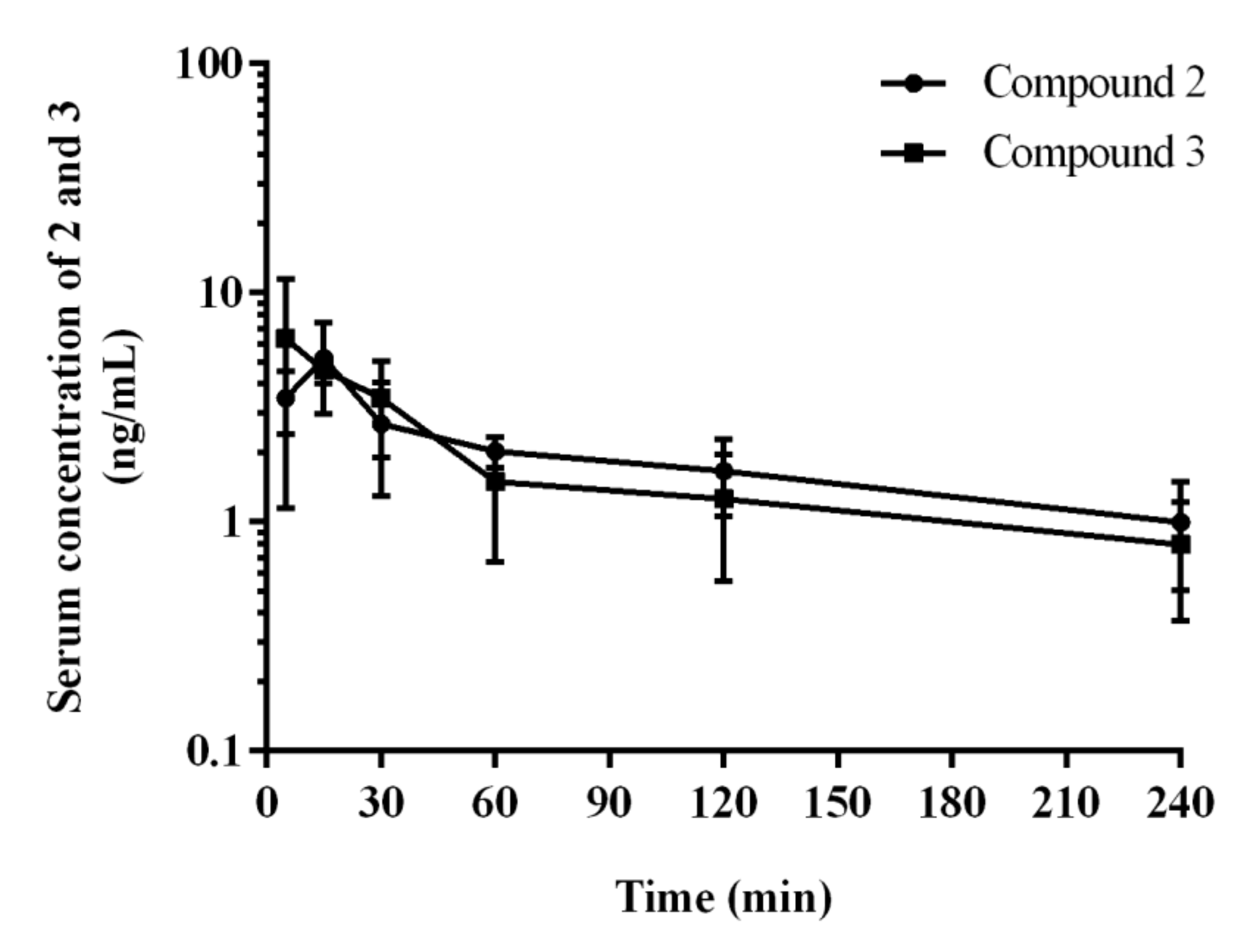
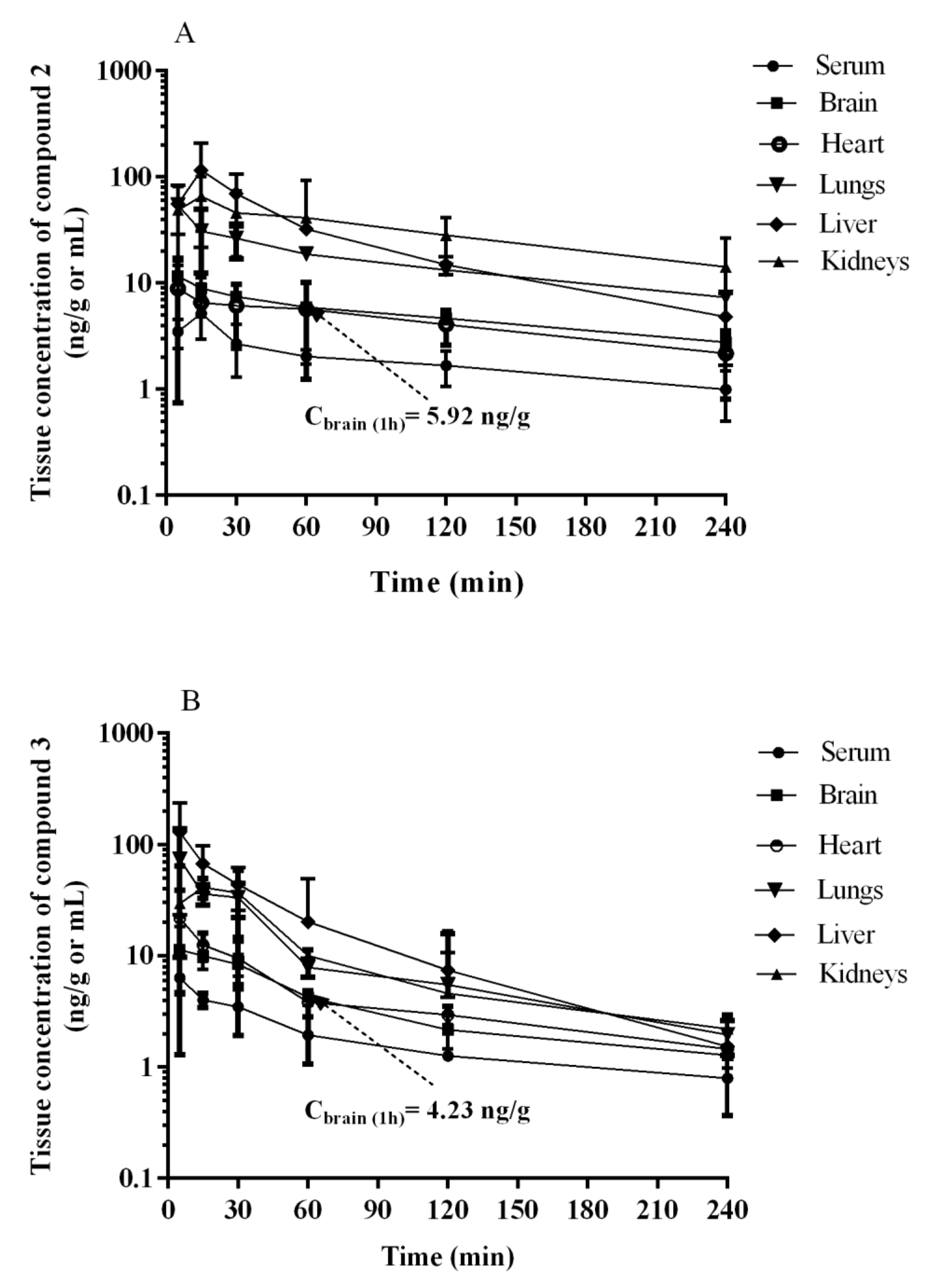
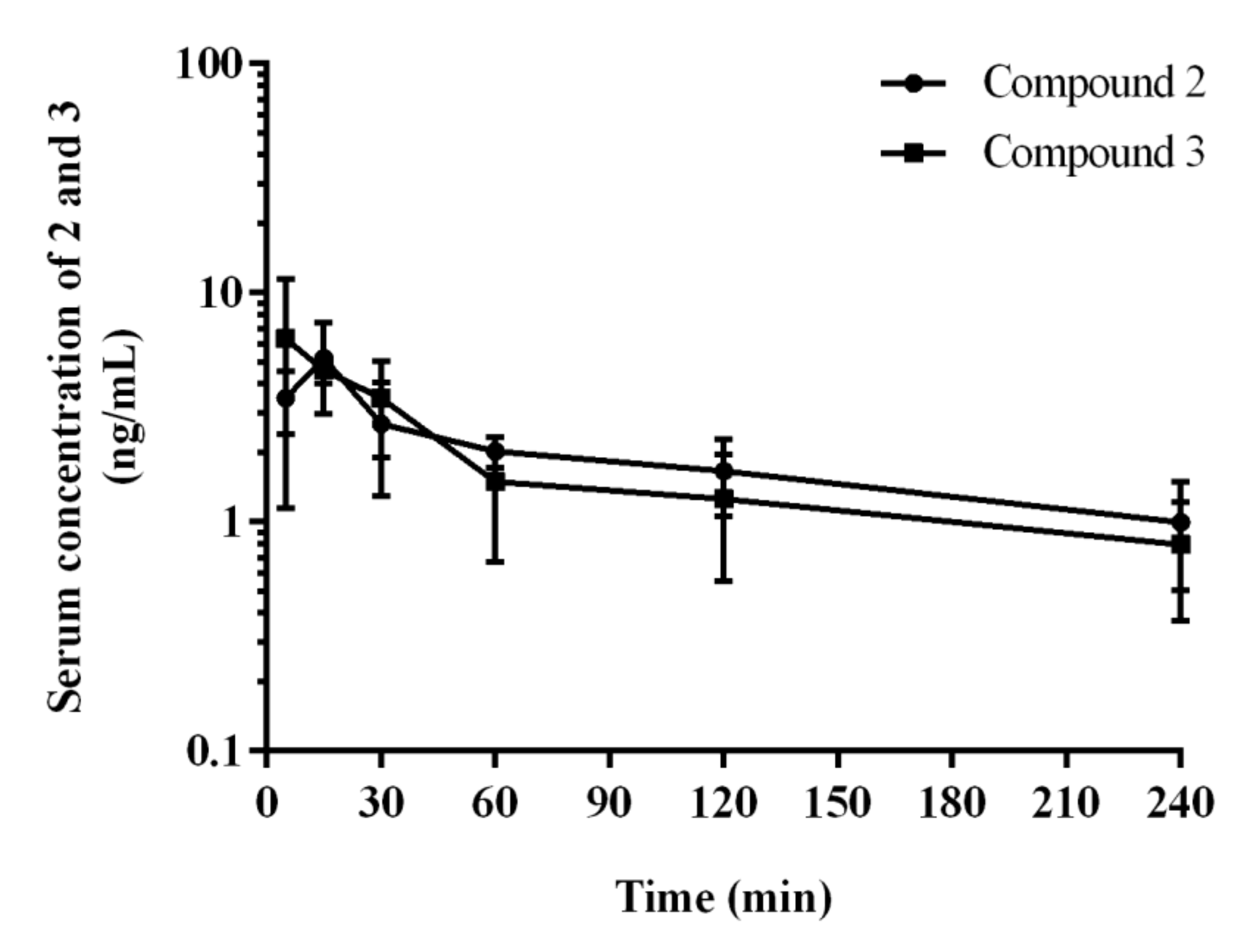
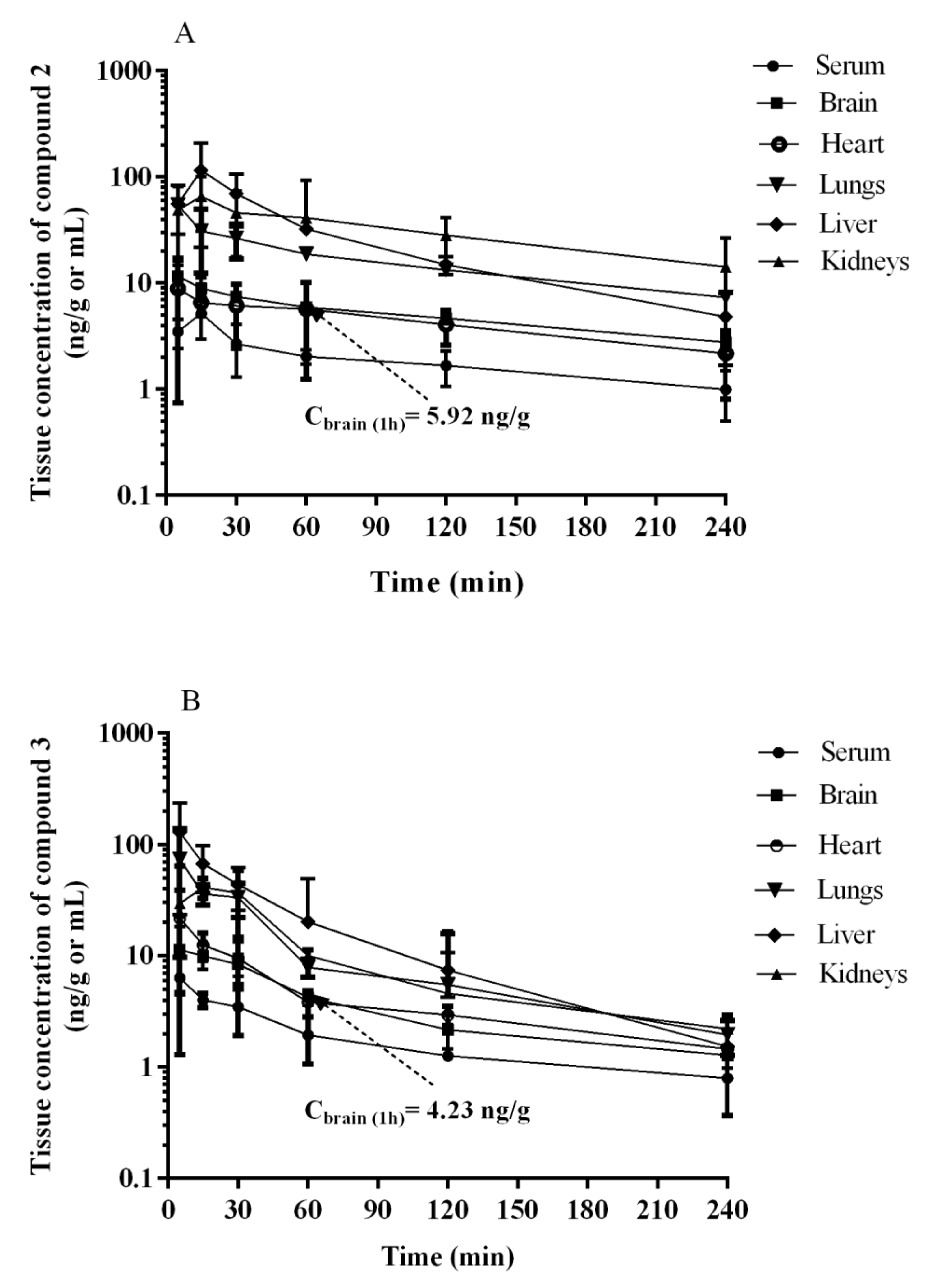
2.3. Pharmacokinetics In Vivo
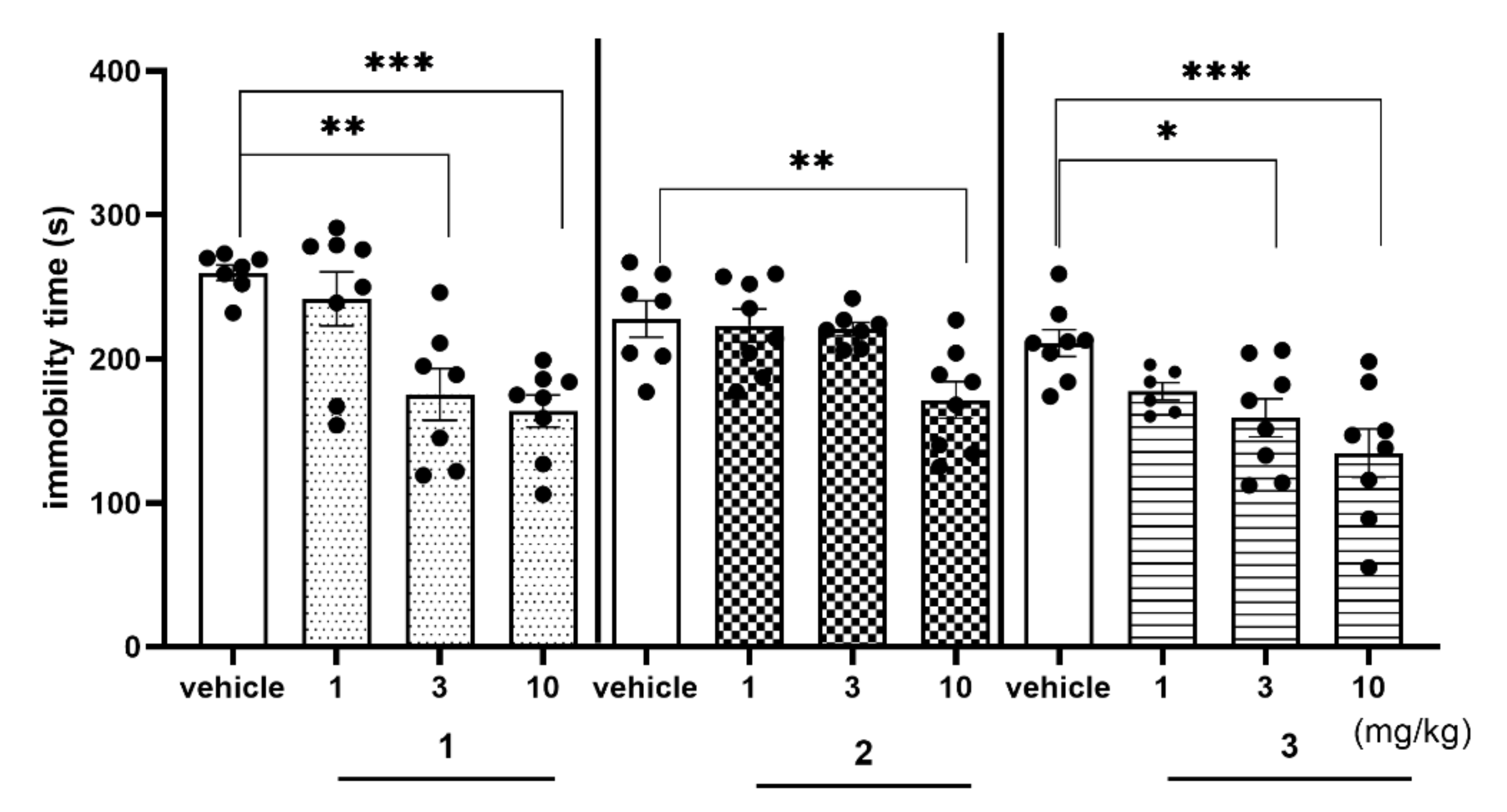
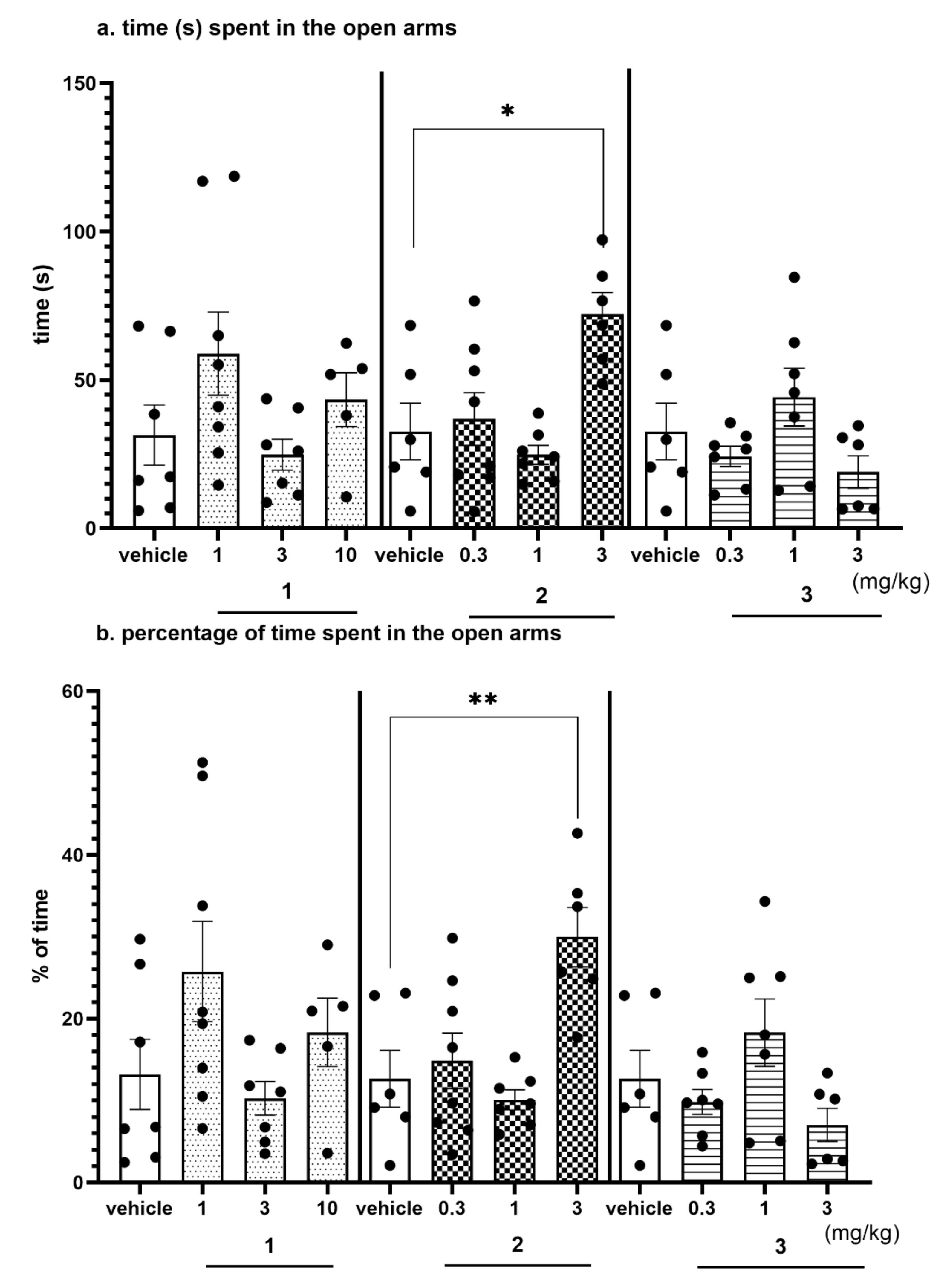
2.4. Behavioral Tests In Vivo
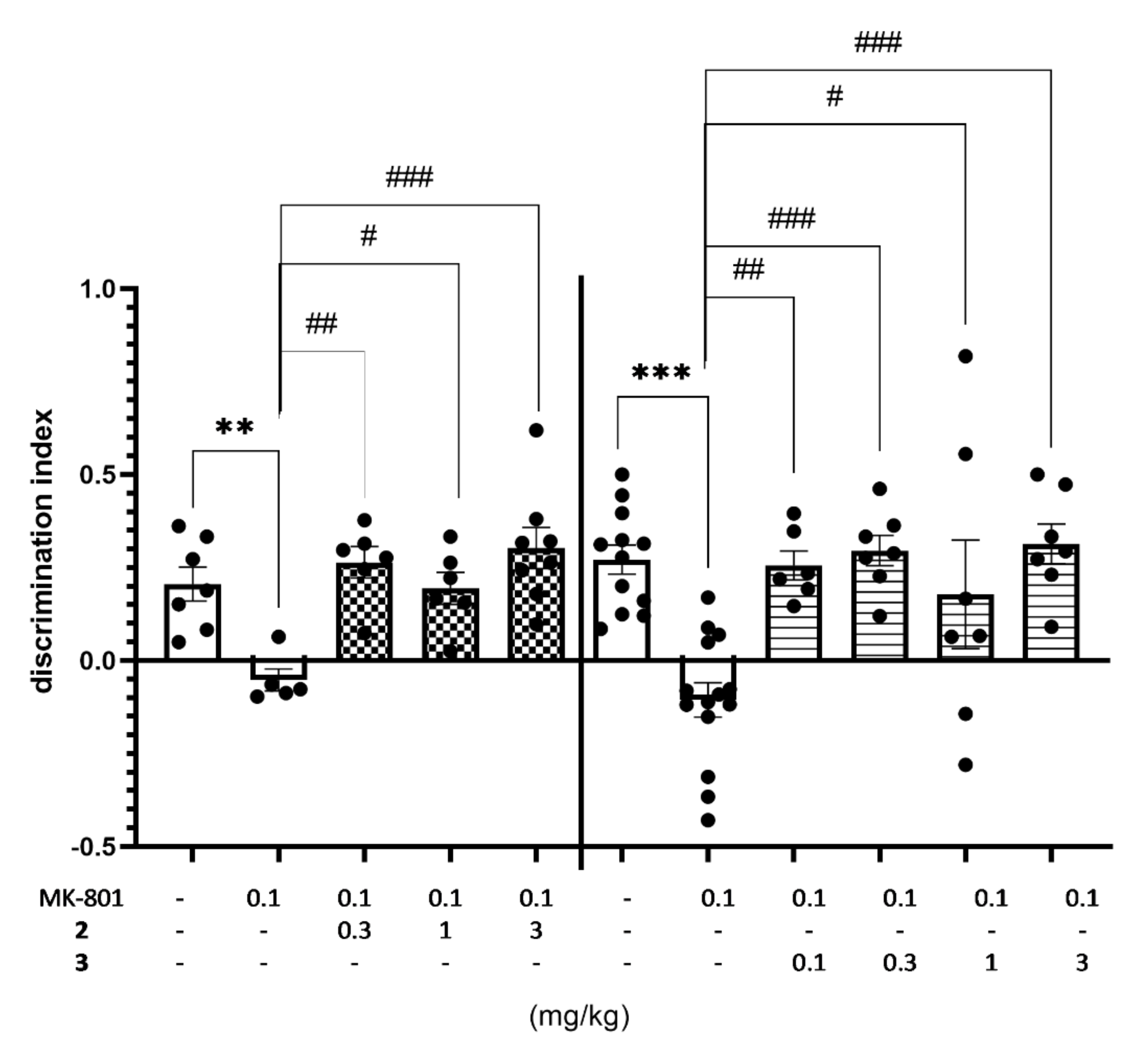
2.4.1. The Impact of Memory Impairment of Investigated Compounds
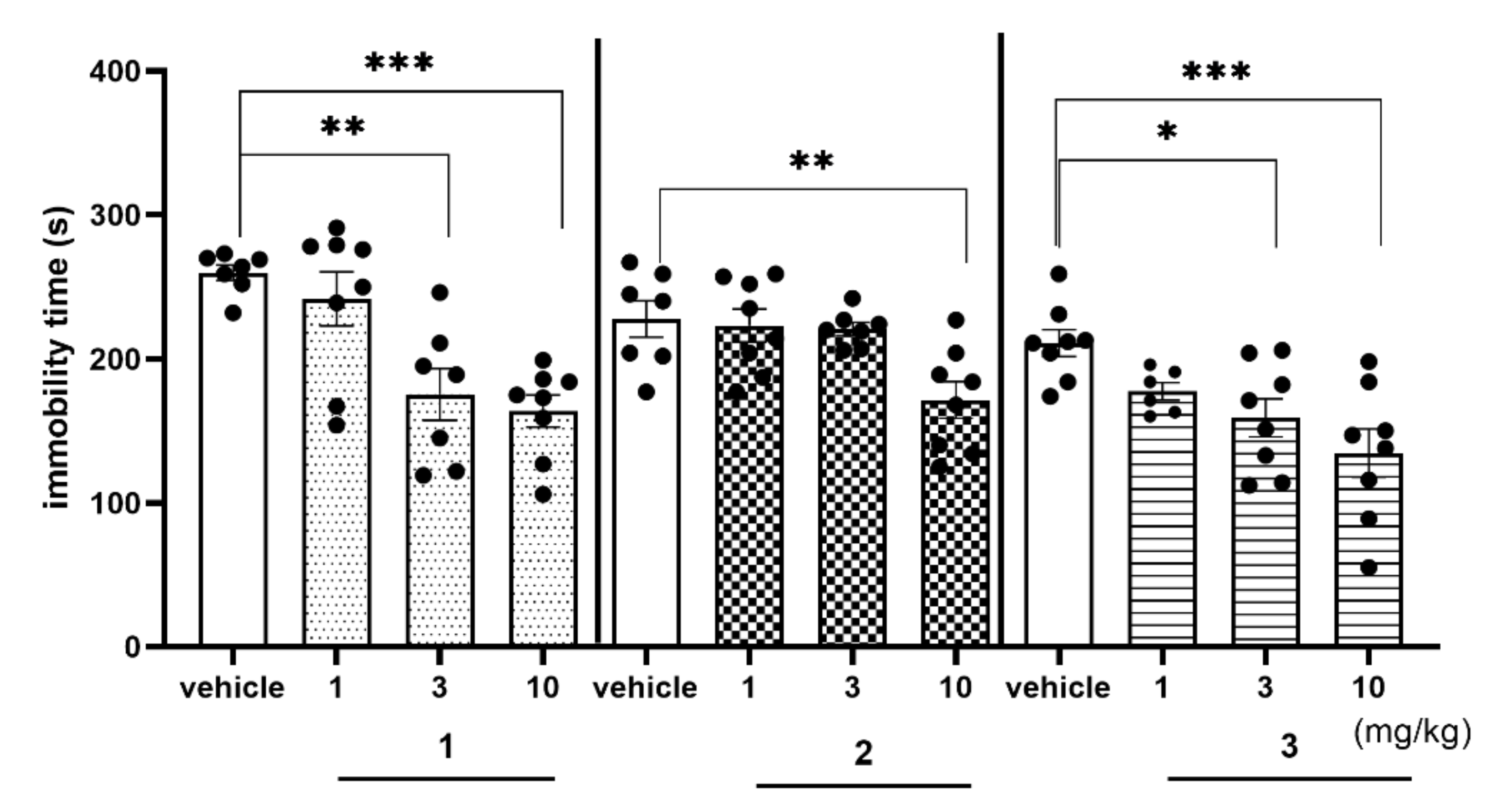
2.4.2. Antidepressant-Like Activity of Compounds 1–3 in the FST
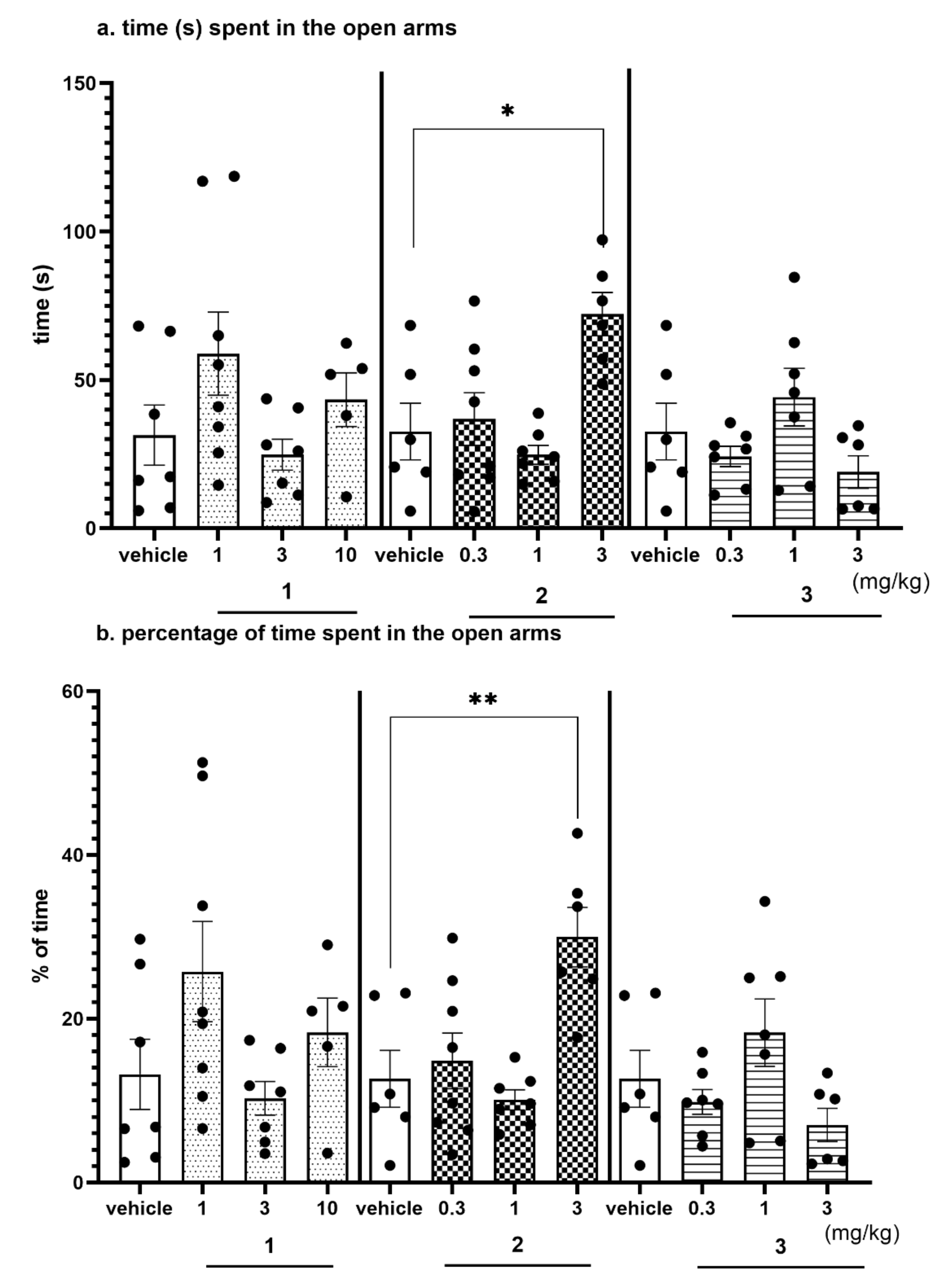
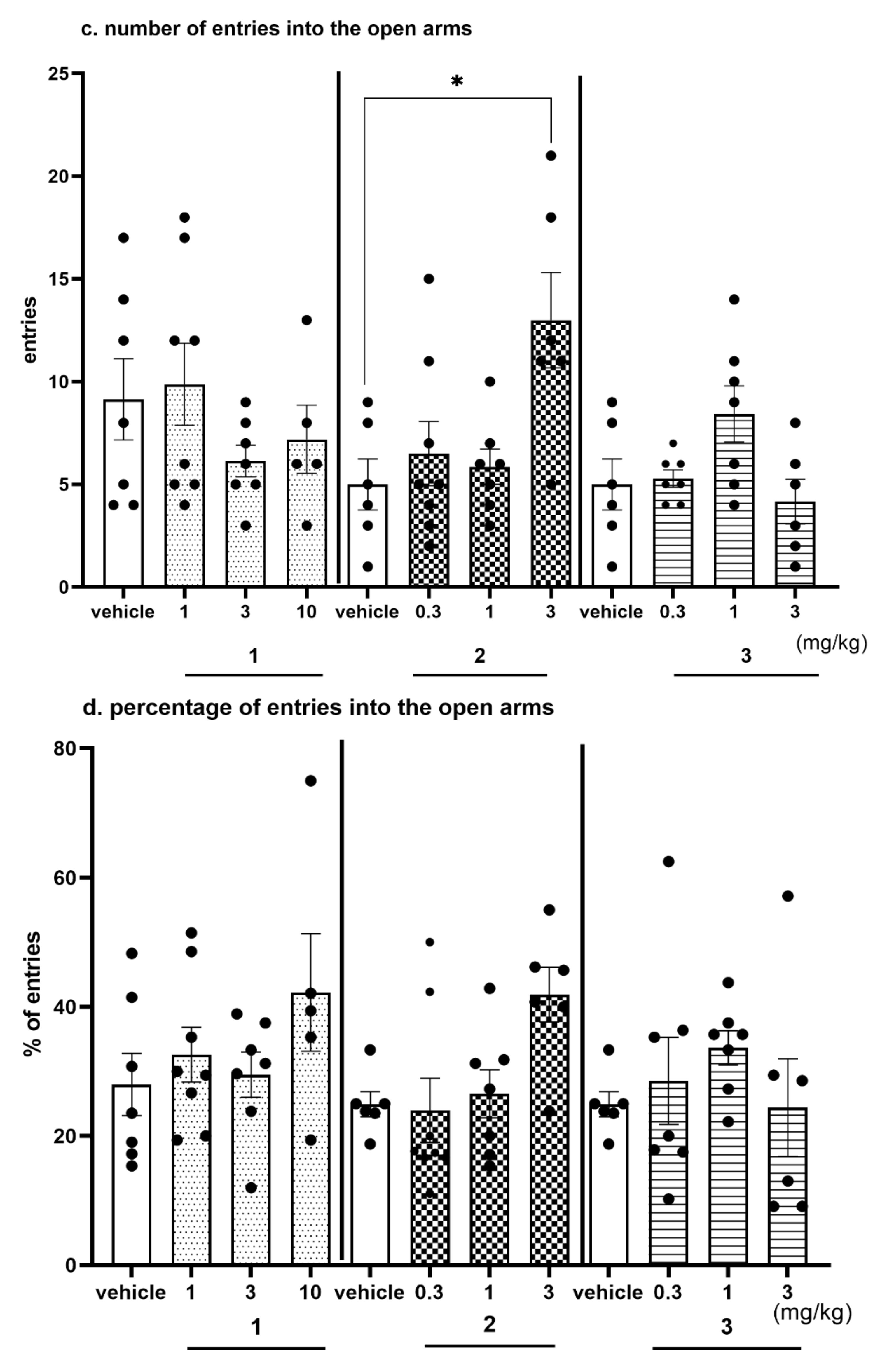
2.4.3. Anxiolytic-Like Activity of Compounds 1–3 in the EPM test
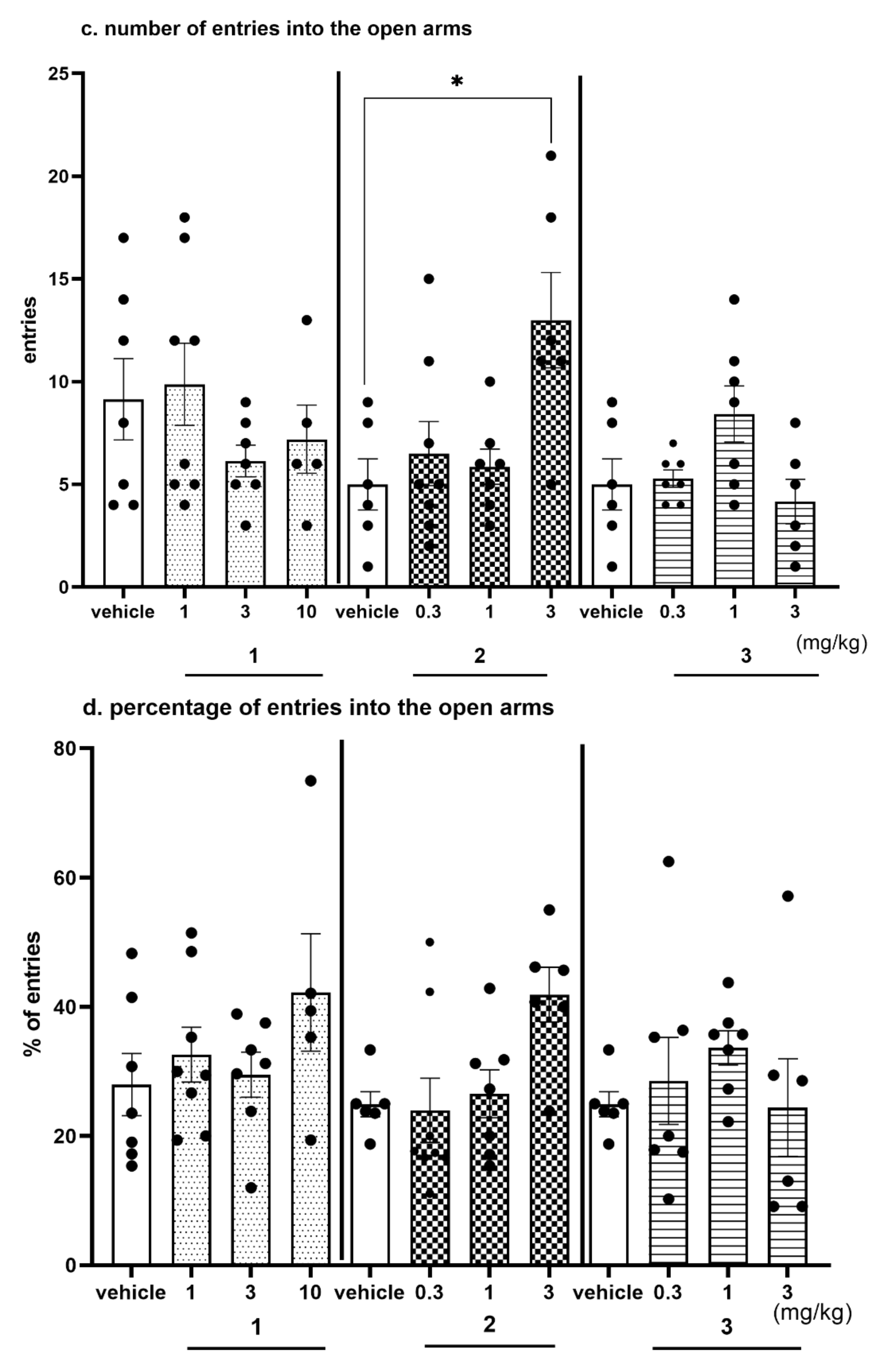
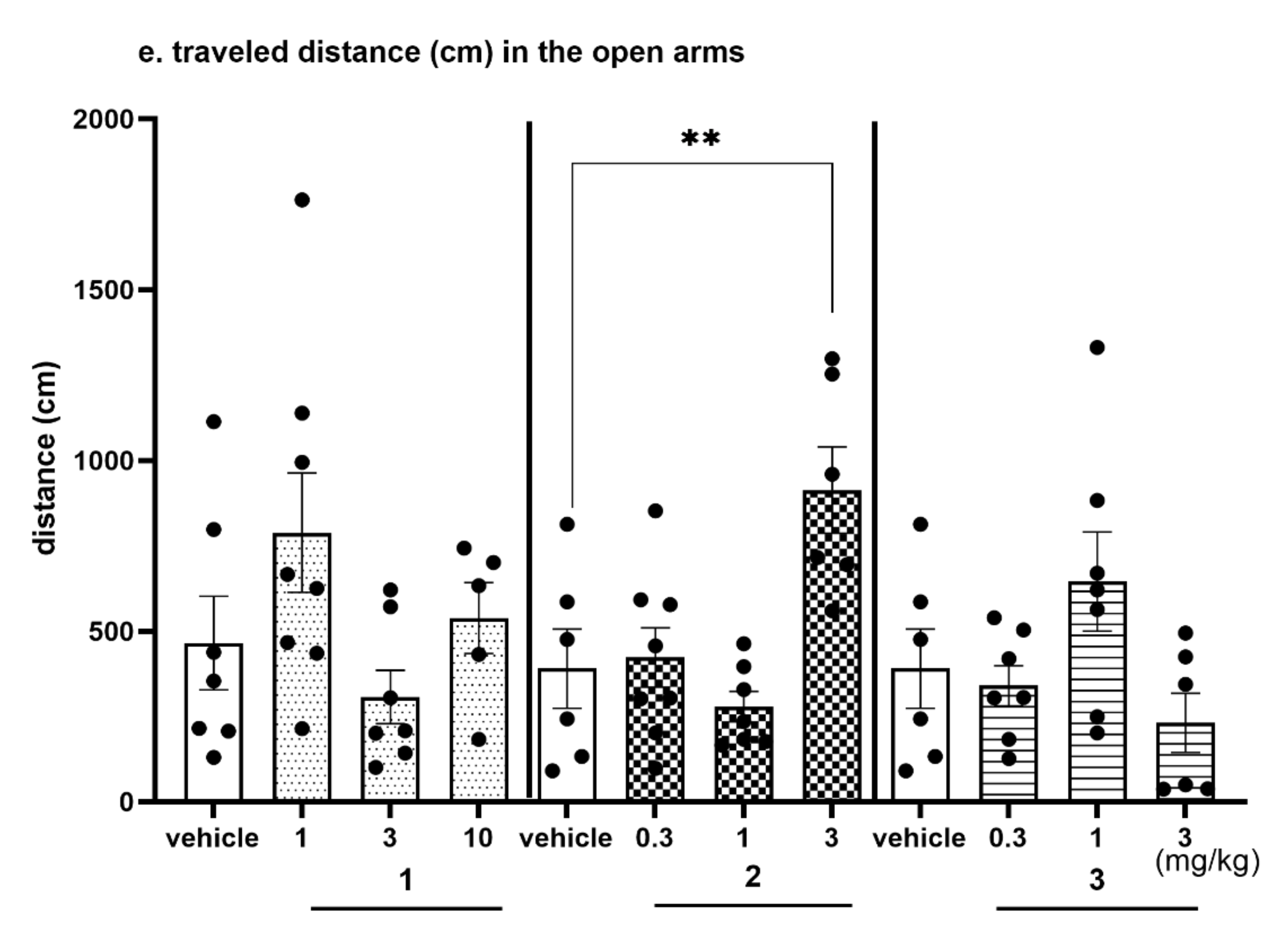
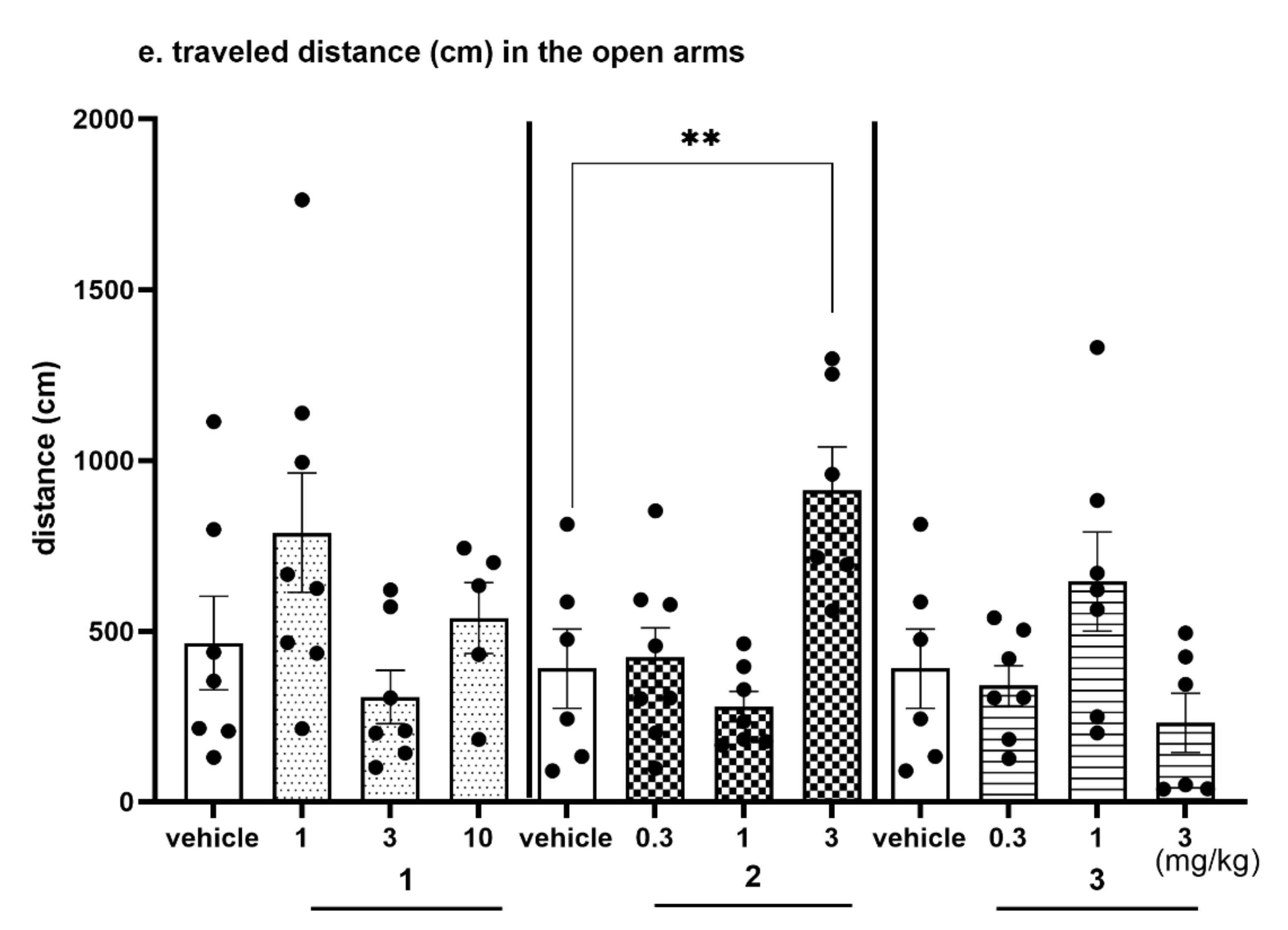
2.4.4. The Exploratory Activity of Investigated Compounds Measured in the EPM Test
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Radioligand Binding Assay
4.3. Functional Assay
4.3.1. Functional Assays for 5-HT6 Receptor
4.3.2. Functional Assays for 5-HT2A Receptor
4.4. ADMET In Vitro
4.4.1. References
4.4.2. Permeability
4.4.3. Caco-2 Permeability Assay
4.4.4. Metabolic Stability
4.4.5. Safety
4.4.6. Neurotoxicity
4.4.7. Determination of Intracellular ROS Levels
4.5. In Vivo Pharmacokinetic Studies
4.5.1. Animals
4.5.2. Application to a Pharmacokinetic Study in Rats
4.5.3. Analytical Method
4.5.4. Determination of Compounds 2 and 3 in Biological Matrices
4.5.5. Pharmacokinetic Analysis
