
Horizontal Transfer of LTR Retrotransposons Contributes to the Genome Diversity of Vitis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
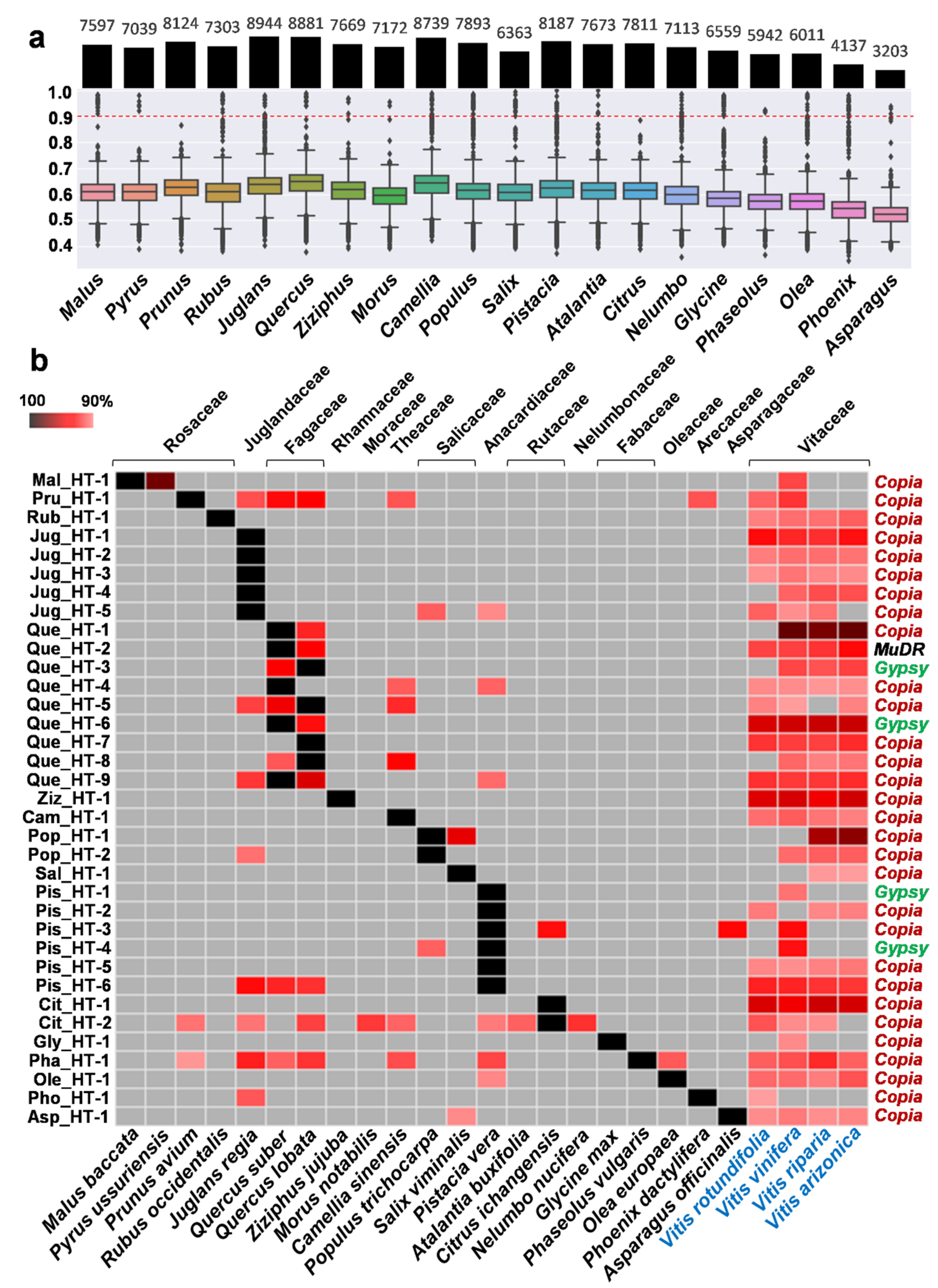
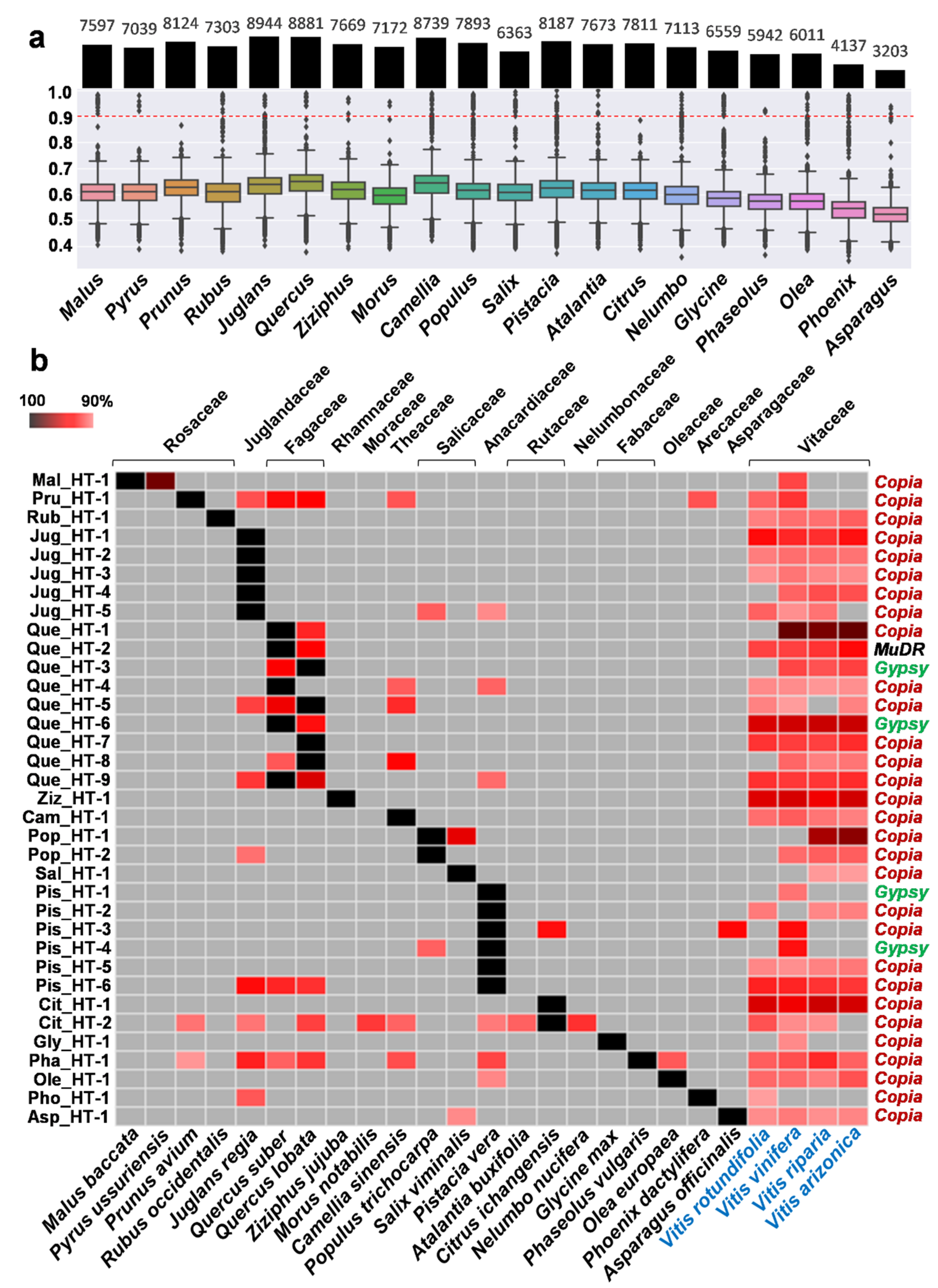
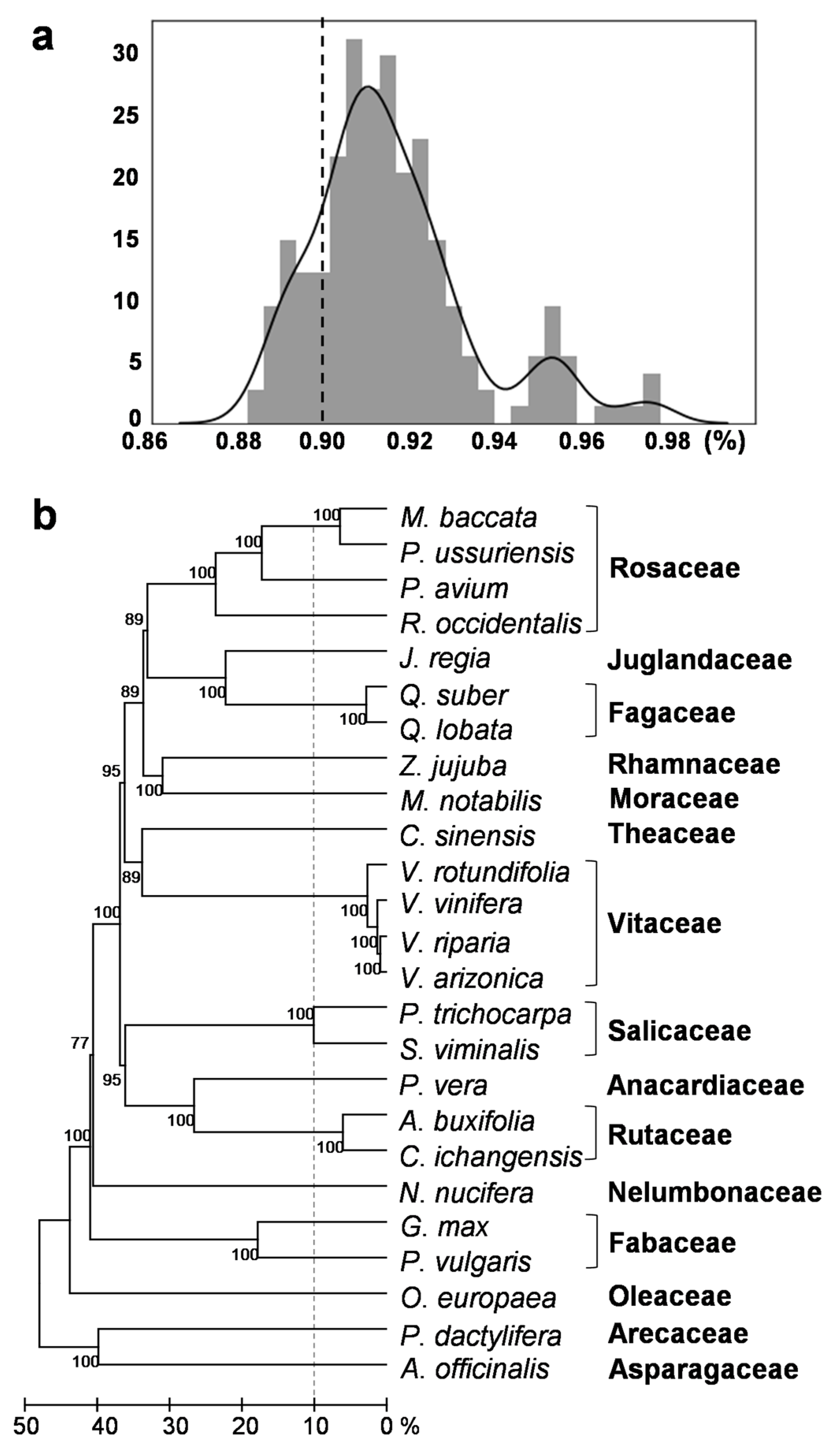
2.1. Investigation of Putative HT Events between Vitis and Other Plant Species
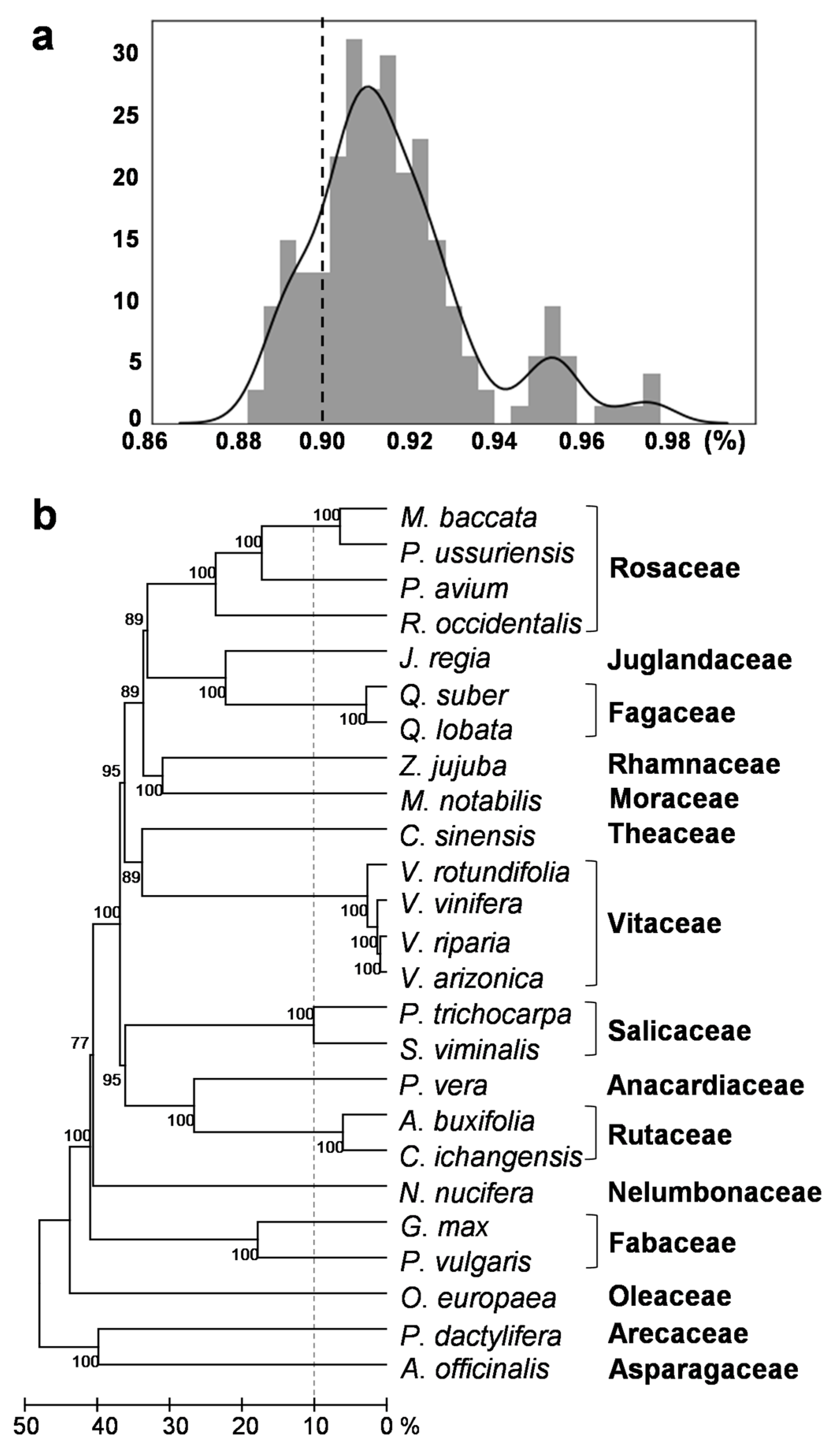
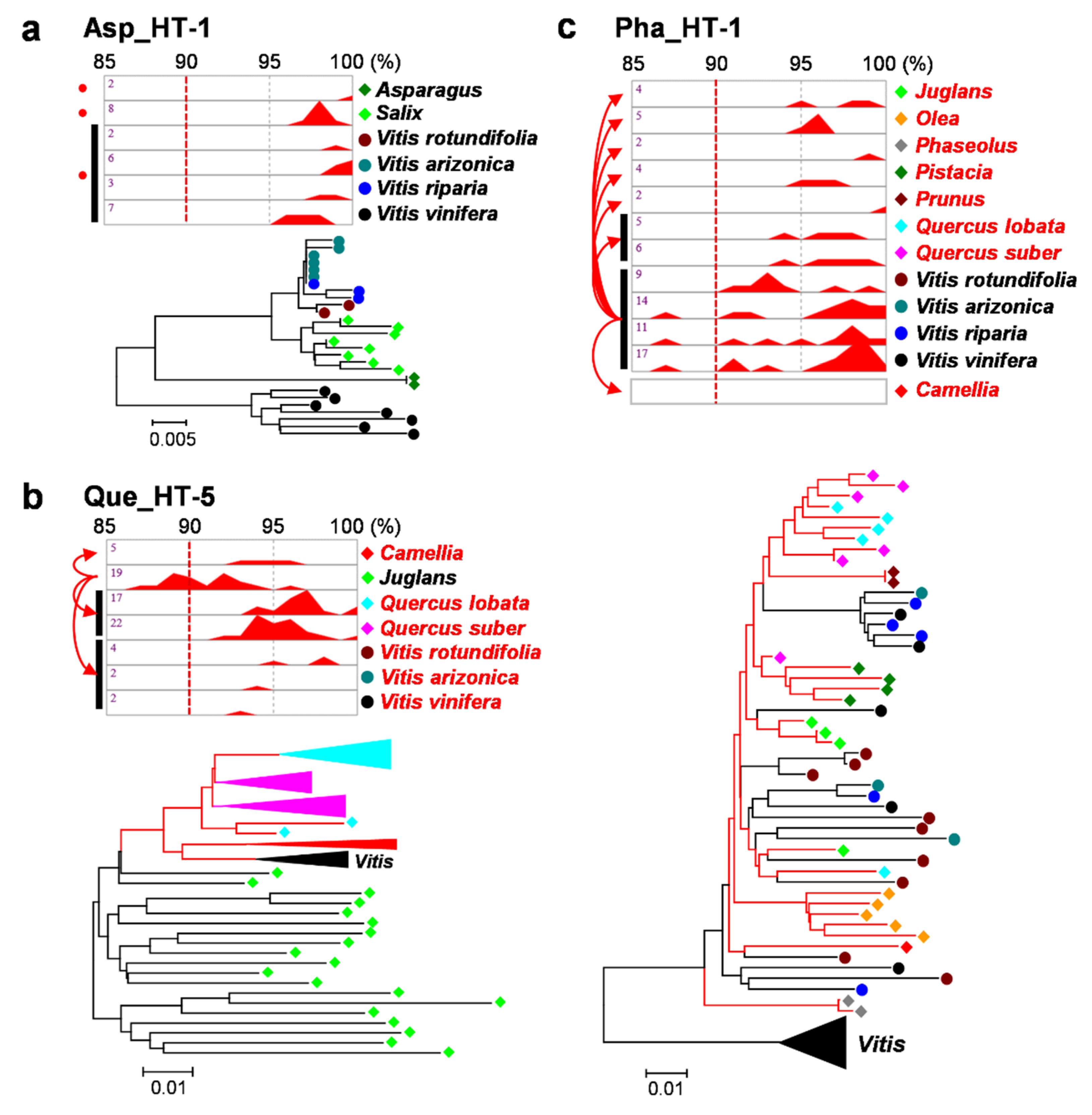
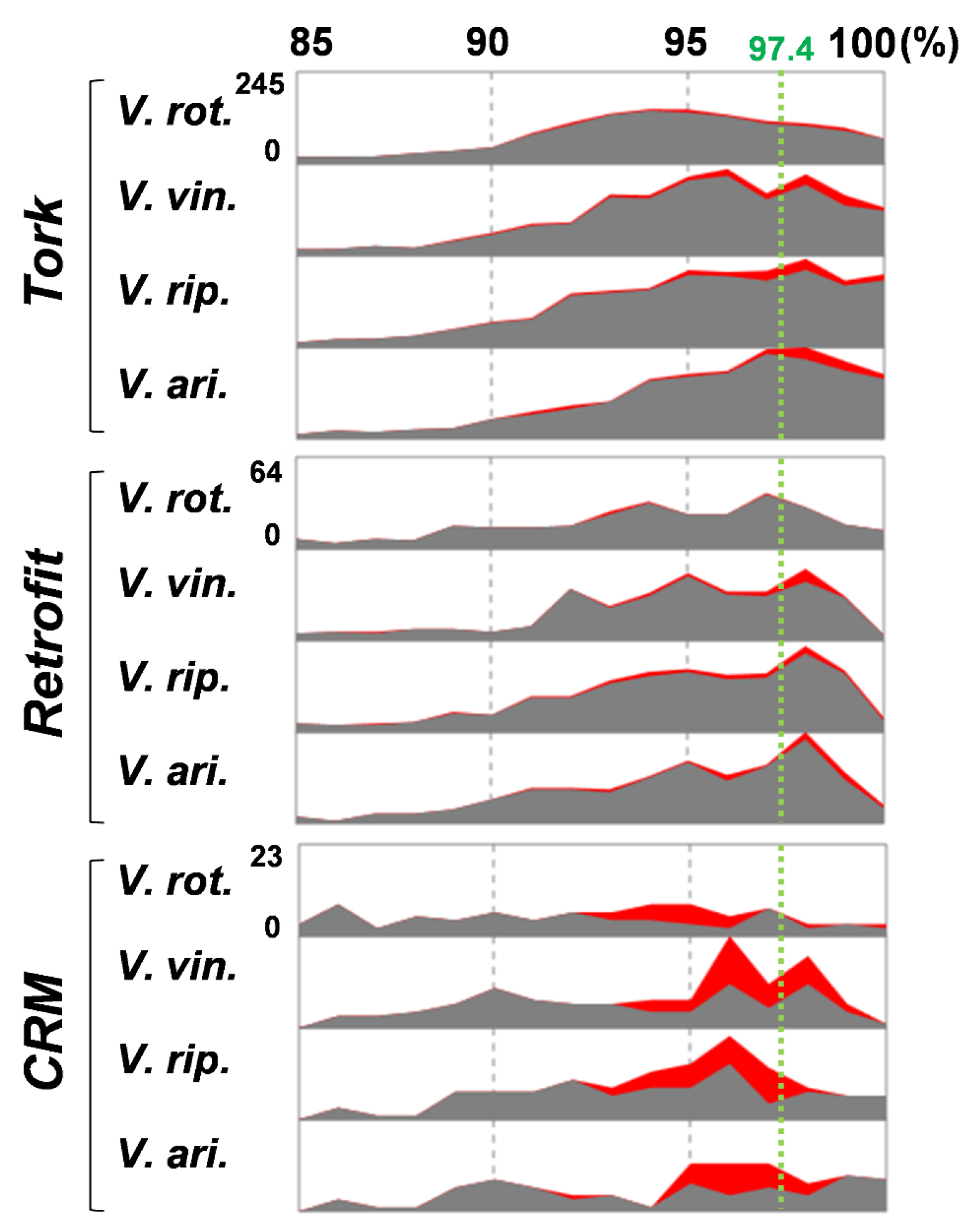
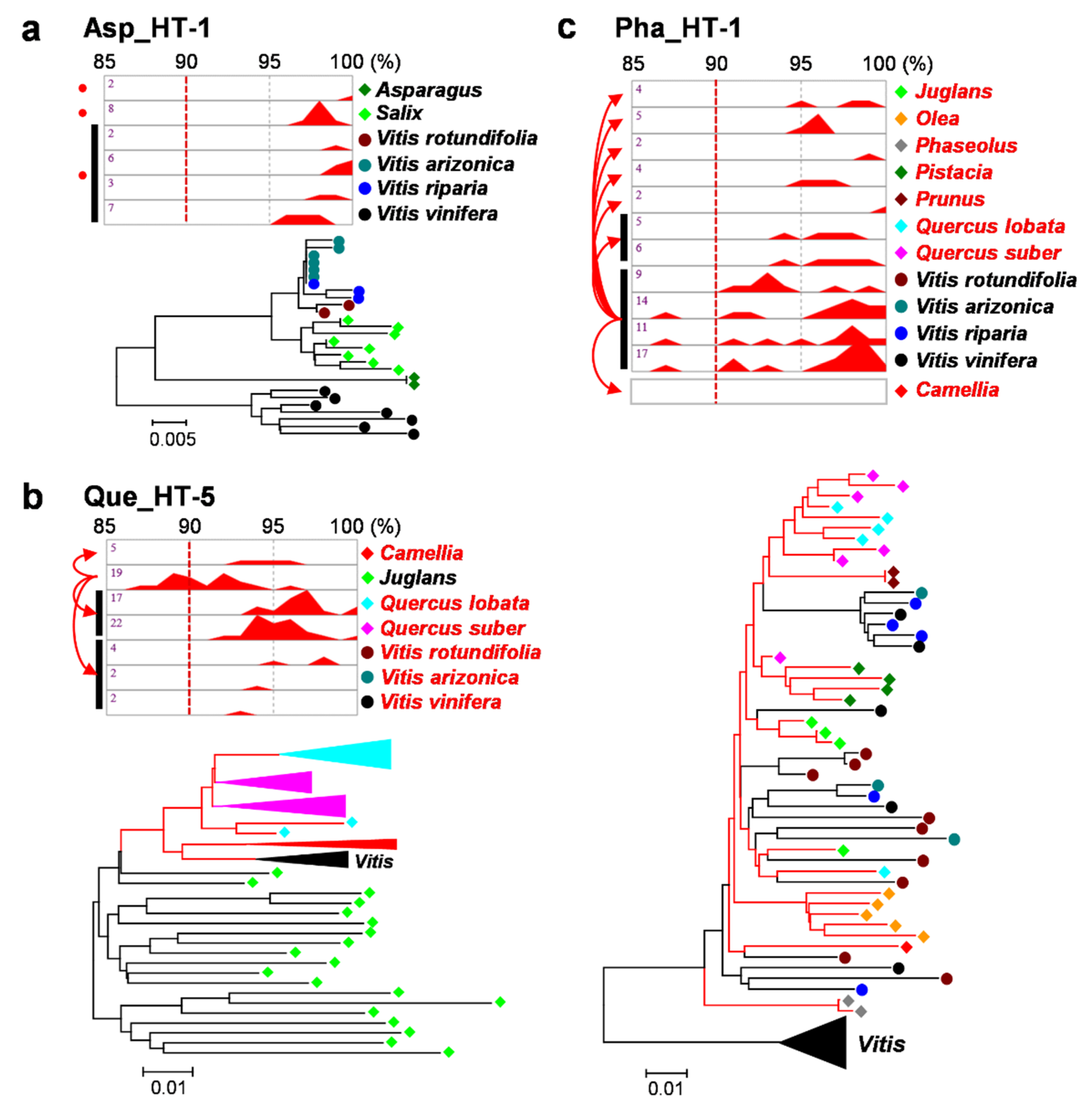
2.2. Verification of HTs
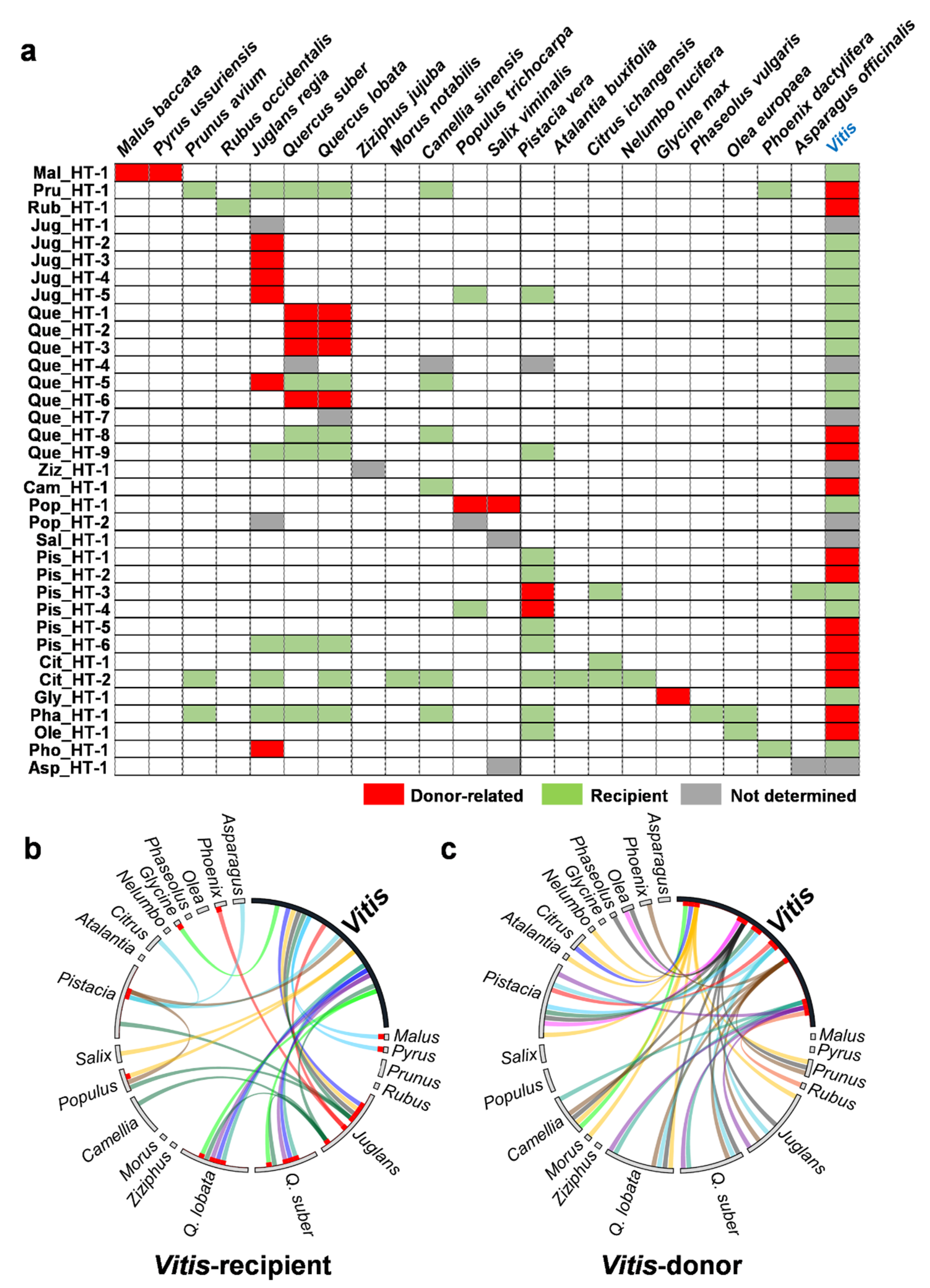
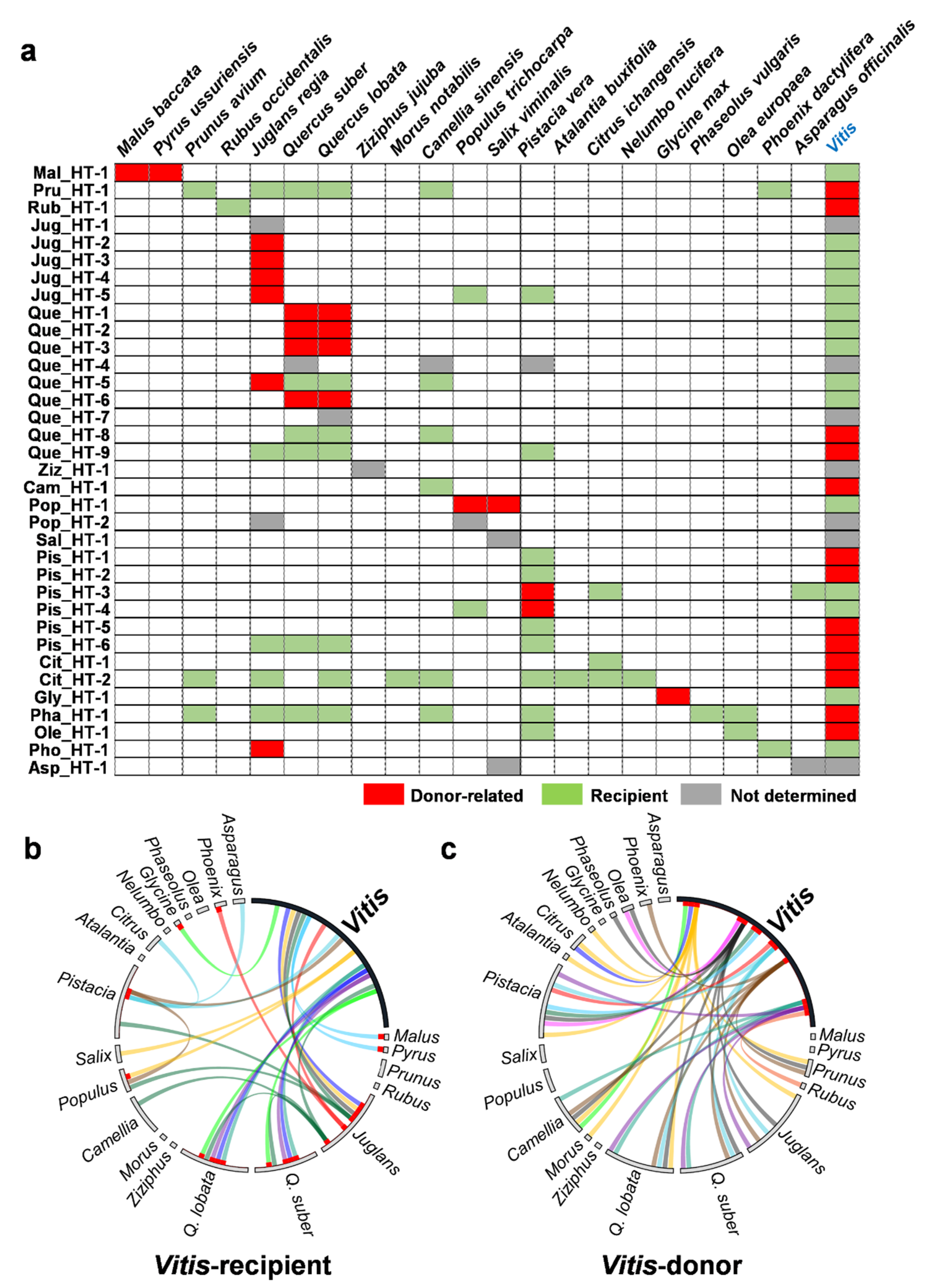
2.3. Donors and Recipients of the HT Events
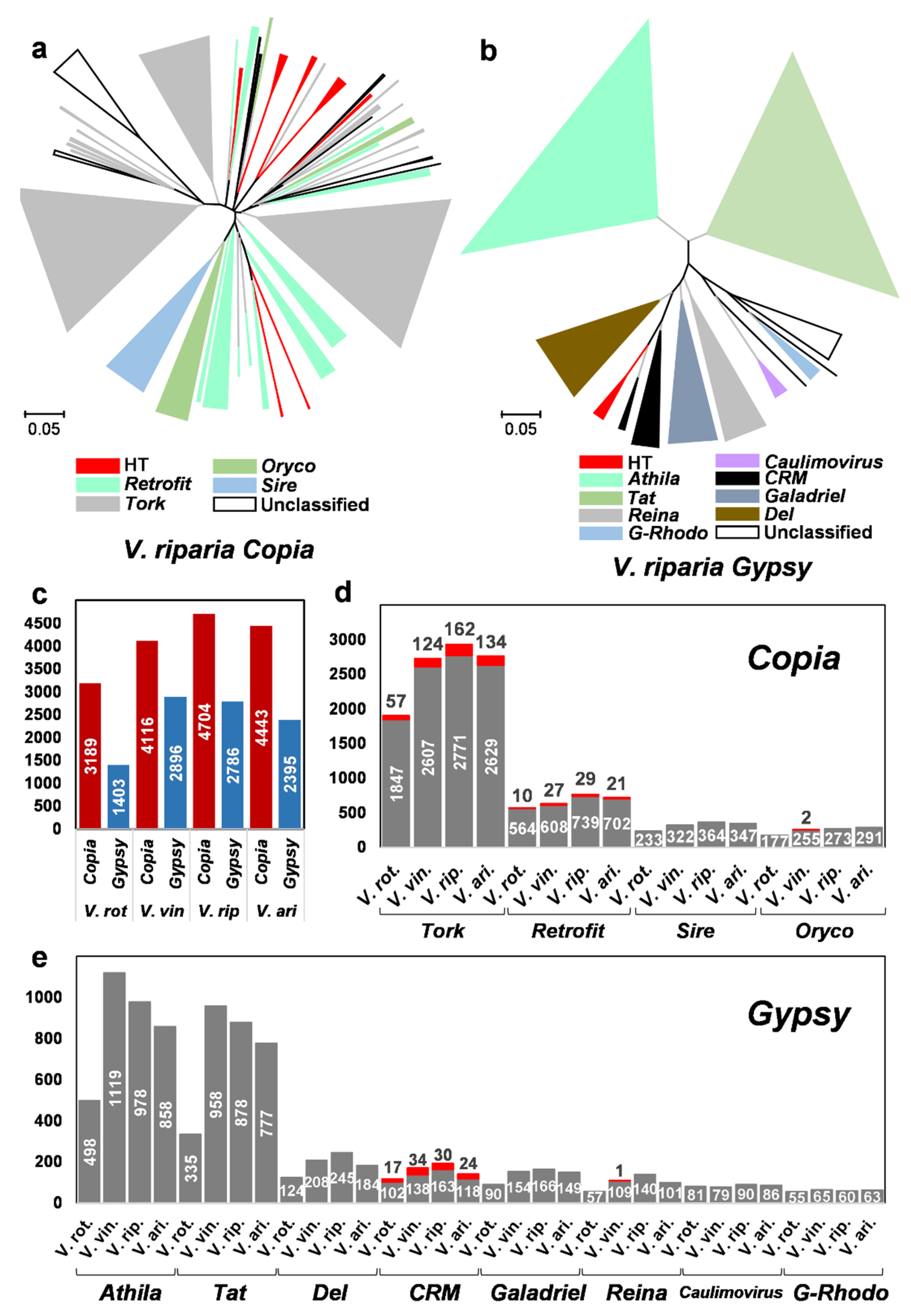
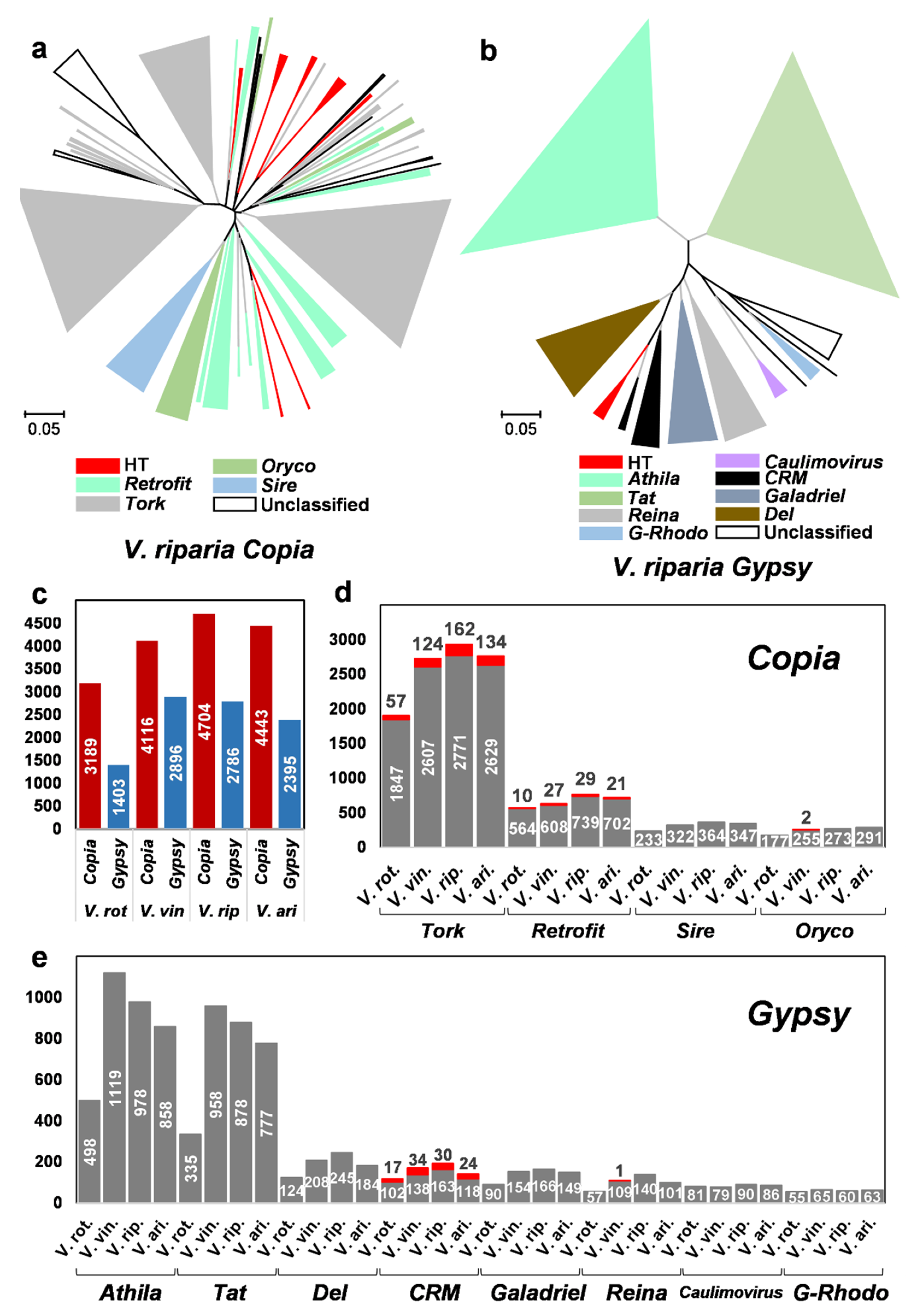
2.4. Horizontally Transferred LTR Retrotransposons in the Vitis Genomes
3. Discussion
3.1. Detection of HTs
3.2. HT of LTR Retrotransposons and Diversification of the Vitis Genomes
3.3. Multiple Recipients of Horizontally Transferred Elements
3.4. Conclusions
4. Materials and Methods
4.1. Screening of Possible HT Cases
4.2. Activity History of TE Paralogs and Phylogenetic Analysis of TE Homologs
4.3. Phylogenetic Tree of the Species Involved in the HT Events
