
Cofilin Signaling in the CNS Physiology and Neurodegeneration



Abstract
:1. Introduction
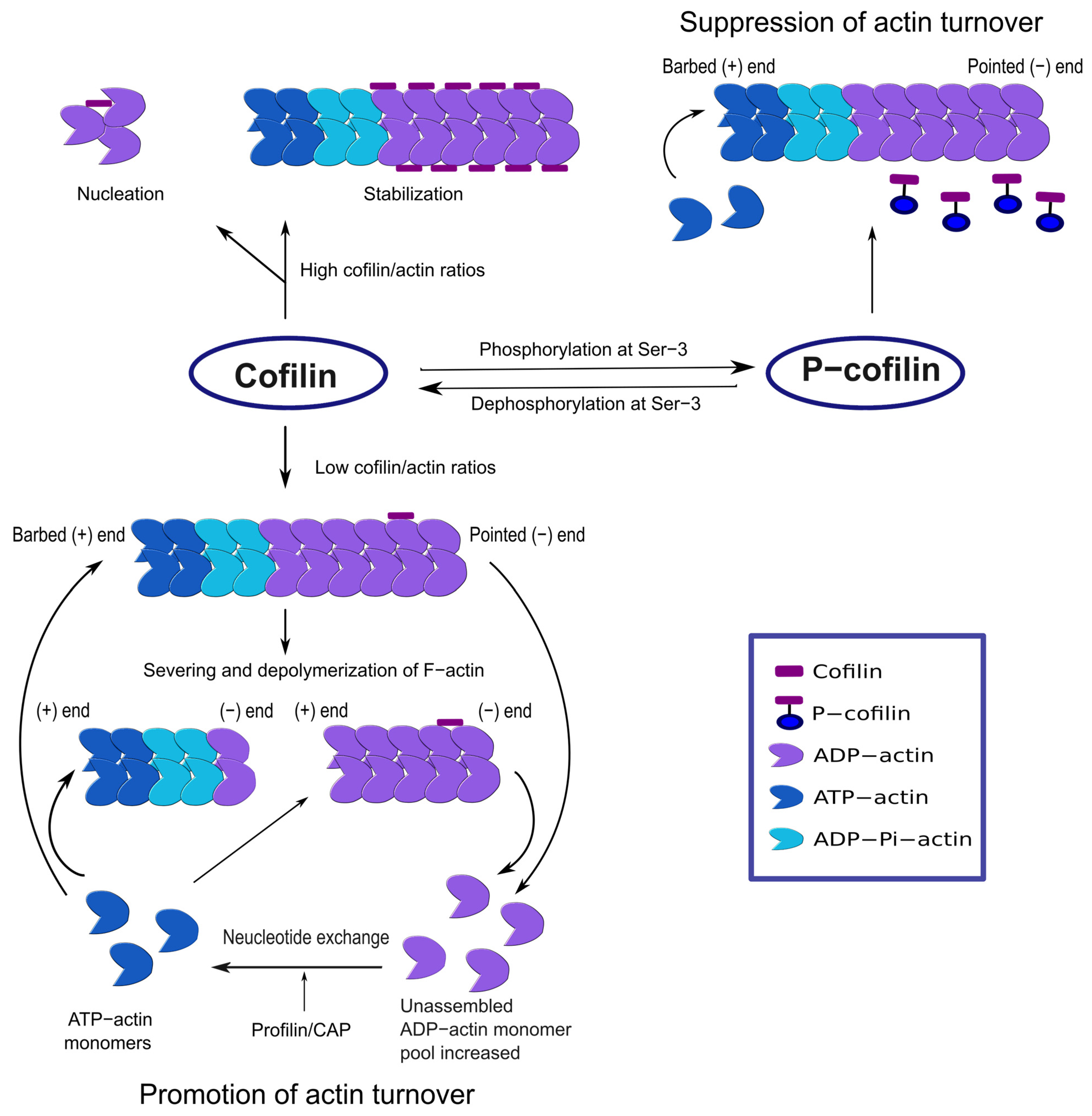
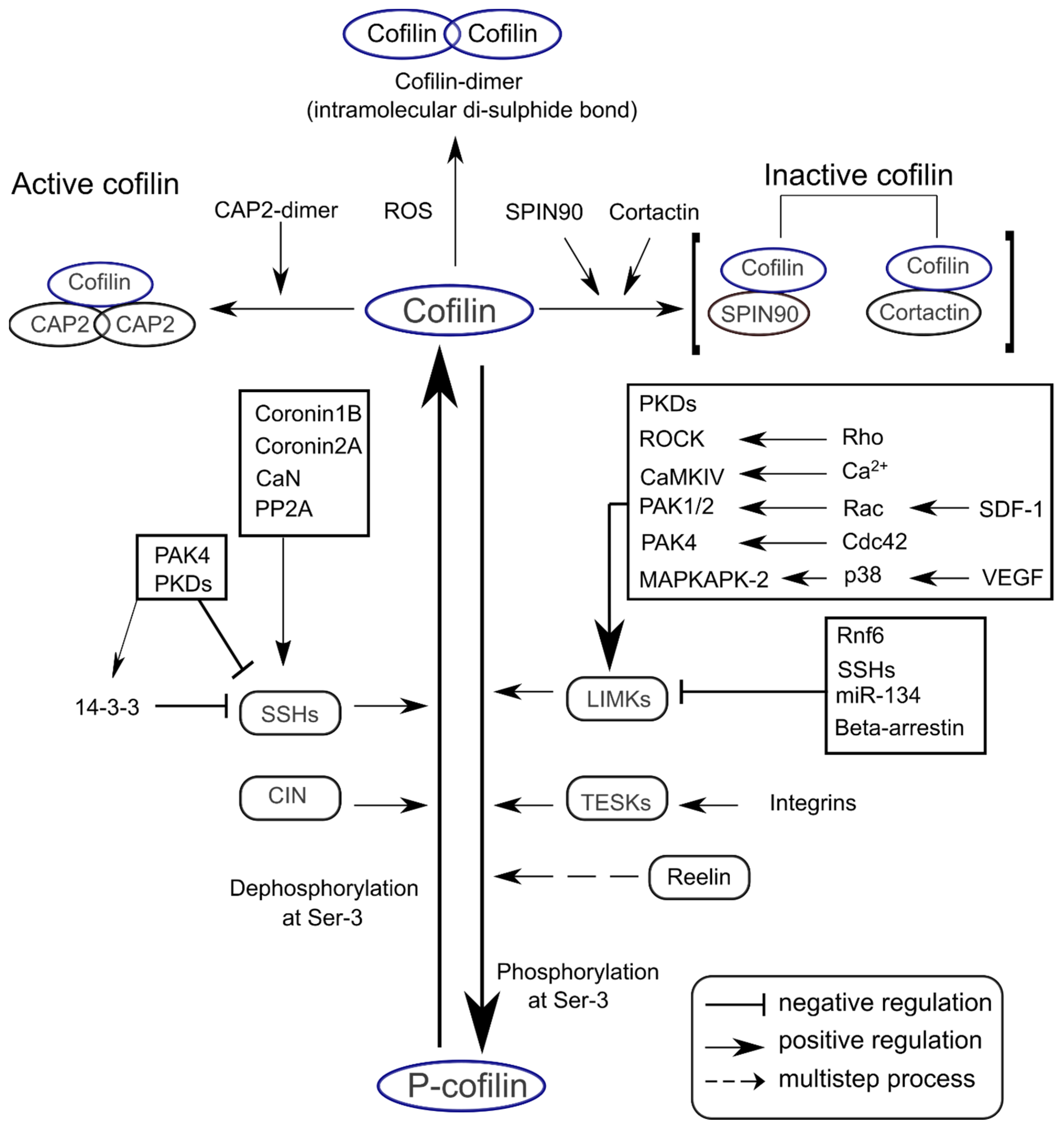
2. Signaling Mechanisms for Cofilin Activation and Inactivation
3. Cofilin Functions in the CNS Development
3.1. Neural Tube Morphogenesis
3.2. Neurite Formation
3.3. Synaptic Plasticity
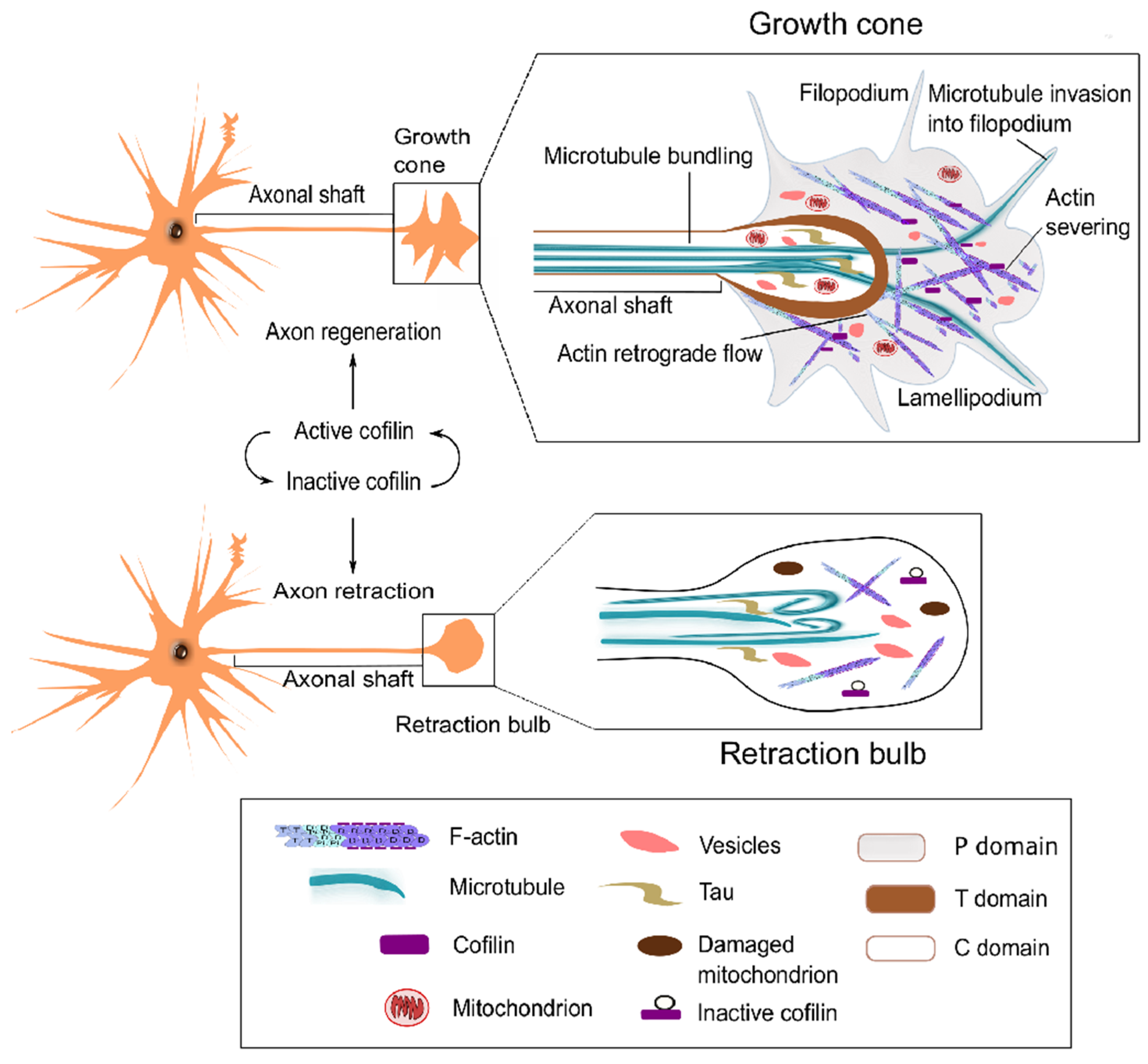
3.4. Axon Regeneration
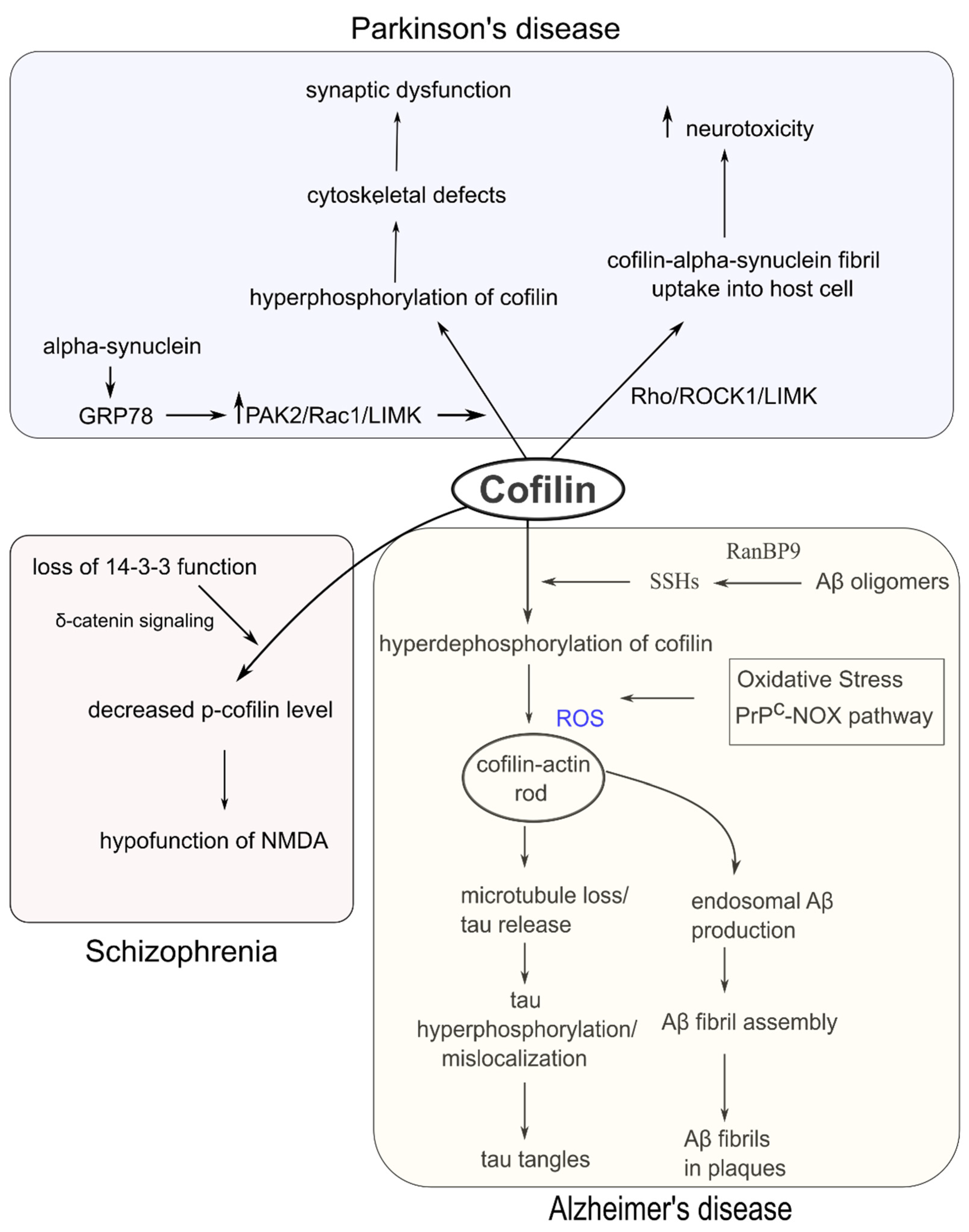
4. Cofilin Dysregulation and Neurodegenerative and Psychiatric Disorders
4.1. Alzheimer’s Disease (AD)
4.2. Schizophrenia
4.3. Ischemic and Hemorrhagic Stroke
4.4. Parkinson’s Disease (PD)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Bamburg, J.R. Proteins of the ADF/Cofilin Family: Essential Regulators of Actin Dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef] [PubMed]
- Maciver, S.K.; Hussey, P.J. The ADF/Cofilin Family: Actin-Remodeling Proteins. Genome Biol. 2002, 3, reviews3007.1. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Harris, H.E.; Weeds, A.G. Partial Purification and Characterization of an Actin Depolymerizing Factor from Brain. FEBS Lett. 1980, 121, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, S.; Nishida, E.; Ohta, Y.; Sakai, H. Isolation of Low Molecular Weight Actin-Binding Proteins from Porcine Brain1. J. Biochem. 1984, 95, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Maekawa, S.; Sakai, H. Cofilin, a Protein in Porcine Brain That Binds to Actin Filaments and Inhibits Their Interactions with Myosin and Tropomyosin. Biochemistry 1984, 23, 5307–5313. [Google Scholar] [CrossRef]
- Ono, S.; Minami, N.; Abe, H.; Obinata, T. Characterization of a Novel Cofilin Isoform That Is Predominantly Expressed in Mammalian Skeletal Muscle. J. Biol. Chem. 1994, 269, 15280–15286. [Google Scholar] [CrossRef]
- Blanchoin, L.; Pollard, T.D. Mechanism of Interaction of Acanthamoeba Actophorin (ADF/Cofilin) with Actin Filaments. J. Biol. Chem. 1999, 274, 15538–15546. [Google Scholar] [CrossRef] [Green Version]
- Hayden, S.M.; Miller, P.S.; Brauweiler, A.; Bamburg, J.R. Analysis of the Interactions of Actin Depolymerizing Factor with G-and F-Actin. Biochemistry 1993, 32, 9994–10004. [Google Scholar] [CrossRef]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin Changes the Twist of F-Actin: Implications for Actin Filament Dynamics and Cellular Function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative Stimuli Induce Persistent ADF/Cofilin–Actin Rods That Disrupt Distal Neurite Function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [CrossRef]
- Carlier, M.F.; Laurent, V.; Santolini, J.; Melki, R.; Didry, D.; Xia, G.X.; Hong, Y.; Chua, N.H.; Pantaloni, D. Actin Depolymerizing Factor (ADF/Cofilin) Enhances the Rate of Filament Turnover: Implication in Actin-Based Motility. J. Cell Biol. 1997, 136, 1307–1322. [Google Scholar] [CrossRef]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef]
- Galkin, V.E.; Orlova, A.; Kudryashov, D.S.; Solodukhin, A.; Reisler, E.; Schröder, G.F.; Egelman, E.H. Remodeling of Actin Filaments by ADF/Cofilin Proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 20568–20572. [Google Scholar] [CrossRef] [Green Version]
- De La Cruz, E.M. Cofilin Binding to Muscle and Non-Muscle Actin Filaments: Isoform-Dependent Cooperative Interactions. J. Mol. Biol. 2005, 346, 557–564. [Google Scholar] [CrossRef]
- Wioland, H.; Guichard, B.; Senju, Y.; Myram, S.; Lappalainen, P.; Jégou, A.; Romet-Lemonne, G. ADF/Cofilin Accelerates Actin Dynamics by Severing Filaments and Promoting Their Depolymerization at Both Ends. Curr. Biol. 2017, 27, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Meyer, G.; Feldman, E.L. Signaling Mechanisms That Regulate Actin-based Motility Processes in the Nervous System. J. Neurochem. 2002, 83, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Samstag, Y.; John, I.; Wabnitz, G.H. Cofilin: A Redox Sensitive Mediator of Actin Dynamics during T-Cell Activation and Migration. Immunol. Rev. 2013, 256, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsick, B.M.; Flynn, K.C.; Santiago-Medina, M.; Bamburg, J.R.; Letourneau, P.C. Activation of ADF/Cofilin Mediates Attractive Growth Cone Turning toward Nerve Growth Factor and Netrin-1. Dev. Neurobiol. 