4.6. Behavioral Studies In Vivo
4.6.1. Animals
4.6.2. Drugs
4.6.3. Behavioral Procedures in Rats
Novel Object Recognition (NOR) Test
Forced Swim Test (FST)
Elevated Plus-Maze Test (EPM Test)
Exploratory Activity Measured in the EPM Test
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ruat, M.; Traiffort, E.; Arrang, J.M.; Tardivel-Lacombe, J.; Diaz, J.; Leurs, R.; Schwartz, J.C. A Novel Rat Serotonin (5-HT6) Receptor: Molecular Cloning, Localization and Stimulation of cAMP Accumulation. Biochem. Biophys. Res. Commun. 1993, 193, 268–276. [Google Scholar] [CrossRef]
- Olde, B.; McCombie, W.R. Molecular cloning and functional expression of a serotonin receptor from Caenorhabditis elegans. J. Mol. Neurosci. 1997, 8, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wicke, K.; Haupt, A.; Bespalov, A. Investigational drugs targeting 5-HT6 receptors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6 Receptor Antagonists for the Treatment of Cognitive Deficiency in Alzheimer’s Disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, A.; Zhang, J.; Lao, J.Z.; White, A.; Metzger, J.M.; Fong, T.M.; Chen, R.Z. Reduced sensitivity to diet-induced obesity in mice carrying a mutant 5-HT6 receptor. Brain Res. 2008, 1236, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Shortall, S.E.; Negm, O.H.; Fowler, M.; Fairclough, L.; Tighe, P.; Wigmore, P.M.; King, M.V. Characterization of Behavioral, Signaling and Cytokine Alterations in a Rat Neurodevelopmental Model for Schizophrenia, and Their Reversal by the 5-HT6 Receptor Antagonist SB-399885. Mol. Neurobiol. 2018, 55, 7413–7430. [Google Scholar] [CrossRef] [Green Version]
