
Ganoderic Acids Prevent Renal Ischemia Reperfusion Injury by Inhibiting Inflammation and Apoptosis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
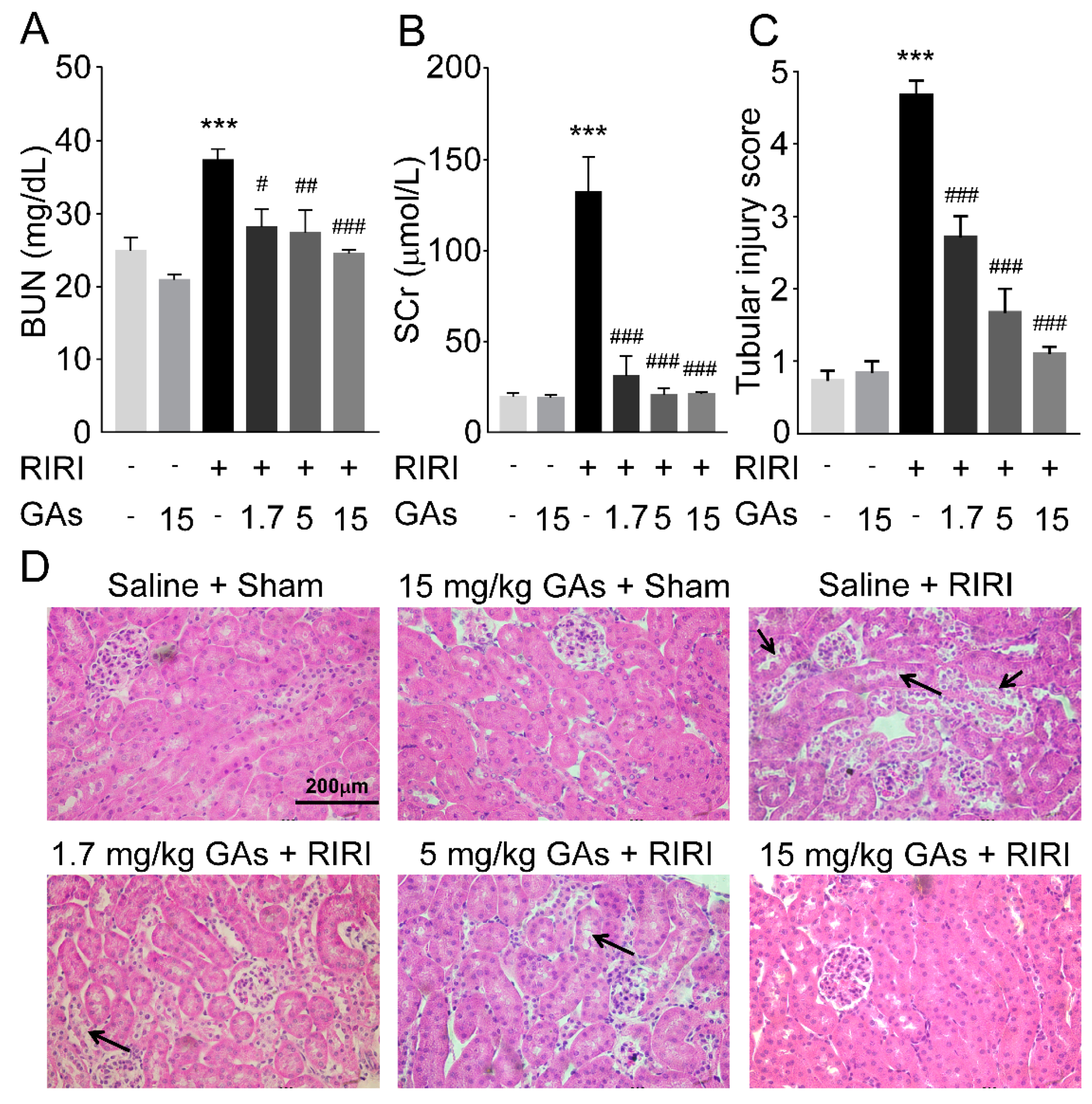
2.1. GAs Protected the Kidney against RIRI
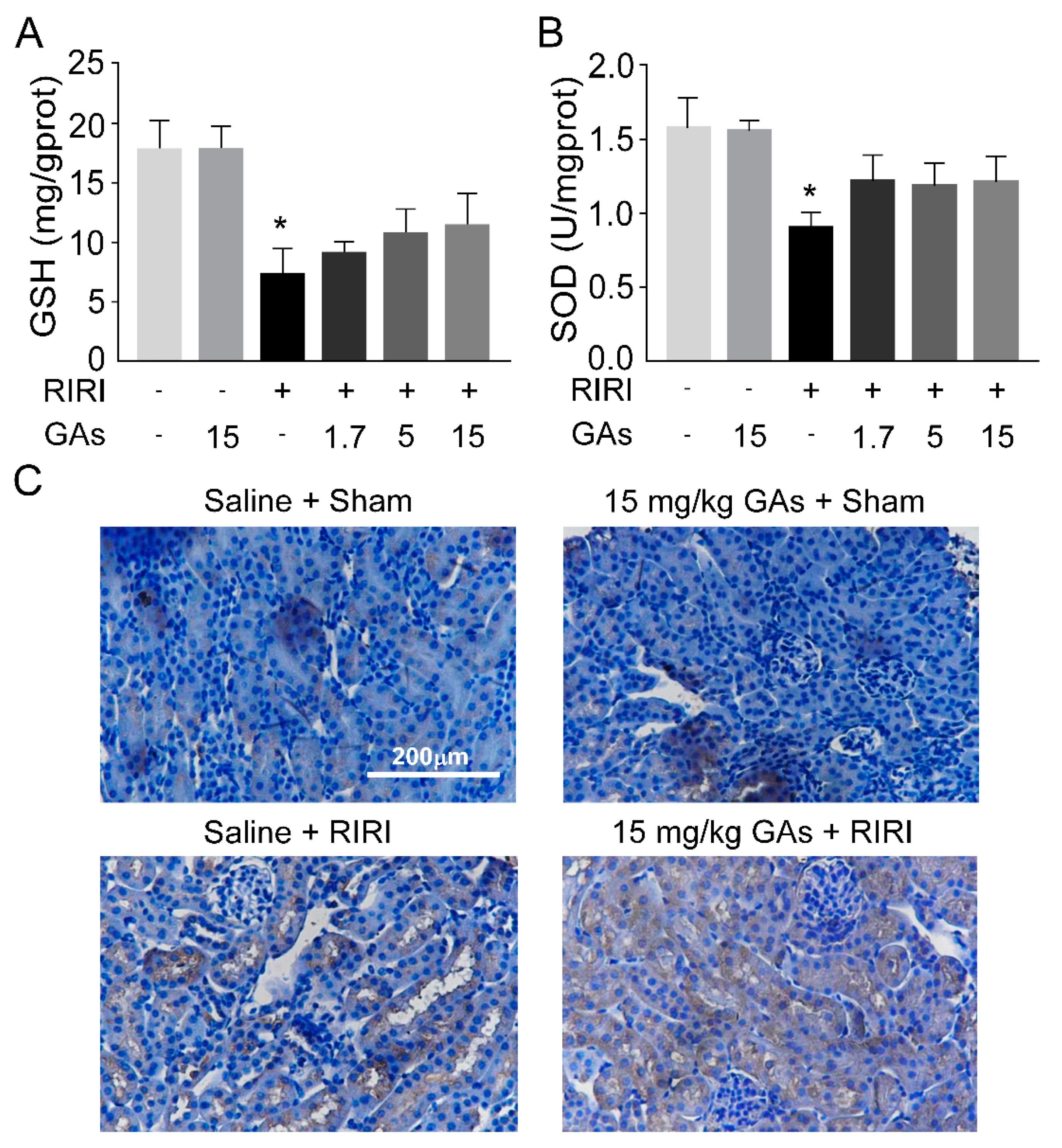
2.2. GAs Had No Significant Effect on Oxidative Stress Induced by RIRI
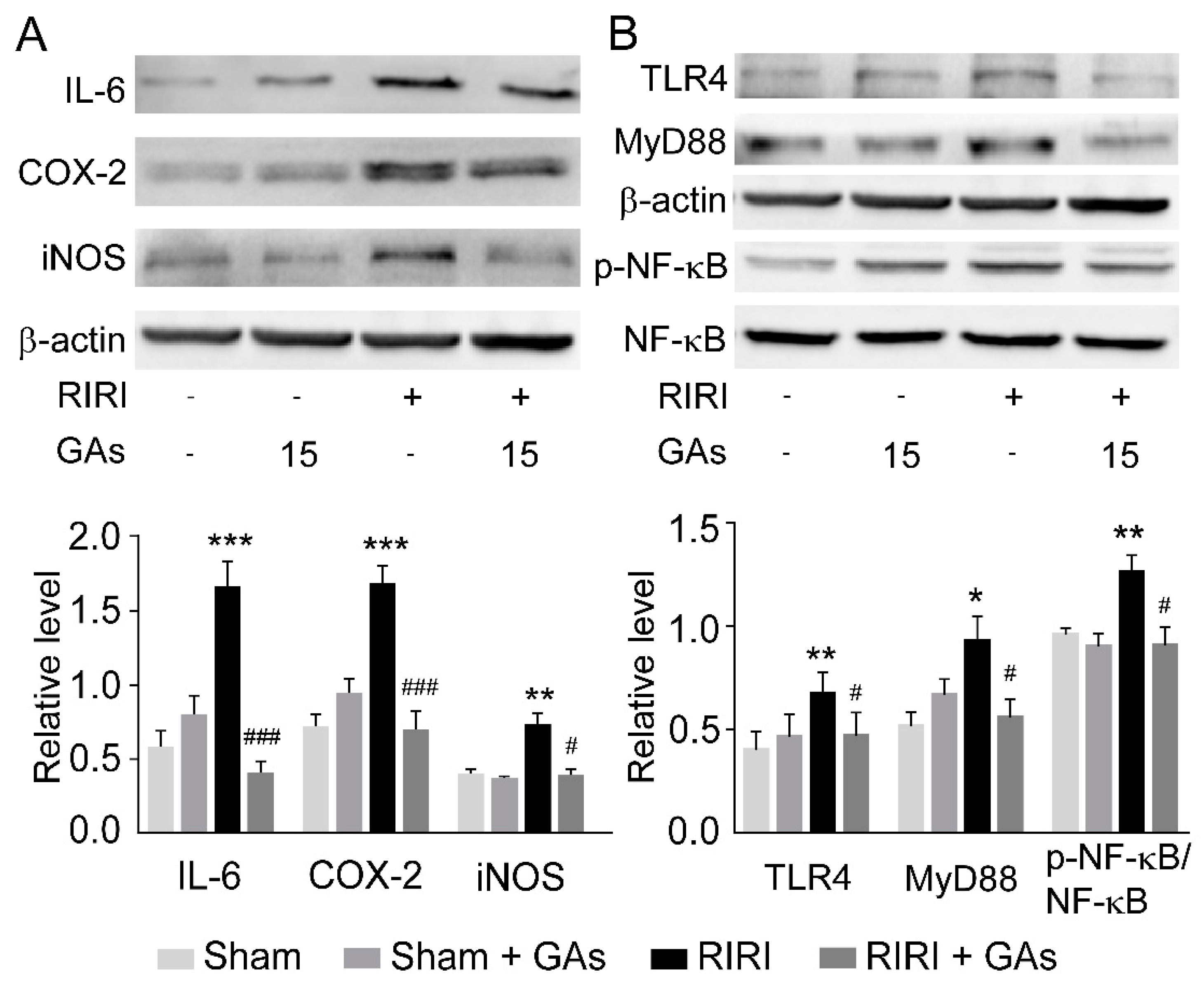
2.3. GAs Inhibited RIRI-Induced Inflammatory Response
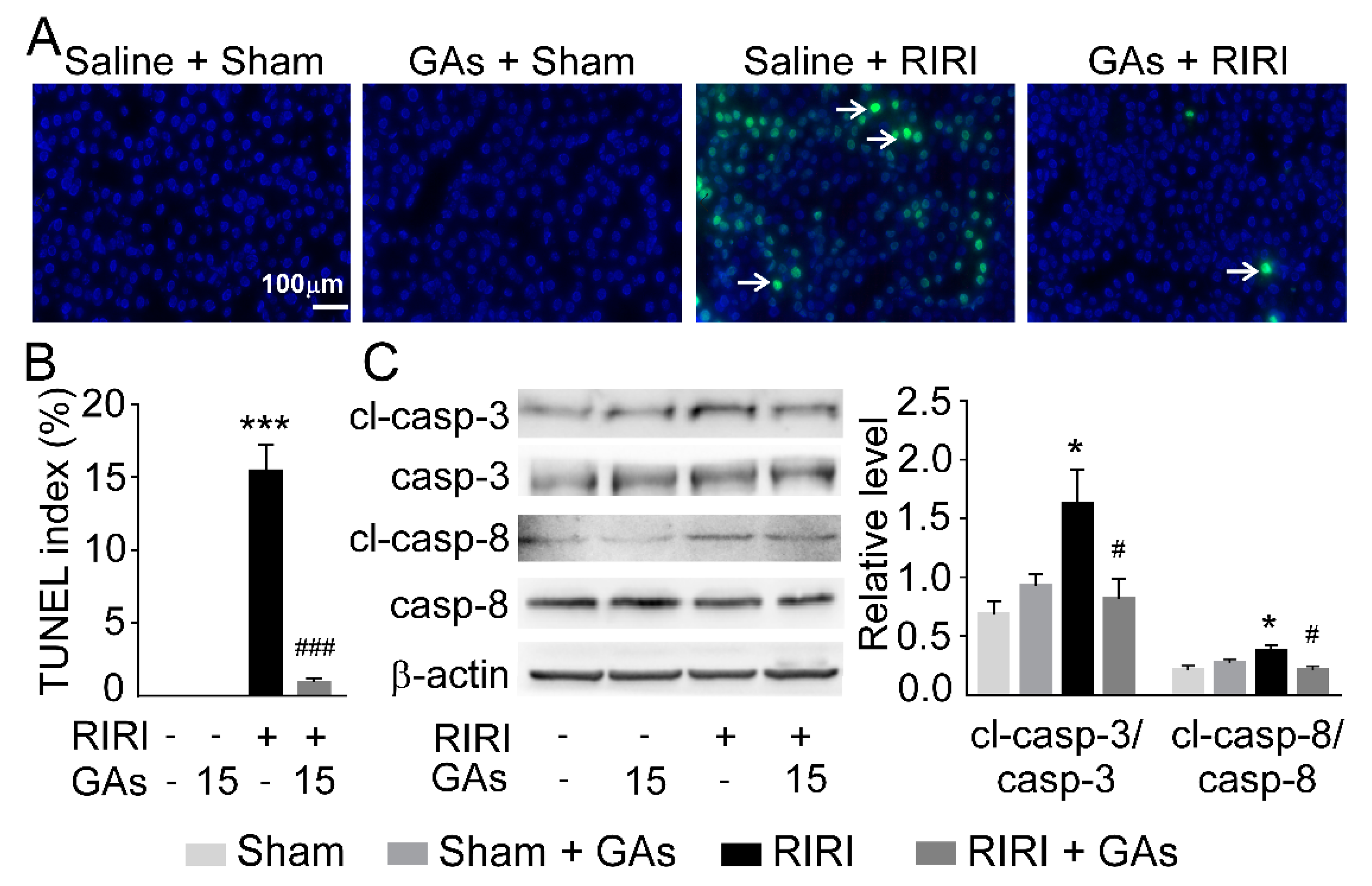
2.4. GAs Ameliorated Apoptosis in the RIRI Model
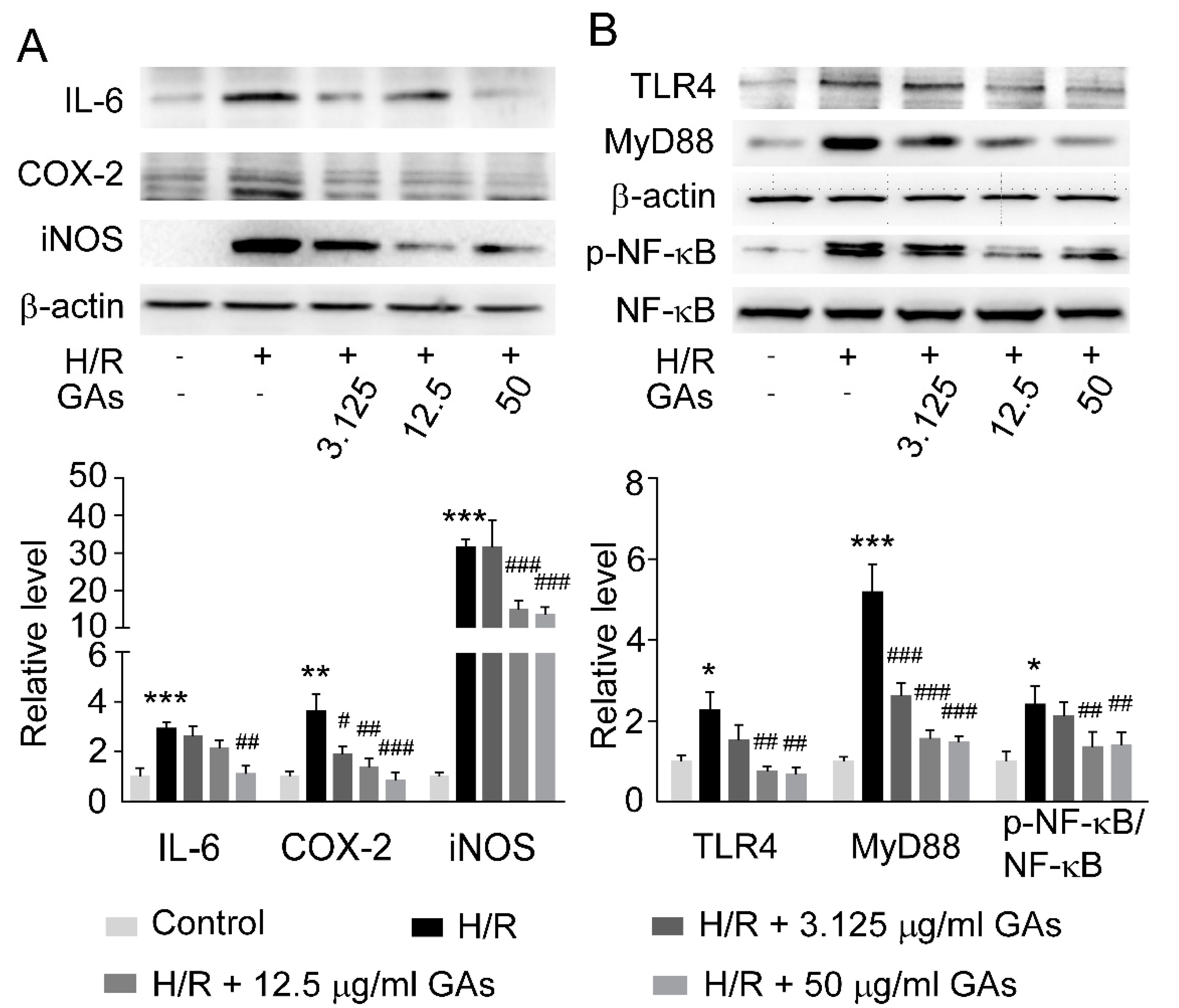
2.5. GAs Suppressed H/R-Induced Inflammatory Response in NRK-52E Cell
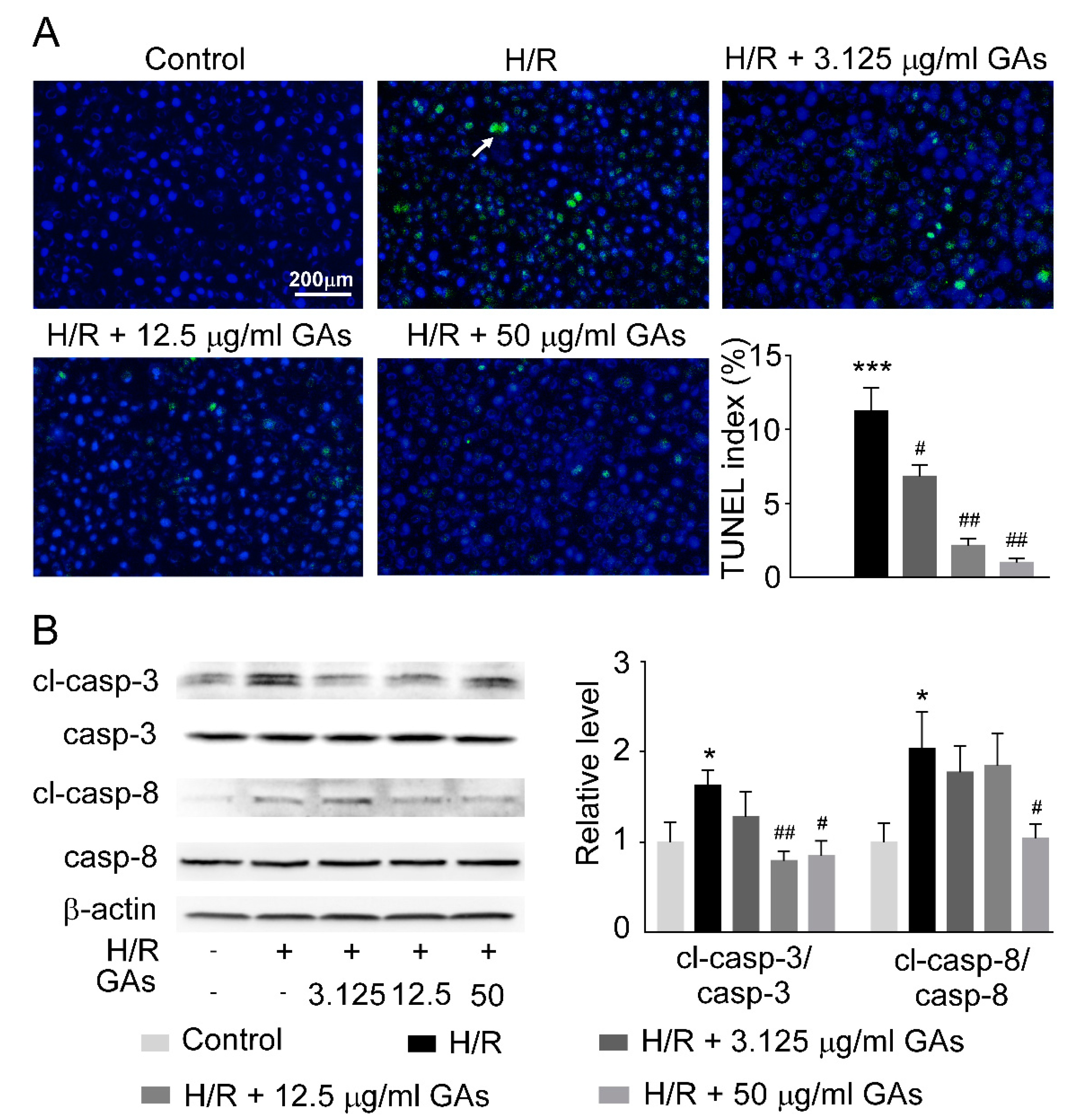
2.6. GAs Reduced Cell Apoptosis Induced by H/R
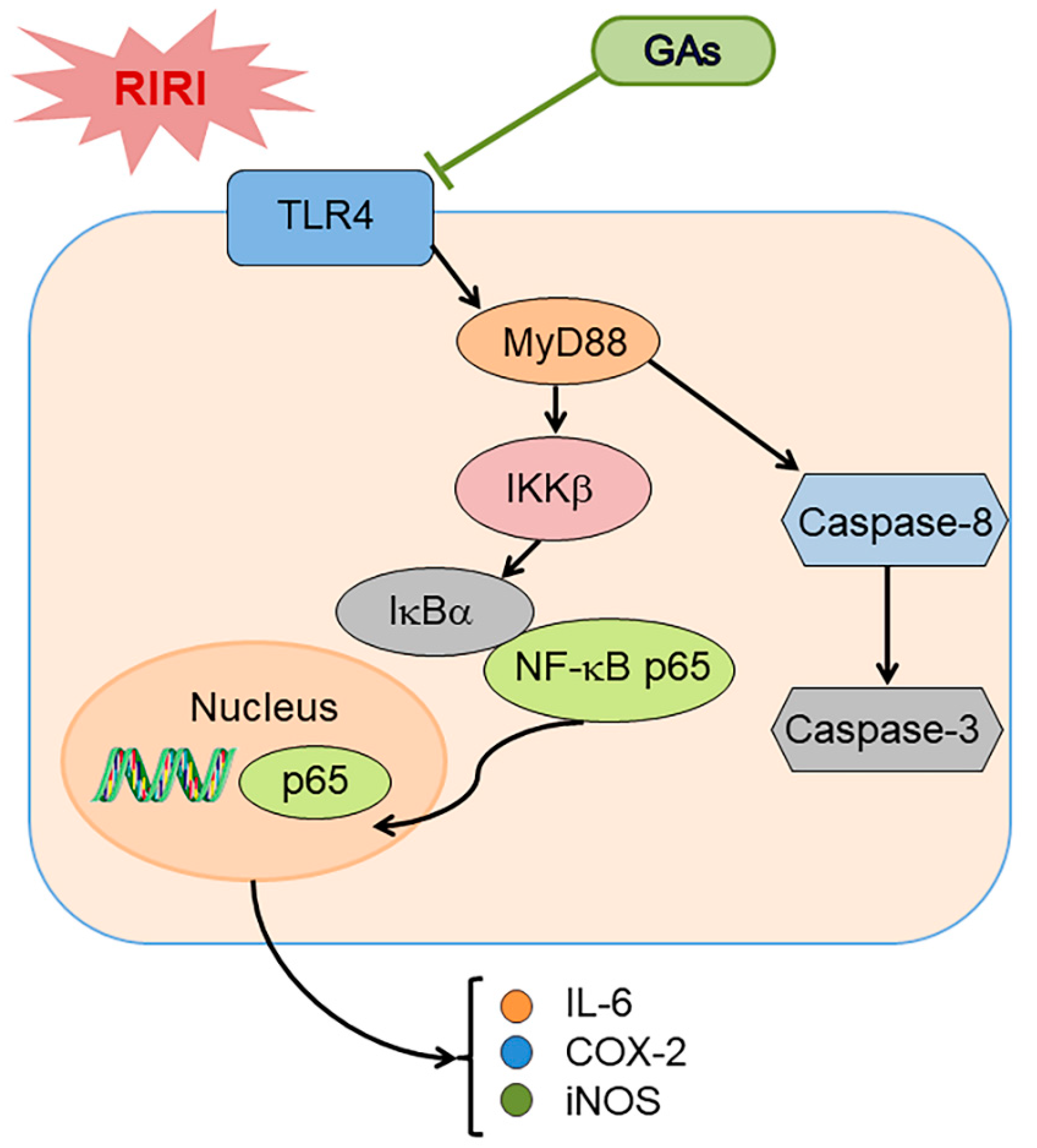
3. Discussion
4. Materials and Methods
4.1. Ganoderic Acids Preparation
4.2. Ethics Statement
4.3. RIRI Mouse Model
4.4. Blood BUN and Creatinine Measurement
4.5. Measurement of GSH and SOD
4.6. Hematoxylin-Eosin Staining and Terminal Deoxynucleotidyl Transferase-Mediated 2′-Deoxyuridine 5′-Triphosphate Nick-End Labeling (TUNEL) Assay
4.7. Cell Culture