4.4. Analysis of LTR Retrotransposons in the Four Vitis Genomes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Park, M.; Yeom, S.-I.; Kim, Y.-M.; Lee, J.M.; Lee, H.-A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.-T.; et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270. [Google Scholar] [CrossRef] [PubMed]
- Wessler, S.R.; Bureau, T.E.; White, S.E. LTR-retrotransposons and mites: Important players in the evolution of plant genomes. Curr. Opin. Genet. Dev. 1995, 5, 814–821. [Google Scholar] [CrossRef]
- Vitte, C.; Bennetzen, J.L. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17638–17643. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Park, J.; Kim, S.; Kwon, J.-K.; Park, H.M.; Bae, I.H.; Yang, T.-J.; Lee, Y.-H.; Kang, B.-C.; Choi, D. Evolution of the large genome in Capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 2012, 69, 1018–1029. [Google Scholar] [CrossRef]
- Park, M.; Jo, S.; Kwon, J.-K.; Park, J.; Ahn, J.H.; Kim, S.; Lee, Y.-H.; Yang, T.-J.; Hur, C.-G.; Kang, B.-C.; et al. Comparative analysis of pepper and tomato reveals euchromatin expansion of pepper genome caused by differential accumulation of Ty3/Gypsy-like elements. BMC Genomics. 2011, 12, 85. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Sugimoto, K.; Otsuki, H.; Hirochika, H. Transcriptional activation of the tobacco retrotransposon Tto1 by wounding and methyl jasmonate. Plant Mol. Biol. 1998, 36, 365–376. [Google Scholar] [CrossRef]