- Kotańska, M.; Lustyk, K.; Bucki, A.; Marcinkowska, M.; Śniecikowska, J.; Kolaczkowski, M. Idalopirdine, a selective 5-HT6 receptor antagonist, reduces food intake and body weight in a model of excessive eating. Metab. Brain Dis. 2018, 33, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Wesołowska, A. Potential role of the 5-HT6 receptor in depression and anxiety: An overview of preclinical data. Pharmacol. Rep. 2010, 62, 564–577. [Google Scholar] [CrossRef]
- Kucwaj-Brysz, K.; Baltrukevich, H.; Czarnota, K.; Handzlik, J. Chemical update on the potential for serotonin 5-HT6 and 5-HT7 receptor agents in the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2021, 49, 128275. [Google Scholar] [CrossRef]
- Woods, S.; Clarke, N.N.; Layfield, R.; Fone, K. 5-HT6 receptor agonists and antagonists enhance learning and memory in a conditioned emotion response paradigm by modulation of cholinergic and glutamatergic mechanisms. Br. J. Pharmacol. 2012, 167, 436–449. [Google Scholar] [CrossRef] [Green Version]
- Schechter, L.E.; Lin, Q.; Smith, D.L.; Zhang, G.; Shan, Q.; Platt, B.; Brandt, M.R.; Dawson, L.A.; Cole, D.; Bernotas, R.; et al. Neuropharmacological Profile of Novel and Selective 5-HT6 Receptor Agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology 2007, 33, 1323–1335. [Google Scholar] [CrossRef]