4.8. Cell Viability Assay
4.9. Cell Hypoxia/Reoxygenation (H/R)
4.10. Western Blot Analysis
4.11. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gueler, F.; Gwinner, W.; Schwarz, A.; Haller, H. Long-term effects of acute ischemia and reperfusion injury. Kidney Int. 2004, 66, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.C.; Pavan, N.; Parekh, D.J. Current Paradigm for Ischemia in Kidney Surgery. J. Urol. 2016, 195, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Biglarnia, A.R.; Huber-Lang, M.; Mohlin, C.; Ekdahl, K.N.; Nilsson, B. The multifaceted role of complement in kidney transplantation. Nat. Rev. Nephrol. 2018, 14, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Investigators, R.R.T.S.; Bellomo, R.; Cass, A.; Cole, L.; Finfer, S.; Gallagher, M.; Lo, S.; McArthur, C.; McGuinness, S.; Myburgh, J.; et al. Intensity of continuous renal-replacement therapy in critically ill patients. N. Engl. J. Med. 2009, 361, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Zhong, D.; Wang, H.; Liu, M.; Li, X.; Huang, M.; Zhou, H.; Lin, S.; Lin, Z.; Yang, B. Ganoderma lucidum polysaccharide peptide prevents renal ischemia reperfusion injury via counteracting oxidative stress. Sci. Rep. 2015, 5, 16910. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Zuk, A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004, 66, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Munshi, R.; Hsu, C.; Himmelfarb, J. Advances in understanding ischemic acute kidney injury. BMC Med. 2011, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef] [Green Version]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, R.; Wang, Y.; Niu, Z.; Chen, T.; Wang, C.; Jin, L.; Huang, Q.; Li, Q.; Wang, X.M.; et al. Regulatory innate lymphoid cells suppress innate immunity and reduce renal ischemia/reperfusion injury. Kidney Int. 2020, 97, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, K.; Han, P.; Qi, X.; Zhang, W.; Niu, T. Octreotide Ameliorates Renal Ischemia/Reperfusion Injury via Antioxidation and Anti-inflammation. Transplant. Proc. 2017, 49, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Tian, D.; Liu, Y.; Li, H.; Zhu, J.; Li, M.; Xin, M.; Xia, J. Review of the molecular mechanisms of Ganoderma lucidum triterpenoids: Ganoderic acids A, C2, D, F, DM, X and Y. Eur. J. Med. Chem. 2019, 174, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Sheng, F.; Zhang, L.; Wang, S.; Yang, L.; Li, P. Deacetyl Ganoderic Acid F Inhibits LPS-Induced Neural Inflammation via NF-kappaB Pathway Both In Vitro and In Vivo. Nutrients 2019, 12, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Xie, P. Ganoderic acid A holds promising cytotoxicity on human glioblastoma mediated by incurring apoptosis and autophagy and inactivating PI3K/AKT signaling pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22392. [Google Scholar] [CrossRef] [PubMed]
- Ren, L. Protective effect of ganoderic acid against the streptozotocin induced diabetes, inflammation, hyperlipidemia and microbiota imbalance in diabetic rats. Saudi J. Biol. Sci. 2019, 26, 1961–1972. [Google Scholar] [CrossRef]
- Gill, B.S.; Navgeet; Mehra, R.; Kumar, V.; Kumar, S. Ganoderic acid, lanostanoid triterpene: A key player in apoptosis. Investig. New Drugs 2018, 36, 136–143. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.F.; Wang, K.; Jia, Z.M.; Guo, Y.J.; Guan, Y.W.; Li, Z.H.; Ai, X. Lentivirus-Mediated Silencing of Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase 2 Inhibits Release of Inflammatory Cytokines and Apoptosis in Renal Tubular Epithelial Cells Via Inhibition of the TLR4/NF-kB Pathway in Renal Ischemia-Reperfusion Injury. Kidney Blood Press. Res. 2018, 43, 1084–1103. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Lorne, E.; Dupont, H.; Abraham, E. Toll-like receptors 2 and 4: Initiators of non-septic inflammation in critical care medicine? Intensive Care Med. 2010, 36, 1826–1835. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Deng, S.; Liu, F.; He, Z. Ursolic Acid Ameliorates Inflammation in Cerebral Ischemia and Reperfusion Injury Possibly via High Mobility Group Box 1/Toll-Like Receptor 4/NFkappaB Pathway. Front. Neurol. 2018, 9, 253. [Google Scholar] [CrossRef]
- Han, F.; Dou, M.; Wang, Y.; Xu, C.; Li, Y.; Ding, X.; Xue, W.; Zheng, J.; Tian, P.; Ding, C. Cordycepin protects renal ischemia/reperfusion injury through regulating inflammation, apoptosis, and oxidative stress. Acta Biochim. Biophys. Sin. 2020, 52, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.F.; Hosgood, S.A.; Nicholson, M.L. Ischemia-reperfusion injury in