2010, 70, 565–588. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Kim, B.; van Golen, C.; Feldman, E.L. Cofilin Activity during Insulin-like Growth Factor I-Stimulated Neuroblastoma Cell Motility. Cell. Mol. Life Sci. 2005, 62, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilve, S.; Difato, F.; Chieregatti, E. Cofilin 1 Activation Prevents the Defects in Axon Elongation and Guidance Induced by Extracellular Alpha-Synuclein. Sci. Rep. 2015, 5, 16524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamburg, J.R.; Bernstein, B.W. Roles of ADF/Cofilin in Actin Polymerization and Beyond. F1000 Biol. Rep. 2010, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; DerMardirossian, C.; Bokoch, G.M. Cofilin Phosphatases and Regulation of Actin Dynamics. Curr. Opin. Cell Biol. 2006, 18, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K. Signaling Mechanisms and Functional Roles of Cofilin Phosphorylation and Dephosphorylation. Cell. Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Ostrowska, Z.; Moraczewska, J. Cofilin—A Protein Controlling Dynamics of Actin Filaments. Postepy Hig. Med. Dosw. (Online) 2017, 71, 339–351. [Google Scholar] [CrossRef]
- Sarmiere, P.D.; Bamburg, J.R. Regulation of the Neuronal Actin Cytoskeleton by ADF/Cofilin. J. Neurobiol. 2004, 58, 103–117. [Google Scholar] [CrossRef]
- Van Troys, M.; Huyck, L.; Leyman, S.; Dhaese, S.; Vandekerkhove, J.; Ampe, C. Ins and Outs of ADF/Cofilin Activity and Regulation. Eur. J. Cell Biol. 2008, 87, 649–667. [Google Scholar] [CrossRef]
- Klejnot, M.; Gabrielsen, M.; Cameron, J.; Mleczak, A.; Talapatra, S.K.; Kozielski, F.; Pannifer, A.; Olson, M.F. Analysis of the Human Cofilin 1 Structure Reveals Conformational Changes Required for Actin Binding. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1780–1788. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, K.; Iida, K.; Yahara, I. Phosphorylation of Ser-3 of Cofilin Regulates Its Essential Function on Actin. Genes Cells 1996, 1, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of Actin Reorganization by Slingshot, a Family of Phosphatases That Dephosphorylate ADF/Cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shibasaki, F.; Mizuno, K. Calcium Signal-Induced Cofilin Dephosphorylation Is Mediated by Slingshot via Calcineurin. J. Biol. Chem. 2005, 280, 12683–12689. [Google Scholar] [CrossRef] [Green Version]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of Actin Dynamics through Phosphorylation of Cofilin by LIM-Kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin Phosphorylation by LIM-Kinase 1 and Its Role in Rac-Mediated Actin Reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Kurita, S.; Gunji, E.; Ohashi, K.; Mizuno, K. Actin Filaments-stabilizing And-bundling Activities of Cofilin-phosphatase Slingshot-1. Genes Cells 2007, 12, 663–676. [Google Scholar] [CrossRef]
- Cai, L.; Marshall, T.W.; Uetrecht, A.C.; Schafer, D.A.; Bear, J.E. Coronin 1B Coordinates Arp2/3 Complex and Cofilin Activities at the Leading Edge. Cell 2007, 128, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Marshall, T.W.; Aloor, H.L.; Bear, J.E. Coronin 2A Regulates a Subset of Focal-Adhesion-Turnover Events through the Cofilin Pathway. J. Cell Sci. 2009, 122, 3061–3069. [Google Scholar] [CrossRef] [Green Version]
- Nagata-Ohashi, K.; Ohta, Y.; Goto, K.; Chiba, S.; Mori, R.; Nishita, M.; Ohashi, K.; Kousaka, K.; Iwamatsu, A.; Niwa, R. A Pathway of Neuregulin-Induced Activation of Cofilin-Phosphatase Slingshot and Cofilin in Lamellipodia. J. Cell Biol. 2004, 165, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.C.; Zivraj, K.H.; Strochlic, L.; Holt, C.E. 14-3-3 Proteins Regulate Retinal Axon Growth by Modulating ADF/Cofilin Activity. Dev. Neurobiol. 2012, 72, 600–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiseler, T.; Döppler, H.; Yan, I.K.; Kitatani, K.; Mizuno, K.; Storz, P. Protein Kinase D1 Regulates Cofilin-Mediated F-Actin Reorganization and Cell Motility through Slingshot. Nat. Cell Biol. 2009, 11, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Ryu, H.J.; Kim, M.-J.; Kim, D.-W.; Kwon, O.-S.; Choi, S.Y.; Kang, T.-C. Pyridoxal-5′-Phosphate Phosphatase/Chronophin Induces Astroglial Apoptosis via Actin-Depolymerizing Factor/Cofilin System in the Rat Brain Following Status Epilepticus. Glia 2010, 58, 1937–1948. [Google Scholar] [CrossRef]