- Zajdel, P.; Marciniec, K.; Satała, G.; Canale, V.; Kos, T.; Partyka, A.; Jastrzębska-Więsek, M.; Wesołowska, A.; Basińska-Ziobroń, A.; Wójcikowski, J.; et al. N1-Azinylsulfonyl-1H-indoles: 5-HT6 Receptor Antagonists with Procognitive and Antidepressant-Like Properties. ACS Med. Chem. Lett. 2016, 7, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Vera, J.A.G.; Medina, R.A.; Martin-Fontecha, M.; Gonzalez, A.; De La Fuente, T.; Vázquez-Villa, H.; Cárceles, J.G.; Botta, J.; McCormick, P.J.; Benhamu, B.; et al. A new serotonin 5-HT6 receptor antagonist with procognitive activity–Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, srep41293. [Google Scholar] [CrossRef] [Green Version]
- Ivanenkov, Y.A.; Majouga, A.G.; Veselov, M.; Chufarova, N.V.; Baranovsky, S.S.; Filkov, G.I. Computational approaches to the design of novel 5-HT6 R ligands. Rev. Neurosci. 2014, 25, 451–467. [Google Scholar] [CrossRef]
- Millan, M.J.; Dekeyne, A.; Gobert, A.; Brocco, M.; la Cour, C.M.; Ortuno, J.-C.; Watson, D.; Fone, K.C. Dual-acting agents for improving cognition and real-world function in Alzheimer’s disease: Focus on 5-HT6 and D3 receptors as hubs. Neuropharmacology 2020, 177, 108099. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Abraham, R.; Benade, V.; Medapati, R.B.; Jayarajan, P.; Bhyrapuneni, G.; Muddana, N.; Mekala, V.R.; Subramanian, R.; Shinde, A.; et al. SUVN-502, a novel, potent, pure, and orally active 5-HT6 receptor antagonist: Pharmacological, behavioral, and neurochemical characterization. Behav. Pharmacol. 2019, 30, 16–35. [Google Scholar] [CrossRef]