renal transplantation: 3 key signaling pathways in tubular epithelial cells. Kidney Int. 2019, 95, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa-Costa, M.; Azevedo, H.; Amano, M.T.; Goncalves, G.M.; Hyane, M.I.; Cenedeze, M.A.; Renesto, P.G.; Pacheco-Silva, A.; Moreira-Filho, C.A.; Camara, N.O. Transcriptome analysis of renal ischemia/reperfusion injury and its modulation by ischemic pre-conditioning or hemin treatment. PLoS ONE 2012, 7, e49569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, A.; Wang, W.; Shi, H.; Wang, J.; Liu, T. Identification of Hub Genes and Pathways in a Rat Model of Renal Ischemia-Reperfusion Injury Using Bioinformatics Analysis of the Gene Expression Omnibus (GEO) Dataset and Integration of Gene Expression Profiles. Med. Sci. Monit. 2019, 25, 8403–8411. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal Inj. Prev. 2015, 4, 20–27. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef] [Green Version]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Tanaka, S.; Okusa, M.D. Crosstalk between the nervous system and the kidney. Kidney Int. 2020, 97, 466–476. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. Inflammatory cytokines in acute renal failure. Kidney Int. Suppl. 2004, 66, S56–S61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xia, J.; Zhang, Y.; Xiao, F.; Wang, J.; Gao, H.; Liu, Y.; Rong, S.; Yao, Y.; Xu, G.; et al. HMGB1-TLR4 signaling participates in renal ischemia reperfusion injury and could be attenuated by dexamethasone-mediated inhibition of the ERK/NF-kappaB pathway. Am. J. Transl. Res. 2016, 8, 4054–4067. [Google Scholar]
- Zi, S.F.; Li, J.H.; Liu, L.; Deng, C.; Ao, X.; Chen, D.D.; Wu, S.Z. Dexmedetomidine-mediated protection against septic liver injury depends on TLR4/MyD88/NF-kappaB signaling downregulation partly via cholinergic anti-inflammatory mechanisms. Int. Immunopharmacol. 2019, 76, 105898. [Google Scholar] [CrossRef]
- Gu, L.; Tao, Y.; Chen, C.; Ye, Y.; Xiong, X.; Sun, Y. Initiation of the inflammatory response after renal ischemia/reperfusion injury during renal transplantation. Int. Urol. Nephrol. 2018, 50, 2027–2035. [Google Scholar] [CrossRef]