- Grandbastien, M.A.; Audeon, C.; Bonnivard, E.; Casacuberta, J.M.; Chalhoub, B.; Costa, A.P.P.; Le, Q.H.; Melayah, D.; Petit, M.; Poncet, C.; et al. Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 2005, 110, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.; Yeom, S.-I.; Kim, Y.-M.; Seo, E.; Kim, K.-T.; Kim, M.-S.; Lee, J.M.; Cheong, K.; Shin, H.-S.; et al. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genom. Biol. 2017, 18, 210. [Google Scholar]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.-A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Estep, M.C.; DeBarry, J.D.; Bennetzen, J.L. The dynamics of LTR retrotransposon accumulation across 25 million years of panicoid grass evolution. Heredity 2013, 110, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Schulman, A.H. Retrotransposon replication in plants. Curr. Opin. Virol. 2013, 3, 604–614. [Google Scholar] [CrossRef]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Fitzpatrick, D.A. Horizontal gene transfer in fungi. FEMS Microbiol. Lett. 2012, 329, 1–8. [Google Scholar] [CrossRef]
- Peccoud, J.; Loiseau, V.; Cordaux, R.; Gilbert, C. Massive horizontal transfer of transposable elements in insects. Proc. Natl. Acad. Sci. USA 2017, 114, 4721–4726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Schaack, S.; Pritham, E.J. Pervasive horizontal transfer of rolling-circle transposons among animals. Genom. Biol. Evol. 2010, 2, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread horizontal transfer of retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Baidouri, M.; Carpentier, M.-C.; Cooke, R.; Gao, D.; Lasserre, E.; Llauro, C.; Mirouze, M.; Picault, N.; Jackson, S.A.; Panaud, O. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 2014, 24, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, L.T.; Olofsson, J.K.; Parisod, C.; Choudhury, R.R.; Moreno-Villena, J.J.; Yang, Y.; Dionora, J.; Quick, W.P.; Park, M.; Bennetzen, J.L.; et al. Lateral transfers of large DNA fragments spread functional genes among grasses. Proc. Natl. Acad. Sci. USA 2019, 116, 4416. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Christin, P.-A.; Bennetzen, J.L. Sample sequence analysis uncovers recurrent horizontal transfers of transposable elements among grasses. Mol. Biol. Evol. 2021, 38, 3664–3675. [Google Scholar] [CrossRef] [PubMed]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, X.; Freeling, M.; Lisch, D. Horizontal transfer of