- Monsma, F.J.; Shen, Y.; Ward, R.P.; Hamblin, M.W.; Sibley, D.R. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol. Pharmacol. 1993, 43, 320–327. [Google Scholar] [PubMed]
- Roth, B.L.; Meltzer, H.; Khan, N. Binding of typical and atypical antipsychotic drugs to multiple neurotransmitter receptors. Adv. Pharmacol. 1997, 42, 482–485. [Google Scholar] [CrossRef]
- Karila, D.; Freret, T.; Bouet, V.; Boulouard, M.; Dallemagne, P.; Rochais, C. Therapeutic Potential of 5-HT6 Receptor Agonists. J. Med. Chem. 2015, 58, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, M.L.; Benhamu, B.; De La Fuente, T.; Sanz, A.; Pardo, L.; Campillo, M. A Three-Dimensional Pharmacophore Model for 5-Hydroxytryptamine6(5-HT6) Receptor Antagonists. J. Med. Chem. 2005, 48, 4216–4219. [Google Scholar] [CrossRef] [PubMed]
- Sudoł, S.; Kucwaj-Brysz, K.; Kurczab, R.; Wilczyńska, N.; Jastrzębska-Więsek, M.; Satała, G.; Latacz, G.; Głuch-Lutwin, M.; Mordyl, B.; Żesławska, E.; et al. Chlorine substituents and linker topology as factors of 5-HT6R activity for novel highly active 1,3,5-triazine derivatives with procognitive properties in vivo. Eur. J. Med. Chem. 2020, 203, 112529. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Ames MPF™ 98/100, Instruction, version 5.01. Test do oceny mutagenności w formacie mikropłytki. [Microplate Format Mutagenicity Assay]. Xenometrix, Swiss Comitment for Bioassays: Allschwil, Switzerland, 2018.