- Kim, J.-E.; Kim, D.-W.; Kwak, S.-E.; Kwon, O.-S.; Choi, S.-Y.; Kang, T.-C. Potential Role of Pyridoxal-5′-Phosphate Phosphatase/Chronopin in Epilepsy. Exp. Neurol. 2008, 211, 128–140. [Google Scholar] [CrossRef]
- Medina, C.; de la Fuente, V.; Tom Dieck, S.; Nassim-Assir, B.; Dalmay, T.; Bartnik, I.; Lunardi, P.; de Oliveira Alvares, L.; Schuman, E.M.; Letzkus, J.J.; et al. LIMK, Cofilin 1 and Actin Dynamics Involvement in Fear Memory Processing. Neurobiol. Learn Mem. 2020, 173, 107275. [Google Scholar] [CrossRef]
- Dan, C.; Kelly, A.; Bernard, O.; Minden, A. Cytoskeletal Changes Regulated by the PAK4 Serine/Threonine Kinase Are Mediated by LIM Kinase 1 and Cofilin. J. Biol. Chem. 2001, 276, 32115–32121. [Google Scholar] [CrossRef] [Green Version]
- Toshima, J.; Toshima, J.Y.; Amano, T.; Yang, N.; Narumiya, S.; Mizuno, K. Cofilin Phosphorylation by Protein Kinase Testicular Protein Kinase 1 and Its Role in Integrin-Mediated Actin Reorganization and Focal Adhesion Formation. MBoC 2001, 12, 1131–1145. [Google Scholar] [CrossRef] [Green Version]
- Toshima, J.; Toshima, J.Y.; Takeuchi, K.; Mori, R.; Mizuno, K. Cofilin Phosphorylation and Actin Reorganization Activities of Testicular Protein Kinase 2 and Its Predominant Expression in Testicular Sertoli Cells. J. Biol. Chem. 2001, 276, 31449–31458. [Google Scholar] [CrossRef] [Green Version]
- Chai, X.; Förster, E.; Zhao, S.; Bock, H.H.; Frotscher, M. Reelin Stabilizes the Actin Cytoskeleton of Neuronal Processes by Inducing N-Cofilin Phosphorylation at Serine3. J. Neurosci. 2009, 29, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.T.; Zhao, S.; Chai, X.; Brunne, B.; Bouché, E.; Bock, H.H.; Frotscher, M. Role for Reelin-Induced Cofilin Phosphorylation in the Assembly of Sympathetic Preganglionic Neurons in the Murine Intermediolateral Column. Eur. J. Neurosci. 2010, 32, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.H.; Lee, M.J.; Kim, D.H.; Kim, B.; Bae, J.; Choi, K.Y.; Kim, S.-M.; Huh, Y.H.; Lee, K.H.; Kim, C.-H.; et al. SPIN90 Dephosphorylation Is Required for Cofilin-Mediated Actin Depolymerization in NMDA-Stimulated Hippocampal Neurons. Cell Mol. Life Sci. 2013, 70, 4369–4383. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.; Yamaguchi, H.; Mader, C.C.; Bravo-Cordero, J.J.; Arias, M.; Chen, X.; Desmarais, V.; van Rheenen, J.; Koleske, A.J.; Condeelis, J. Cortactin Regulates Cofilin and N-WASp Activities to Control the Stages of Invadopodium Assembly and Maturation. J. Cell Biol. 2009, 186, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Kotila, T.; Wioland, H.; Enkavi, G.; Kogan, K.; Vattulainen, I.; Jégou, A.; Romet-Lemonne, G.; Lappalainen, P. Mechanism of Synergistic Actin Filament Pointed End Depolymerization by Cyclase-Associated Protein and Cofilin. Nat. Commun. 2019, 10, 5320. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Ohashi, K.; Sasaki, Y.; Goshima, Y.; Niwa, R.; Uemura, T.; Mizuno, K. Control of Growth Cone Motility and Morphology by LIM Kinase and Slingshot via Phosphorylation and Dephosphorylation of Cofilin. J. Neurosci. 2003, 23, 2527–2537. [Google Scholar] [CrossRef]
- Kurita, S.; Watanabe, Y.; Gunji, E.; Ohashi, K.; Mizuno, K. Molecular Dissection of the Mechanisms of Substrate Recognition and F-Actin-Mediated Activation of Cofilin-Phosphatase Slingshot-1. J. Biol. Chem. 2008, 283, 32542–32552. [Google Scholar] [CrossRef] [Green Version]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between Components of a Novel LIM Kinase–Slingshot Phosphatase Complex Regulates Cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Singla, B.; Lin, H.-P.; Ghoshal, P.; Cherian-Shaw, M.; Csányi, G. PKCδ Stimulates Macropinocytosis via Activation of SSH1-Cofilin Pathway. Cell. Signal. 2019, 53, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Halasz, E.; Townes-Anderson, E. Actin Dynamics, Regulated by RhoA-LIMK-Cofilin Signaling, Mediates Rod Photoreceptor Axonal Retraction After Retinal Injury. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2274–2285. [Google Scholar] [CrossRef]