a plant transposon. PLoS Biol. 2006, 4, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulin, A.; Piegu, B.; Wing, R.A.; Panaud, O. Evidence of multiple horizontal transfers of the long terminal repeat retrotransposon RIRE1 within the genus Oryza. Plant J. 2008, 53, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [PubMed]
- Girollet, N.; Rubio, B.; Lopez-Roques, C.; Valière, S.; Ollat, N.; Bert, P.-F. De novo phased assembly of the Vitis Ripar. Grape Genome. Sci. Data 2019, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Massonnet, M.; Cochetel, N.; Minio, A.; Vondras, A.M.; Lin, J.; Muyle, A.; Garcia, J.F.; Zhou, Y.; Delledonne, M.; Riaz, S.; et al. The genetic basis of sex determination in grapes. Nat. Commun. 2020, 11, 2902. [Google Scholar] [CrossRef] [PubMed]
- Cochetel, N.; Minio, A.; Massonnet, M.; Vondras, A.M.; Figueroa-Balderas, R.; Cantu, D. Diploid chromosome-scale assembly of the Muscadinia rotundifolia genome supports chromosome fusion and disease resistance gene expansion during Vitis and Muscadinia divergence. G3 Bethesda 2021, 11, jkab033. [Google Scholar] [CrossRef]