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Marć, M.A.; Satała, G.; Wilczyńska, D.; Kotańska, M.; Więcek, M.; Kamińska, K.; et al. The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latacz, G.; Hogendorf, A.S.; Hogendorf, A.; Lubelska, A.; Wierońska, J.M.; Wozniak, M.; Cieślik, P.; Kieć-Kononowicz, K.; Handzlik, J.; Bojarski, A.J. Search for a 5-CT alternative. In vitro and in vivo evaluation of novel pharmacological tools: 3-(1-alkyl-1H-imidazol-5-yl)-1H-indole-5-carboxamides, low-basicity 5-HT7 receptor agonists. MedChemComm 2018, 9, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a Novel Pyrrolidine-2,5-dione Derived Broad-Spectrum Anticonvulsant: Its Antiepileptogenic, Antinociceptive Properties and in Vitro Characterization. ACS Chem. Neurosci. 2018, 10, 636–648. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Kucwaj-Brysz, K.; Wesołowska, A.; Kiec-Kononowicz, K.; Handzlik, J. MF-8, a novel promising arylpiperazine-hydantoin based 5-HT 7 receptor antagonist: In vitro drug-likeness studies and in vivo pharmacological evaluation. Bioorganic Med. Chem. Lett. 2018, 28, 878–883. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Sobiło, A.; Olejarz-Maciej, A.; Kucwaj-Brysz, K.; Satała, G.; Bojarski, A.; Wesołowska, A.; et al. In the search for a lead structure among series of potent and selective hydantoin 5-HT7R agents: The drug-likeness in vitro study. Chem. Biol. Drug Des. 2017, 90, 1295–1306. [Google Scholar] [CrossRef]
- Fone, K.C. An update on the role of the 5-hydroxytryptamine6 receptor in cognitive function. Neuropharmacology 2008, 55, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Kemp, J.A.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef] [Green Version]
- Ellison, G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res. Rev. 1995, 20, 250–267. [Google Scholar] [CrossRef]
- Bubeníková-Valešová, V.; Horáček, J.; Vrajová, M.; Höschl, C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci. Biobehav. Rev. 2008, 32, 1014–1023. [Google Scholar] [CrossRef]
- Mitchell, E.S. 5-HT6 Receptor Ligands as Antidementia Drugs. Int. Rev. Neurobiol. 2011, 96, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Codony, X.; Vela, J.M.; Ramírez, M.J. 5-HT6 receptor and cognition. Curr. Opin. Pharmacol. 2011, 11, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Rychtyk, J.; Partyka, A.; Gdula-Argasińska, J.; Mysłowska, K.; Wilczyńska, N.; Jastrzębska-Więsek, M.; Wesołowska, A. 5-HT6 receptor agonist and antagonist improve memory impairments and hippocampal BDNF signaling alterations induced by MK-801. Brain Res. 2019, 1722, 146375. [Google Scholar] [CrossRef] [PubMed]
- Pellow, S.; Chopin, P.; File, S.E.; Briley, M. Validation of open: Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 1985, 14, 149–167. [Google Scholar] [CrossRef]
- Adell, A.; Jiménez-Sánchez, L.; López-Gil, X.; Romón, T. Is the Acute NMDA Receptor Hypofunction a Valid Model of Schizophrenia? Schizophr. Bull. 2011, 38, 9–14. [Google Scholar] [CrossRef]
- Van der Staay, F.J.; Rutten, K.; Erb, C.; Blokland, A. Effects of the cognition impairer MK-801 on learning and memory in mice and rats. Behav. Brain Res. 2011, 220, 215–229. [Google Scholar] [CrossRef]
- Boess, F.G.; Hendrix, M.; van der Staay, F.J.; Erb, C.; Schreiber, R.; van Staveren, W.; de Vente, J.; Prickaerts, J.; Blokland, A.; Koenig, G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 2004, 47, 1081–1092. [Google Scholar] [CrossRef]
- Boess, F.G.; De Vry, J.; Erb, C.; Flessner, T.; Hendrix, M.; Luithle, J.; Methfessel, C.; Riedl, B.; Schnizler, K.; van der Staay, F.-J.; et al. The Novel α7 Nicotinic Acetylcholine Receptor Agonist N-[(3R)-1-Azabicyclo [2.2.2] oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxamide Improves Working and Recognition Memory in Rodents. J. Pharmacol. Exp. Ther. 2007, 321, 716–725. [Google Scholar] [CrossRef]
- De Bruin, N.; Kruse, C. 5-HT6 Receptor Antagonists: Potential Efficacy for the Treatment of Cognitive Impairment in Schizophrenia. Curr. Pharm. Des. 2015, 21, 3739–3759. [Google Scholar] [CrossRef]
- De Jong, I.E.; Mørk, A. Antagonism of the 5-HT 6 receptor–Preclinical rationale for the treatment of Alzheimer’s disease. Neuropharmacology 2017, 125, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Upton, N.; Chuang, T.T.; Hunter, A.J.; Virley, D.J. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Woolley, M.L.; Marsden, C.A.; Fone, K.C. 5-ht6 Receptors. Curr. Drug Target CNS Neurol. Disord. 2004, 3, 59–79. [Google Scholar] [CrossRef]
- Xiao, Q.-Y.; Ye, T.-Y.; Wang, X.-L.; Han, L.; Wang, T.-X.; Qi, D.-M.; Cheng, X.-R.; Wang, S.-Q. A network pharmacology-based study on key pharmacological pathways and targets of Qi Fu Yin acting on Alzheimer’s disease. Exp. Gerontol. 2021, 149, 111336. [Google Scholar] [CrossRef] [PubMed]
- Kołaczkowski, M.; Marcinkowska, M.; Bucki, A.; Pawłowski, M.; Mitka, K.; Jaśkowska, J.; Kowalski, P.; Kazek, G.; Siwek, A.; Wasik, A.; et al. Novel Arylsulfonamide Derivatives with 5-HT6/5-HT7 Receptor Antagonism Targeting Behavioral and Psychological Symptoms of Dementia. J. Med. Chem. 2014, 57, 4543–4557. [Google Scholar] [CrossRef] [PubMed]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Press, B. Permeability for Intestinal Absorption: Caco-2 Assay and Related Issues. Curr. Drug Metab. 2008, 9, 893–900. [Google Scholar] [CrossRef]
- Tetz, L.M.; Kamau, P.W.; Cheng, A.A.; Meeker, J.; Loch-Caruso, R. Troubleshooting the dichlorofluorescein assay to avoid artifacts in measurement of toxicant-stimulated cellular production of reactive oxidant species. J. Pharmacol. Toxicol. Methods 2013, 67, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Committee for Medicinal Products for Human Use; European Medicines Agency. Guideline on bioanalytical method validation. 2011. Available online: https://www.google.com/search?q=Committee+for+Medicinal+Products+for+Human+Use%2C+European+Medicines+Agency.+Guideline+on+bioanalytical+method+validation%2C+2011&client=firefox-b-d&ei=Z8NVYfGxLMntkgX8h6vQBQ&oq=Committee+for+Medicinal+Products+for+Human+Use%2C+European+Medicines+Agency.+Guideline+on+bioanalytical+method+validation%2C+2011&gs_lcp=Cgdnd3Mtd2l6EAMyBwgAEEcQsAMyBwgAEEcQsAMyBwgAEEcQsAMyBwgAEEcQsAMyBwgAEEcQsAMyBwgAEEcQsANKBAhBGABQ3C9Y5TNg0jloAHADeACAAQCIAQCSAQCYAQCgAQHIAQbAAQE&sclient=gws-wiz&ved=0ahUKEwixgZan7qbzAhXJtqQKHfzDCloQ4dUDCA0&uact=5 (accessed on 20 September 2021).