- Lou, D.; Sun, B.; Wei, H.; Deng, X.; Chen, H.; Xu, D.; Li, G.; Xu, H.; Wang, Y. Spatiotemporal Expression of Testicular Protein Kinase 1 after Rat Sciatic Nerve Injury. J. Mol. Neurosci. 2012, 47, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Buckley, A.F.; Spurney, R.F. Regulation of Cofilin Phosphorylation in Glomerular Podocytes by Testis Specific Kinase 1 (TESK1). Sci. Rep. 2018, 8, 12286. [Google Scholar] [CrossRef] [PubMed]
- Ono, S. The Role of Cyclase-Associated Protein in Regulating Actin Filament Dynamics—More than a Monomer-Sequestration Factor. J. Cell Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schröder, R.; Schleicher, M.; Holak, T.A.; Clemen, C.S.; Ramanath-Y, B.; Pfitzer, G.; et al. CAP2, Cyclase-Associated Protein 2, Is a Dual Compartment Protein. Cell Mol. Life Sci. 2007, 64, 2702–2715. [Google Scholar] [CrossRef]
- Kumar, A.; Paeger, L.; Kosmas, K.; Kloppenburg, P.; Noegel, A.A.; Peche, V.S. Neuronal Actin Dynamics, Spine Density and Neuronal Dendritic Complexity Are Regulated by CAP2. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Cameron, J.M.; Gabrielsen, M.; Chim, Y.H.; Munro, J.; McGhee, E.J.; Sumpton, D.; Eaton, P.; Anderson, K.I.; Yin, H.; Olson, M.F. Polarized Cell Motility Induces Hydrogen Peroxide to Inhibit Cofilin via Cysteine Oxidation. Curr. Biol. 2015, 25, 1520–1525. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.W.; Shaw, A.E.; Minamide, L.S.; Pak, C.W.; Bamburg, J.R. Incorporation of Cofilin into Rods Depends on Disulfide Intermolecular Bonds: Implications for Actin Regulation and Neurodegenerative Disease. J. Neurosci. 2012, 32, 6670–6681. [Google Scholar] [CrossRef]
- Pfannstiel, J.; Cyrklaff, M.; Habermann, A.; Stoeva, S.; Griffiths, G.; Shoeman, R.; Faulstich, H. Human Cofilin Forms Oligomers Exhibiting Actin Bundling Activity. J. Biol. Chem. 2001, 276, 49476–49484. [Google Scholar] [CrossRef] [Green Version]
- Yuskaitis, C.J.; Pomeroy, S.L. Development of the Nervous System. In Fetal and Neonatal Physiology; Elsevier: Philadelphia, PA, USA, 2017; pp. 1294–1313.e2. ISBN 978-0-323-35214-7. [Google Scholar]
- Esteves, M.; Almeida, A.; Leite-Almeida, H. Insights on nervous system biology and anatomy. In Handbook of Innovations in Central Nervous System Regenerative Medicine; Elsevier: Philadelphia, PA, USA, 2020; pp. 1–28. ISBN 978-0-12-818084-6. [Google Scholar]
- Gurniak, C.B.; Perlas, E.; Witke, W. The Actin Depolymerizing Factor N-Cofilin Is Essential for Neural Tube Morphogenesis and Neural Crest Cell Migration. Dev. Biol. 2005, 278, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Escuin, S.; Vernay, B.; Savery, D.; Gurniak, C.B.; Witke, W.; Greene, N.D.E.; Copp, A.J. Rho-Kinase-Dependent Actin Turnover and Actomyosin Disassembly Are Necessary for Mouse Spinal Neural Tube Closure. J. Cell Sci. 2015, 128, 2468–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grego-Bessa, J.; Hildebrand, J.; Anderson, K.V. Morphogenesis of the Mouse Neural Plate Depends on Distinct Roles of Cofilin 1 in Apical and Basal Epithelial Domains. Development 2015, 142, 1305–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; García-García, M.J. Secretory Pathway Calcium ATPase 1 (SPCA1) Controls Mouse Neural Tube Closure by Regulating Cytoskeletal Dynamics. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, E.W.; Kalil, K. Axon Branching Requires Interactions between Dynamic Microtubules and Actin Filaments. J. Neurosci. 2001, 21, 9757–9769. [Google Scholar] [CrossRef]
- Mallavarapu, A.; Mitchison, T. Regulated Actin Cytoskeleton Assembly at Filopodium Tips Controls Their Extension and Retraction. J. Cell Biol. 1999, 146, 1097–1106. [Google Scholar] [CrossRef]
- Star, E.N.; Kwiatkowski, D.J.; Murthy, V.N. Rapid Turnover of Actin in Dendritic Spines and Its Regulation by Activity. Nat. Neurosci. 2002, 5, 239–246. [Google Scholar] [CrossRef]
- Fass, J.; Gehler, S.; Sarmiere, P.; Letourneau, P.; Bamburg, J.R. Regulating Filopodial Dynamics through Actin-Depolymerizing Factor/Cofilin. Anato. Sci. Int. 2004, 79, 173. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Wakatsuki, S.; Ishii, A.; Moriyama, K.; Sasaki, Y.; Ohashi, K.; Sekine-Aizawa, Y.; Sehara-Fujisawa, A.; Mizuno, K.; Goshima, Y. Phosphorylation of Cofilin by LIM-Kinase Is Necessary for Semaphorin 3A-Induced Growth Cone Collapse. Nat. Neurosci. 2001, 4, 367–373. [Google Scholar] [CrossRef]