- Loreto, E.; Carareto, C.; Capy, P. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity 2008, 100, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Cavallini, A.; Natali, L.; Zuccolo, A.; Giordani, T.; Jurman, I.; Ferrillo, V.; Vitacolonna, N.; Sarri, V.; Cattonaro, F.; Ceccarelli, M.; et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus l.) genome. Theor. Appl. Genet. 2010, 120, 491–508. [Google Scholar] [CrossRef]
- Cossu, R.M.; Buti, M.; Giordani, T.; Natali, L.; Cavallini, A. A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genet. Genomes 2012, 8, 61–75. [Google Scholar] [CrossRef]
- Lockton, S.; Gaut, B.S. The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana. J. Mol. Evol. 2009, 68, 80–89. [Google Scholar] [CrossRef] [Green Version]
- González, L.G.; Deyholos, M.K. Identification, characterization and distribution of transposable elements in the flax (Linum usitatissimum l.) genome. BMC Genomics 2012, 13, 644. [Google Scholar] [CrossRef] [Green Version]
- Pouteau, S.; Grandbastien, M.-A.; Boccara, M. Microbial elicitors of plant defence responses activate transcription of a retrotransposon. Plant J. 1994, 5, 535–542. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.; Lucas, H.; Morel, J.-B.; Mhiri, C.; Vernhettes, S.; Casacuberta, J.M. The expression of the tobacco Tnt1 retrotransposon is linked to plant defense responses. Genetica 1997, 100, 241–252. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.; Spielmann, A.; Caboche, M. Tnt1, a mobile retroviral-like transposable element of tobacco isolated by plant cell genetics. Nature 1989, 337, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Melayah, D.; Bonnivard, E.; Chalhoub, B.; Audeon, C.; Grandbastien, M.-A. The mobility of the tobacco Tnt1 retrotransposon correlates with its transcriptional activation by fungal factors. Plant J. 2001, 28, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, L.D.; Manchester, S.R.; Judd, W.S. An extinct genus of Salicaceae based on twigs with attached flowers, fruits, and foliage from the Eocene Green River Formation of Utah and Colorado, USA. Am. J. Bot. 2003, 90, 1389–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berggren, W.A.; Prothero, D.R. Eocene-Oligocene Climatic and Biotic Evolution; Princeton University Press: Princeton, CA, USA, 2014; pp. 1–28. [Google Scholar]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Ranadive, K.; Jagtap, N.; Vaidya, J. Host diversity of genus Phellinus from world Elixir. Appl. Bot. 2012, 52, 11402–11408. [Google Scholar]
- Cai, L.; Arnold, B.J.; Xi, Z.; Khost, D.E.; Patel, N.; Hartmann, C.B.; Manickam, S.; Sasirat, S.; Nikolov, L.A.; Mathews, S.; et al. Deeply altered genome architecture in the endoparasitic flowering plant Sapria himalayana griff. (Rafflesiaceae). Curr. Biol. 2021, 31, 1002–1011.e1009. [Google Scholar] [PubMed]