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Zajdel, P.; Kos, T.; Marciniec, K.; Satała, G.; Canale, V.; Kaminski, K.; Holuj, M.; Lenda, T.; Koralewski, R.; Bednarski, M.; et al. Novel multi-target azinesulfonamides of cyclic amine derivatives as potential antipsychotics with pro-social and pro-cognitive effects. Eur. J. Med. Chem. 2018, 145, 790–804. [Google Scholar] [CrossRef]
- Csernansky, J.G.; Martin, M.; Shah, R.; Bertchume, A.; Colvin, J.; Dong, H. Cholinesterase Inhibitors Ameliorate Behavioral Deficits Induced by MK-801 in Mice. Neuropsychopharmacology 2005, 30, 2135–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. Behavioral Despair in Mice: A Primary Screening Test for Antidepressants. Arch. Int. Pharmacodyn. Thér. 1977, 229, 327–336. [Google Scholar]
- Pellow, S.; File, S.E. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze: A novel test of anxiety in the rat. Pharmacol. Biochem. Behav. 1986, 24, 525–529. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
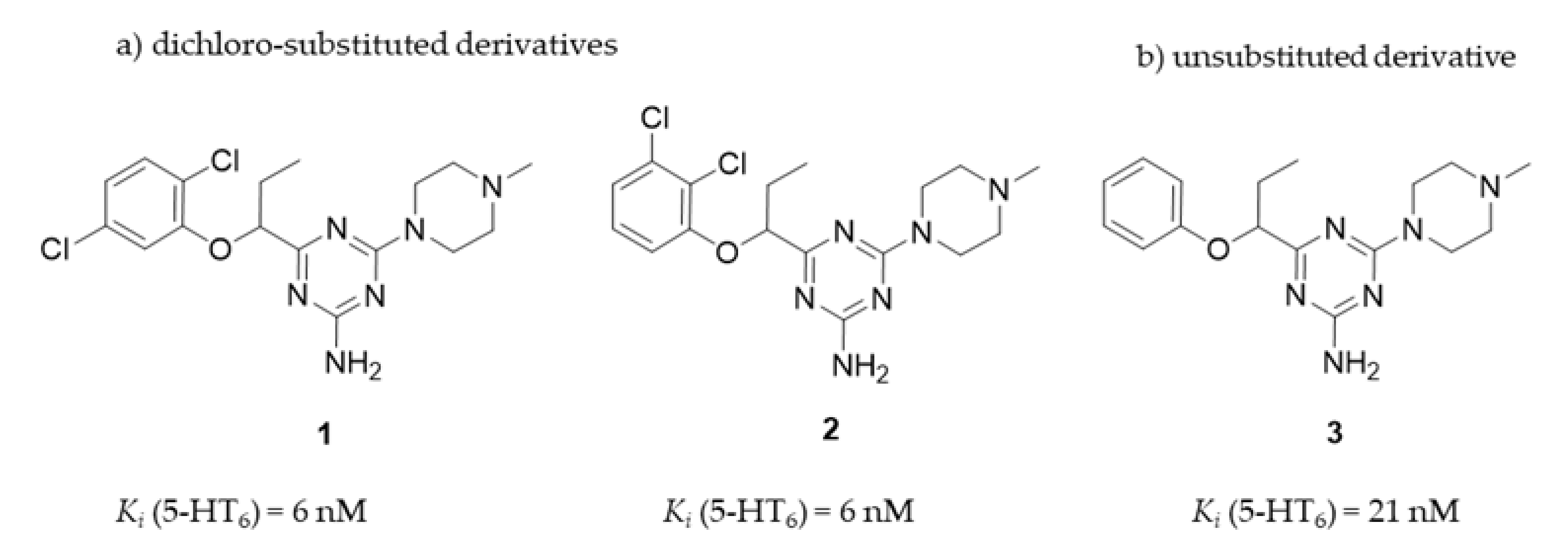
| Cpd | Ki (nM) ± SD | ||||
|---|---|---|---|---|---|
| a 5-HT6 | a D2 | 5-HT1A | a 5-HT2A | a 5-HT7 | |
| 1 2 3 | 6 ± 1 6 ± 2 21 ± 5 | 320 ± 19 421 ± 29 1506 ± 338 | 4760 ± 1191 521 ± 73 3643 ± 816 | 484 ± 77 209 ± 19 5047 ± 752 | 5706 ± 967 5202 ± 1247 19,940 ± 2357 |
| Ref b | 7 ± 1 | 9 ± 2 | - | - | - |
| Target | 5-HT2AR | 5-HT6R | ||
|---|---|---|---|---|
| Compound | Agonist Mode * Emax [%] ± SEM | Antagonist Mode ** pKb ± SEM | Agonist Mode * Emax [%] ± SEM | Antagonist Mode *** pKb ± SEM |
| Serotonin | 100.00 ± 0.30 | N.C. | 100.00 ± 0.5 | N.C. |
| Mianserin | 1.00 ± 0.30 | 9.70 ± 0.12 | 2.00 ± 0.50 | 6.08 ± 0.05 |
| SB258585 | N.T. | N.T. | 2.00 ± 0.50 | 8.92 ± 0.10 |
| 1 | 58.00 ± 9.70 | N.C. | 4.00 ± 0.00 | 10.57 ± 0.07 |
| 2 | 21.00 ± 0.60 | 8.61 ± 0.03 | 5.00 ± 1.10 | 8.19 ± 0.33 |
| 3 | N.T. | N.T. | 9.00 ± 1.40 | 7.67 ± 0.20 |
| Cpd | a,bPe (10−6 cm/s) ± SD |
|---|---|
| CFN | 15.1 ± 0.4 |
| 1 | 18.9 ± 0.9 c |
| 2 | 20.5 ± 5.4 |
| 3 | 15.1 ± 1.0 |
| Cpd | aPapp (10−6 cm/s) ± SD | b Efflux Ratio | |
|---|---|---|---|
| A-B | B-A | ||
| CFN | 15.6 ± 0.55 | 17.9 ± 1.9 | 1.14 |
| 1 | 14.2 ± 0.36 | 22.2 ± 1.92 | 1.56 |
| 2 | 50.3 ± 6.22 | 21.1 ± 2.14 | 0.42 |
| 3 | 3.16 ± 1.35 | 10.1 ± 0.15 | 3.20 |
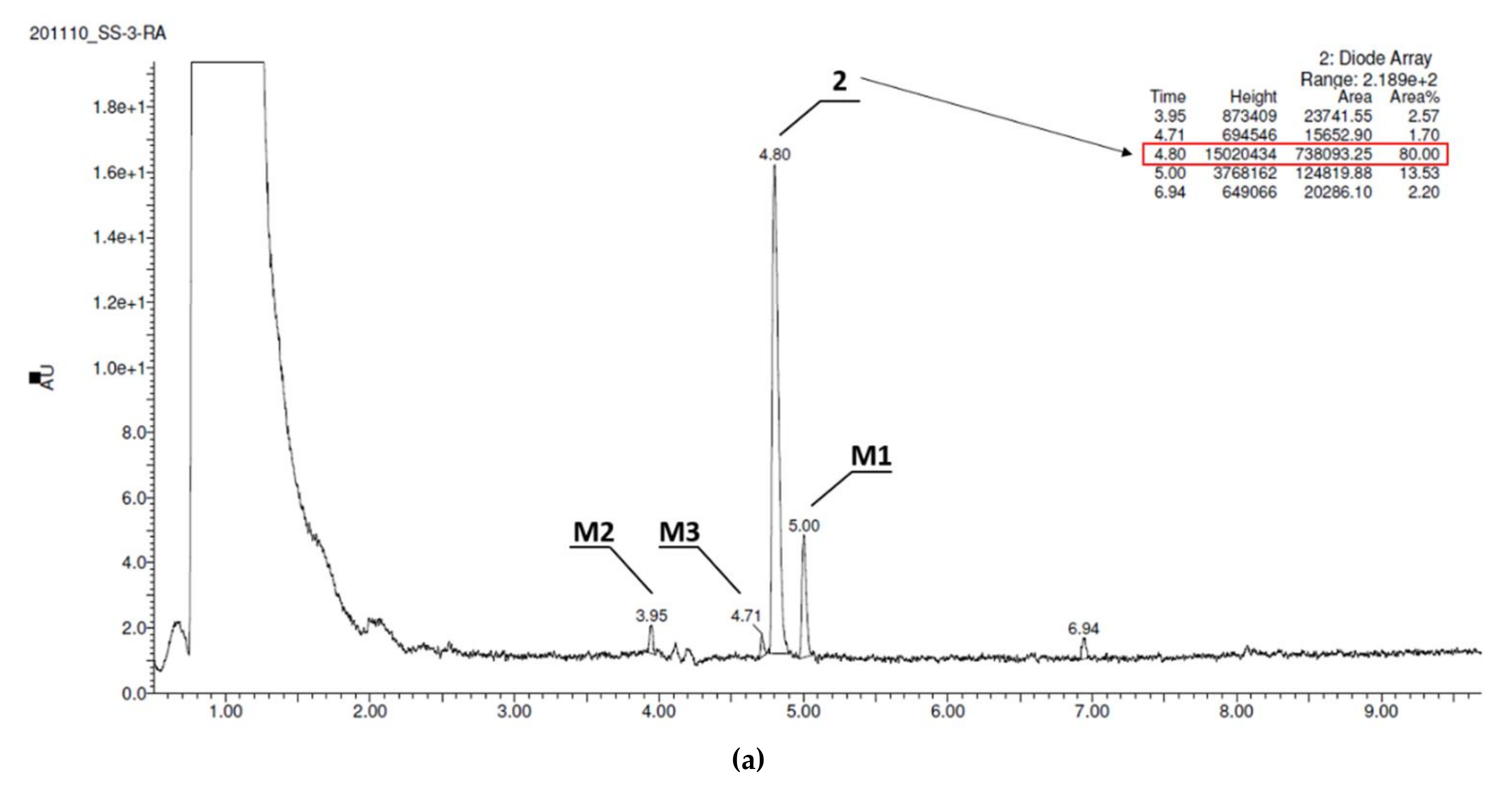
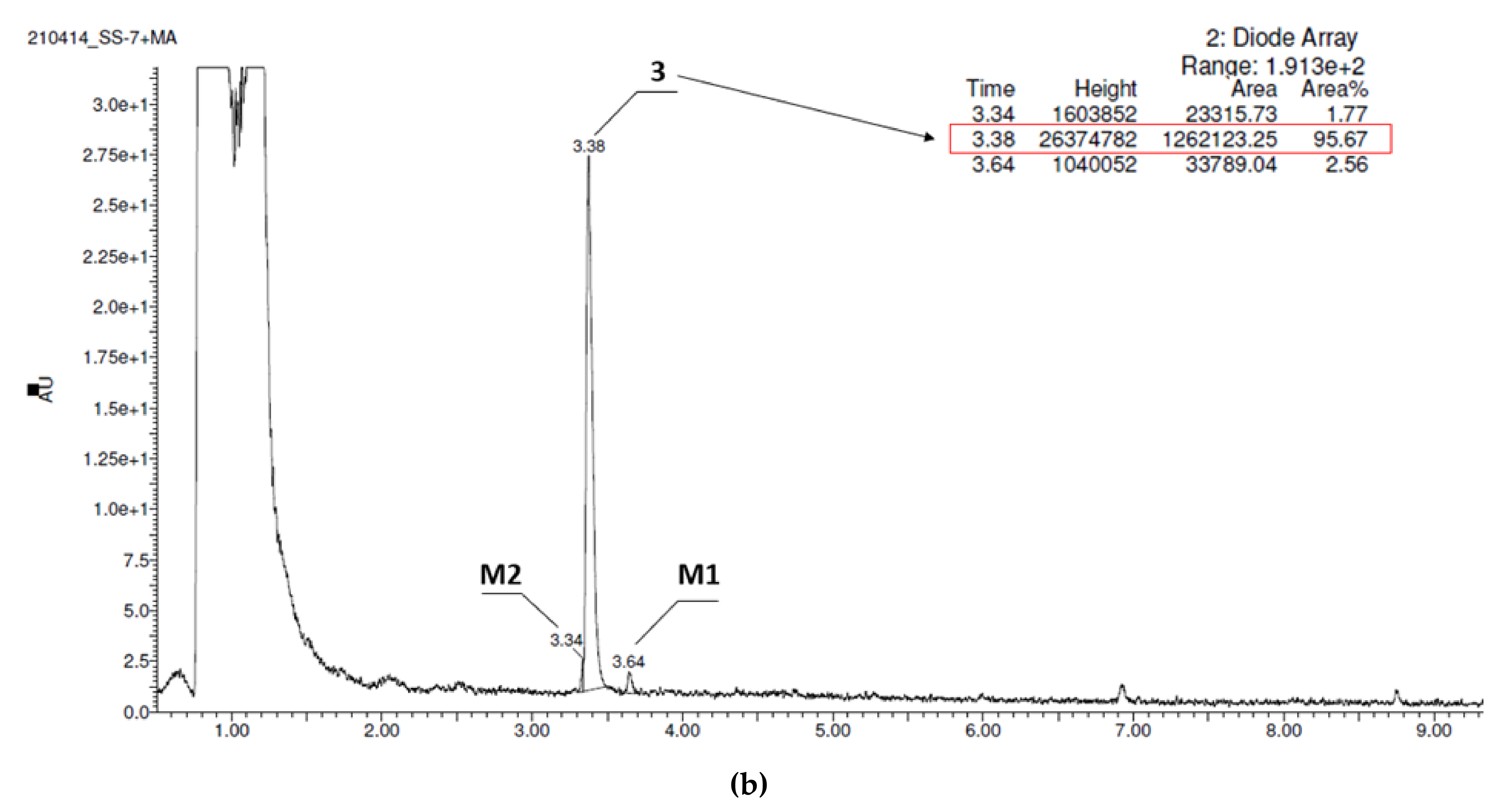
| Substrate | Molecular Mass (m/z) | Amount of Metabolites | Molecular Mass of the Metabolite (m/z) | Content in Reaction Mixture (%) | Metabolic Pathway * |
|---|---|---|---|---|---|
| 2 | 397.31 | 3 | 413.07 (M1) 413.07 (M2) 383.03 (M3) | 13.53 2.57 1.70 | hydroxylation hydroxylation demethylation |
| 3 | 328.42 | 2 | 279.22 (M1) 345.28 (M2) | 2.56 1.77 | decomposition and triple hydroxylation hydroxylation |
| Conc. (µM) | Compound | Fold Increase over MCB a | Binomial B-Value b | Number of Revertants ± SD |
|---|---|---|---|---|
| 0.5 | NQNO | 3.23 | 1.0000 | 48.00 ± 0.00 |
| 1 | 2 | 0.81 | 0.2601 | 12.00 ± 7.55 |
| 10 | 2 | 0.90 | 0.5425 | 13.33 ± 2.31 |
| 0.25 | NQNO | 7.02 | 1.0000 | 40.33 ± 4.04 |
| 1 | 1 | 0.35 | 0.0707 | 2.00 ± 1.00 |
| 10 | 1 | 0.35 | 0.0707 | 2.00 ± 1.00 |
| 1 | 3 | 0.06 | 0.0001 | 0.33 ± 0.58 |
| 10 | 3 | 0.29 | 0.0324 | 1.67 ± 1.53 |
| Pharmacokinetic Parameter 1 | 2 | 3 |
|---|---|---|
| Cmax (ng/mL) | 5.20 ± 2.24 | 6.32 ± 5.03 |
| Tmax (min) | 15 | 5 |
| AUC0-t (ng∙min/mL) | 453.40 ± 145.83 | 423.91 ± 216.75 |
| AUC0-∞ (ng∙min/mL) | 530.79 ± 150.52 | 511.88 ± 262.32 |
| Vz/F (L/kg) | 80.99 a ± 18.22 | 64.81 ± 21.78 |
| CL/F (L/h/kg) | 30.87 ± 9.88 | 35.16 ± 17.92 |
| t1/2λz (min) | 116.18 a ± 17.14 | 76.65 ± 18.07 |
| MRT (min) | 90.08 ± 7.02 | 79.33 ± 6.56 |