- Montani, L.; Gerrits, B.; Gehrig, P.; Kempf, A.; Dimou, L.; Wollscheid, B.; Schwab, M.E. Neuronal Nogo-A Modulates Growth Cone Motility via Rho-GTP/LIMK1/Cofilin in the Unlesioned Adult Nervous System. J. Biol. Chem. 2009, 284, 10793–10807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, K.C.; Hellal, F.; Neukirchen, D.; Jacob, S.; Tahirovic, S.; Dupraz, S.; Stern, S.; Garvalov, B.K.; Gurniak, C.; Shaw, A.E. ADF/Cofilin-Mediated Actin Retrograde Flow Directs Neurite Formation in the Developing Brain. Neuron 2012, 76, 1091–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, K.C.; Pak, C.W.; Shaw, A.E.; Bradke, F.; Bamburg, J.R. Growth Cone-like Waves Transport Actin and Promote Axonogenesis and Neurite Branching. Dev. Neurobiol. 2009, 69, 761–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, L.A.; Goda, Y. Actin in Action: The Interplay between the Actin Cytoskeleton and Synaptic Efficacy. Nat. Rev. Neurosci. 2008, 9, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and Molecular Remodeling of Dendritic Spine Substructures during Long-Term Potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef] [Green Version]
- Rex, C.S.; Chen, L.Y.; Sharma, A.; Liu, J.; Babayan, A.H.; Gall, C.M.; Lynch, G. Different Rho GTPase-Dependent Signaling Pathways Initiate Sequential Steps in the Consolidation of Long-Term Potentiation. J. Cell Biol. 2009, 186, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R. ADF/Cofilin-Mediated Actin Dynamics Regulate AMPA Receptor Trafficking during Synaptic Plasticity. Nat. Neurosci. 2010, 13, 1208. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.B.; Gurniak, C.B.; Renner, M.; Vara, H.; Morando, L.; Görlich, A.; Sassoè-Pognetto, M.; Banchaabouchi, M.A.; Giustetto, M.; Triller, A. Learning, AMPA Receptor Mobility and Synaptic Plasticity Depend on N-cofilin-mediated Actin Dynamics. EMBO J. 2010, 29, 1889–1902. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Homma, K.J.; Poo, M. Shrinkage of Dendritic Spines Associated with Long-Term Depression of Hippocampal Synapses. Neuron 2004, 44, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Malinow, R.; Malenka, R.C. AMPA Receptor Trafficking and Synaptic Plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dong, Q.; Xu, X.-F.; Feng, X.; Xin, J.; Wang, D.-D.; Yu, H.; Tian, T.; Chen, Z.-Y. Phosphorylation of Cofilin Regulates Extinction of Conditioned Aversive Memory via AMPAR Trafficking. J. Neurosci. 2013, 33, 6423–6433. [Google Scholar] [CrossRef]
- Hilton, B.J.; Bradke, F. Can Injured Adult CNS Axons Regenerate by Recapitulating Development? Development 2017, 144, 3417–3429. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Caroni, P. Mechanisms of Axon Degeneration: From Development to Disease. Prog. Neurobiol. 2007, 83, 174–191. [Google Scholar] [CrossRef]
- Hill, C.E.; Beattie, M.S.; Bresnahan, J.C. Degeneration and Sprouting of Identified Descending Supraspinal Axons after Contusive Spinal Cord Injury in the Rat. Exp. Neurol. 2001, 171, 153–169. [Google Scholar] [CrossRef]
- Stern, S.; Haverkamp, S.; Sinske, D.; Tedeschi, A.; Naumann, U.; Giovanni, S.D.; Kochanek, S.; Nordheim, A.; Knöll, B. The Transcription Factor Serum Response Factor Stimulates Axon Regeneration through Cytoplasmic Localization and Cofilin Interaction. J. Neurosci. 2013, 33, 18836–18848. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Douglas, M.R.; Read, M.L.; Berry, M.; Logan, A. Citron Kinase Regulates Axon Growth through a Pathway That Converges on Cofilin Downstream of RhoA. Neurobiol. Dis. 2011, 41, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, A.; Dupraz, S.; Curcio, M.; Laskowski, C.J.; Schaffran, B.; Flynn, K.C.; Santos, T.E.; Stern, S.; Hilton, B.J.; Larson, M.J.E.; et al. ADF/Cofilin-Mediated Actin Turnover Promotes Axon Regeneration in the Adult CNS. Neuron 2019, 103, 1073–1085.e6. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Zimmermann, A.-M.; Görlich, A.; Gurniak, C.B.; Sassoè-Pognetto, M.; Friauf, E.; Witke, W.; Rust, M.B. ADF/Cofilin Controls Synaptic Actin Dynamics and Regulates Synaptic Vesicle Mobilization and Exocytosis. Cereb Cortex 2015, 25, 2863–2875. [Google Scholar] [CrossRef]