- Legendre, P.; Legendre, L. Numerical Ecology; Elsevier Science: Amsterdam, The Netherlands, 1998; pp. 319–321. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Sarkhosh, A.; Tsolova, V.; El-Sharkawy, I. Horizontal Transfer of LTR Retrotransposons Contributes to the Genome Diversity of Vitis. Int. J. Mol. Sci. 2021, 22, 10446. https://doi.org/10.3390/ijms221910446
Park M, Sarkhosh A, Tsolova V, El-Sharkawy I. Horizontal Transfer of LTR Retrotransposons Contributes to the Genome Diversity of Vitis. International Journal of Molecular Sciences. 2021; 22(19):10446. https://doi.org/10.3390/ijms221910446
Chicago/Turabian StylePark, Minkyu, Ali Sarkhosh, Violeta Tsolova, and Islam El-Sharkawy. 2021. "Horizontal Transfer of LTR Retrotransposons Contributes to the Genome Diversity of Vitis" International Journal of Molecular Sciences 22, no. 19: 10446. https://doi.org/10.3390/ijms221910446
APA StylePark, M., Sarkhosh, A., Tsolova, V., & El-Sharkawy, I. (2021). Horizontal Transfer of LTR Retrotransposons Contributes to the Genome Diversity of Vitis. International Journal of Molecular Sciences, 22(19), 10446. https://doi.org/10.3390/ijms221910446