| 2 | ||||
|---|---|---|---|---|
| Tissue | Tmax (min) | Cmax (ng/mL or ng/g) | AUC0-t (ng·min/mL or ng·min/g) | Kp |
| Serum | 15 | 5.20 ± 2.24 | 453.40 ± 145.83 | - |
| Brain | 5 | 11.63 ± 3.13 | 1219.94 ± 368.79 | 2.69 |
| Heart | 5 | 8.90 ± 8.15 | 1040.74 ± 448.65 | 2.30 |
| Lungs | 5 | 54.20 ± 21.61 | 3870.66 ± 680.07 | 8.54 |
| Liver * | 15 | 115.22 ± 93.46 | 6523.60 ± 4997.28 | 14.39 |
| Kidneys | 15 | 65.73 ± 34.94 | 7488.06 ± 4372.92 | 16.52 |
| 3 | ||||
| Serum | 5 | 6.32 ± 5.04 | 423.91 ± 216.75 | - |
| Brain | 5 | 11.35 ± 6.93 | 859.08 ± 312.87 | 2.03 |
| Heart | 5 | 21.83 ± 17.16 | 1057.37 ± 285.41 | 2.49 |
| Lungs | 5 | 73.65 ± 63.94 | 2715.09 ± 1000.42 | 6.41 |
| Liver * | 5 | 129.62 ± 106.23 ** | 4467.18 ± 2249.99 | 10.54 |
| Kidneys | 15 | 41.25 ± 8.41 | 2557.08 ± 1084.42 | 6.03 |
| Treatment | Dose (mg/kg) | Total Exploratory Time in T2 Session (s) |
|---|---|---|
| Vehicle + vehicle | 0 + 0 | 58.71 ± 3.38 |
| MK-801 + vehicle | 0.1 + 0 | 42.00 ± 3.13 |
| 2 + MK-801 | 0.3 + 0.1 | 52.67 ± 5.37 |
| 1 + 0.1 | 40.33 ± 6.66 | |
| 3 + 0.1 | 42.88 ± 2.10 F(4,27) = 3.5450; p < 0.05 | |
| vehicle | 0 + 0 | 45.00 ± 4.30 |
| MK-801 + vehicle | 0.1 + 0 | 47.07 ± 3.89 |
| 3 + MK-801 | 0.1 + 0.1 | 47.33 ± 4.79 |
| 0.3 + 0.1 | 38.14 ± 4.18 | |
| 1 + 0.1 | 35.57 ± 3.76 | |
| 3 + 0.1 | 34.14 ± 1.22; F(5,47) = 1.9394; NS |
| Treatment | Dose (mg/kg) | Total Distance (cm) | X Ambulation | Y Ambulation |
|---|---|---|---|---|
| Vehicle | 0 | 4600 ± 140 | 175 ± 7 | 104 ± 10 |
| 1 | 1 | 4520 ± 168 | 172 ± 6 | 99 ± 6 |
| 3 | 3995 ± 218 | 146 ± 10 | 82 ± 7 | |
| 10 | 3681 ± 256; p < 0.05 F(3,24) = 4.5291; p < 0.05 | 140 ± 11 F(3,24) = 3.8035; p < 0.05 | 83 ± 8 F(3,24) = 1.9215; NS | |
| Vehicle | 0 | 3639 ± 327 | 137 ± 19 | 81 ± 12 |
| 2 | 0.3 | 4372 ± 132 | 180 ± 9 | 101 ± 9 |
| 1 | 3987 ± 281 | 149 ± 18 | 78 ± 9 | |
| 3 | 4595± 191 F(3,23) = 2.9772; NS | 168 ± 6 F(3,23) = 1.9417; NS | 115 ± 7 F(3,23) = 3.1727; NS | |
| Vehicle | 0 | 3639 ± 327 | 137 ± 19 | 81 ± 12 |
| 3 | 0.3 | 4033 ± 340 | 151 ± 16 | 80 ± 8 |
| 1 | 4102 ± 291 | 136 ± 15 | 86 ± 10 | |
| 3 | 3545 ± 136 F(3,22) = 0.9071; NS | 123 ± 5 F(3,22) = 0.6336; NS | 73 ± 5 F(3,22) = 0.3805; NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudoł, S.; Cios, A.; Jastrzębska-Więsek, M.; Honkisz-Orzechowska, E.; Mordyl, B.; Wilczyńska-Zawal, N.; Satała, G.; Kucwaj-Brysz, K.; Partyka, A.; Latacz, G.; et al. The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. Int. J. Mol. Sci. 2021, 22, 10773. https://doi.org/10.3390/ijms221910773
Sudoł S, Cios A, Jastrzębska-Więsek M, Honkisz-Orzechowska E, Mordyl B, Wilczyńska-Zawal N, Satała G, Kucwaj-Brysz K, Partyka A, Latacz G, et al. The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. International Journal of Molecular Sciences. 2021; 22(19):10773. https://doi.org/10.3390/ijms221910773
Chicago/Turabian StyleSudoł, Sylwia, Agnieszka Cios, Magdalena Jastrzębska-Więsek, Ewelina Honkisz-Orzechowska, Barbara Mordyl, Natalia Wilczyńska-Zawal, Grzegorz Satała, Katarzyna Kucwaj-Brysz, Anna Partyka, Gniewomir Latacz, and et al. 2021. "The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases" International Journal of Molecular Sciences 22, no. 19: 10773. https://doi.org/10.3390/ijms221910773
APA StyleSudoł, S., Cios, A., Jastrzębska-Więsek, M., Honkisz-Orzechowska, E., Mordyl, B., Wilczyńska-Zawal, N., Satała, G., Kucwaj-Brysz, K., Partyka, A., Latacz, G., Olejarz-Maciej, A., Wesołowska, A., & Handzlik, J. (2021). The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. International Journal of Molecular Sciences, 22(19), 10773. https://doi.org/10.3390/ijms221910773