- Heredia, L.; Helguera, P.; de Olmos, S.; Kedikian, G.; Vigo, F.S.; LaFerla, F.; Staufenbiel, M.; de Olmos, J.; Busciglio, J.; Cáceres, A.; et al. Phosphorylation of Actin-Depolymerizing Factor/Cofilin by LIM-Kinase Mediates Amyloid β-Induced Degeneration: A Potential Mechanism of Neuronal Dystrophy in Alzheimer’s Disease. J. Neurosci. 2006, 26, 6533–6542. [Google Scholar] [CrossRef] [PubMed]
- Maloney, M.T.; Minamide, L.S.; Kinley, A.W.; Boyle, J.A.; Bamburg, J.R. Beta-Secretase-Cleaved Amyloid Precursor Protein Accumulates at Actin Inclusions Induced in Neurons by Stress or Amyloid Beta: A Feedforward Mechanism for Alzheimer’s Disease. J. Neurosci. 2005, 25, 11313–11321. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.P.; Minamide, L.S.; Kane, S.J.; Shaw, A.E.; Brown, D.R.; Pulford, B.; Zabel, M.D.; Lambeth, J.D.; Kuhn, T.B.; Bamburg, J.R. Amyloid-β and Proinflammatory Cytokines Utilize a Prion Protein-Dependent Pathway to Activate NADPH Oxidase and Induce Cofilin-Actin Rods in Hippocampal Neurons. PLoS ONE 2014, 9, e95995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, J.M.; Seward, M.E.; Bloom, G.S. Alzheimer Disease: A Tale of Two Prions. Prion 2013, 7, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Rahman, T.; Davies, D.S.; Tannenberg, R.K.; Fok, S.; Shepherd, C.; Dodd, P.R.; Cullen, K.M.; Goldsbury, C. Cofilin Rods and Aggregates Concur with Tau Pathology and the Development of Alzheimer’s Disease. J. Alzheimers Dis. 2014, 42, 1443–1460. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Zhao, X.; Khan, H.; Penn, C.; Wang, X.; Joly-Amado, A.; Weeber, E.; Morgan, D.; Kang, D.E. Slingshot-Cofilin Activation Mediates Mitochondrial and Synaptic Dysfunction via Aβ Ligation to Β1-Integrin Conformers. Cell Death Differ. 2015, 22, 921–934. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.A.; Boggess, T.; Uhlar, C.; Wang, X.; Khan, H.; Cappos, G.; Joly-Amado, A.; De Narvaez, E.; Majid, S.; Minamide, L.S.; et al. RanBP9 at the Intersection between Cofilin and Aβ Pathologies: Rescue of Neurodegenerative Changes by RanBP9 Reduction. Cell Death Dis. 2015, 6, 1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.A.; Jung, A.R.; Lakshmana, M.K.; Bedrossian, A.; Lim, Y.; Bu, J.H.; Park, S.A.; Koo, E.H.; Mook-Jung, I.; Kang, D.E. Pivotal Role of the RanBP9-Cofilin Pathway in Aβ-Induced Apoptosis and Neurodegeneration. Cell Death Differ. 2012, 19, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Whiteman, I.T.; Gervasio, O.L.; Cullen, K.M.; Guillemin, G.J.; Jeong, E.V.; Witting, P.K.; Antao, S.T.; Minamide, L.S.; Bamburg, J.R.; Goldsbury, C. Activated Actin-Depolymerizing Factor/Cofilin Sequesters Phosphorylated Microtubule-Associated Protein during the Assembly of Alzheimer-like Neuritic Cytoskeletal Striations. J. Neurosci. 2009, 29, 12994–13005. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.-A.A.; Liu, T.; Fang, C.C.; Cazzaro, S.; Kee, T.; LePochat, P.; Yrigoin, K.; Penn, C.; Zhao, X.; Wang, X.; et al. Activated Cofilin Exacerbates Tau Pathology by Impairing Tau-Mediated Microtubule Dynamics. Commun. Biol. 2019, 2, 112. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Liu, H.; Tran, K.C.; Leng, A.; Massa, S.M.; Longo, F.M. Small-Molecule Modulation of the P75 Neurotrophin Receptor Inhibits a Wide Range of Tau Molecular Pathologies and Their Sequelae in P301S Tauopathy Mice. Acta Neuropathol. Commun. 2020, 8, 156. [Google Scholar] [CrossRef]
- Pelucchi, S.; Vandermeulen, L.; Pizzamiglio, L.; Aksan, B.; Yan, J.; Konietzny, A.; Bonomi, E.; Borroni, B.; Padovani, A.; Rust, M.B.; et al. Cyclase-Associated Protein 2 Dimerization Regulates Cofilin in Synaptic Plasticity and Alzheimer’s Disease. Brain Commun. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Joyce, E.M.; Roiser, J.P. Cognitive Heterogeneity in Schizophrenia. Curr. Opin. Psychiatry 2007, 20, 268. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular Mechanisms Contributing to Dendritic Spine Alterations in the Prefrontal Cortex of Subjects with Schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Ide, M.; Lewis, D.A. Altered Cortical CDC42 Signaling Pathways in Schizophrenia: Implications for Dendritic Spine Deficits. Biol. Psychiatry 2010, 68, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Hachisuka, A.; Nakajima, O.; Yamazaki, T.; Sawada, J. Developmental Expression of Opioid-Binding Cell Adhesion Molecule (OBCAM) in Rat Brain. Dev. Brain Res. 2000, 122, 183–191. [Google Scholar] [CrossRef]
- Schol-Gelok, S.; Janssens, A.C.J.; Tiemeier, H.; Liu, F.; Lopez-Leon, S.; Zorkoltseva, I.V.; Axenovich, T.I.; van Swieten, J.C.; Uitterlinden, A.G.; Hofman, A. A Genome-Wide Screen for Depression in Two Independent Dutch Populations. Biol. Psychiatry 2010, 68, 187–196. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, M.; Li, Q.; You, Y.; Yu, H.; Ma, Y.; Mei, L.; Sun, X.; Wang, L.; Yue, W. The Schizophrenia Susceptibility Gene OPCML Regulates Spine Maturation and Cognitive Behaviors through Eph-Cofilin Signaling. Cell Rep. 2019, 29, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Foote, M.; Qiao, H.; Graham, K.; Wu, Y.; Zhou, Y. Inhibition of 14-3-3 Proteins Leads to Schizophrenia-Related Behavioral Phenotypes and Synaptic Defects in Mice. Biol. Psychiatry 2015, 78, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, Z.-H. Molecular and Cellular Mechanisms of Excitotoxic Neuronal Death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, T.; Kitagawa, K.; Kuwabara, K.; Takasawa, K.; Ohtsuki, T.; Xia, Z.; Storm, D.; Yanagihara, T.; Hori, M.; Matsumoto, M. Phosphorylation of CAMP Response Element-Binding Protein in Hippocampal Neurons as a Protective Response after Exposure to Glutamate in Vitro and Ischemia in Vivo. J. Neurosci. 2001, 21, 9204–9213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Jiang, M.; Zhou, M.; Chen, L.; Liu, X.; Wang, X.; Wang, Y. Both NMDA and Non-NMDA Receptors Mediate Glutamate Stimulation Induced Cofilin Rod Formation in Cultured Hippocampal Neurons. Brain Res. 2012, 1486, 1–13. [Google Scholar] [CrossRef]
- Shu, L.; Chen, B.; Chen, B.; Xu, H.; Wang, G.; Huang, Y.; Zhao, Y.; Gong, H.; Jiang, M.; Chen, L.; et al. Brain Ischemic Insult Induces Cofilin Rod Formation Leading to Synaptic Dysfunction in Neurons. J. Cereb. Blood Flow Metab. 2019, 39, 2181–2195. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Minnella, A.M.; Wu, L.; Eun, C.H.; Rome, E.; Herson, P.S.; Shaw, A.E.; Bamburg, J.R.; Swanson, R.A. Cofilin-Actin Rod Formation in Neuronal Processes after Brain Ischemia. PLoS ONE 2018, 13, e0198709. [Google Scholar] [CrossRef] [Green Version]
- Posadas, I.; Pérez-Martínez, F.C.; Guerra, J.; Sánchez-Verdú, P.; Ceña, V. Cofilin Activation Mediates Bax Translocation to Mitochondria during Excitotoxic Neuronal Death. J. Neurochem. 2012, 120, 515–527. [Google Scholar] [CrossRef]
- Chen, B.; Lin, W.; Qi, W.; Li, S.; Hong, Z.; Zhao, H. Cofilin Inhibition by Limk1 Reduces Rod Formation and Cell Apoptosis after Ischemic Stroke. Neuroscience 2020, 444, 64–75. [Google Scholar] [CrossRef]
- Juurlink, B.H.; Thorburne, S.K.; Hertz, L. Peroxide-scavenging Deficit Underlies Oligodendrocyte Susceptibility to Oxidative Stress. Glia 1998, 22, 371–378. [Google Scholar] [CrossRef]
- Wabnitz, G.H.; Goursot, C.; Jahraus, B.; Kirchgessner, H.; Hellwig, A.; Klemke, M.; Konstandin, M.H.; Samstag, Y. Mitochondrial Translocation of Oxidized Cofilin Induces Caspase-Independent Necrotic-like Programmed Cell Death of T Cells. Cell Death Dis. 2010, 1, e58. [Google Scholar] [CrossRef] [Green Version]
- Alhadidi, Q.; Bin Sayeed, M.S.; Shah, Z.A. Cofilin as a Promising Therapeutic Target for Ischemic and Hemorrhagic Stroke. Transl. Stroke Res. 2016, 7, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Han, Y.; Lee, J.E.; Yenari, M.A. The 70-KDa Heat Shock Protein (Hsp70) as a Therapeutic Target for Stroke. Expert Opin. Ther. Targets 2018, 22, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurisu, K.; You, J.; Zheng, Z.; Won, S.J.; Swanson, R.A.; Yenari, M.A. Cofilin-Actin Rod Formation in Experimental Stroke Is Attenuated by Therapeutic Hypothermia and Overexpression of the Inducible 70 KD Inducible Heat Shock Protein (Hsp70). Brain Circ. 2019, 5, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-Y.; Englund, E.; Widner, H.; Rehncrona, S.; Björklund, A.; Lindvall, O.; Brundin, P. Characterization of Lewy Body Pathology in 12- and 16-Year-Old Intrastriatal Mesencephalic Grafts Surviving in a Patient with Parkinson’s Disease. Mov. Disord. 2010, 25, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Agid, Y.; Dürr, A.; Brice, A.; Pollak, P. Causal Relation between α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced α-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 Clustering at the Cell Surface of Neurons Transduces the Action of Exogenous Alpha-Synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Yan, M.; Meng, L.; Dai, L.; Zhang, X.; Chen, G.; Zheng, Y.; Zha, Y.; Zeng, Y.; Zhang, Z. Cofilin 1 Promotes the Aggregation and Cell-to-Cell Transmission of α-Synuclein in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2020, 529, 1053–1060. [Google Scholar] [CrossRef]
- Zhong, Z.; Grasso, L.; Sibilla, C.; Stevens, T.J.; Barry, N.; Bertolotti, A. Prion-like Protein Aggregates Exploit the RHO GTPase to Cofilin-1 Signaling Pathway to Enter Cells. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Latouche, M.; Lohmann, E.; Rawal, N.; De Michele, G.; Ricard, S.; Teive, H.; Fraix, V.; Vidailhet, M.; Nicholl, D.; et al. Parkin Mutations Are Frequent in Patients with Isolated Early-onset Parkinsonism. Brain 2003, 126, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.K.; Kawamura, T.; Ohsawa, Y.; Ohtsubo, M.; Asakawa, S.; Takayanagi, A.; Shimizu, N. Parkin Interacts with LIM Kinase 1 and Reduces Its Cofilin-Phosphorylation Activity via Ubiquitination. Exp. Cell Res. 2007, 313, 2858–2874. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Gene Name | Isoforms/Alternative Names | Roles in Cofilin Regulation Pathway | References |
|---|---|---|---|---|
| Slingshot phosphatase | SSH | SSH1 SSH2 SSH3 | Dephosphorylate cofilin; dephosphorylate coronins | [31,34] |
| Coronins | CORO | Coronin 1A Coronin 1B Coronin 2A | Induce cofilin activation via SSH1 dephosphorylation, Interact with 14-3-3zeta protein | [35,36] |
| 14-3-3zeta protein | YWHAZ | Protein kinase C inhibitor protein 1 (KCIP-1) | Downregulates cofilin activity via SSH1 deactivation and LIMK activation | [37,38] |
| Protein kinase D enzymes (PKDs) | PRKD | PKD1 PKD2 PKD3 | Decrease cofilin dephosphorylation by promote 14-3-3zeta protein binding with SSH1 and by inducing LIMK1 activation | [39] |
| Chronophin (CIN) | PDXP | Pyridoxal phosphate phosphatase | Interacts and inhibits Hsp90-mediated LIMK activation, hence induce cofilin phosphorylation | [40,41] |
| LIM kinases | LIMK | LIMK1 LIMK2 | Phosphorylate and inactivate cofilin | [42,43] |
| TES kinases | TESK | TESK1 TESK2 | Phosphorylate and inactivate cofilin | [44,45] |
| Reelin | RELN | Isoform 1 Isoform 2 Isoform 3 | Increase cofilin phosphorylation by inducing LIMK1 activation | [46,47] |
| SH3 protein interacting with Nck, 90 kDa (SPIN90) | KCKIPSD | 54 kDa vimentin-interacting protein (VIP54) | Inhibits cofilin activity by binding | [48] |
| Cortactin | CTTN | Amplaxin; oncogene EMS1 | Downregulates cofilin activity by binding | [49] |
| Adenylyl cyclase-associated proteins (CAPs) | CAP | CAP1 CAP2 | Enhance cofilin activity by synergizing cofilin activity | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namme, J.N.; Bepari, A.K.; Takebayashi, H. Cofilin Signaling in the CNS Physiology and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 10727. https://doi.org/10.3390/ijms221910727
Namme JN, Bepari AK, Takebayashi H. Cofilin Signaling in the CNS Physiology and Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(19):10727. https://doi.org/10.3390/ijms221910727
Chicago/Turabian StyleNamme, Jannatun Nayem, Asim Kumar Bepari, and Hirohide Takebayashi. 2021. "Cofilin Signaling in the CNS Physiology and Neurodegeneration" International Journal of Molecular Sciences 22, no. 19: 10727. https://doi.org/10.3390/ijms221910727
APA StyleNamme, J. N., Bepari, A. K., & Takebayashi, H. (2021). Cofilin Signaling in the CNS Physiology and Neurodegeneration. International Journal of Molecular Sciences, 22(19), 10727. https://doi.org/10.3390/ijms221910727