- Chen, J.; Hartono, J.R.; John, R.; Bennett, M.; Zhou, X.J.; Wang, Y.; Wu, Q.; Winterberg, P.D.; Nagami, G.T.; Lu, C.Y. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011, 80, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Ting, J.; Alfonso, Z.; Strem, B.M.; Fraser, J.K.; Rutenberg, J.; Kuo, H.C.; Pinkernell, K. Fresh and cryopreserved, uncultured adipose tissue-derived stem and regenerative cells ameliorate ischemia-reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 2010, 25, 3874–3884. [Google Scholar] [CrossRef] [PubMed]
- Gennip, A.; Broers, N.J.H.; Meulen, K.J.T.; Canaud, B.; Christiaans, M.H.L.; Cornelis, T.; Gelens, M.; Hermans, M.M.H.; Konings, C.; Net, J.B.V.; et al. Endothelial dysfunction and low-grade inflammation in the transition to renal replacement therapy. PLoS ONE 2019, 14, e0222547. [Google Scholar] [CrossRef] [Green Version]
- Satou, R.; Gonzalez-Villalobos, R.A.; Miyata, K.; Ohashi, N.; Urushihara, M.; Acres, O.W.; Navar, L.G.; Kobori, H. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol. Cell Endocrinol. 2009, 311, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lan, S.; Dieude, M.; Sabo-Vatasescu, J.P.; Karakeussian-Rimbaud, A.; Turgeon, J.; Qi, S.; Gunaratnam, L.; Patey, N.; Hebert, M.J. Caspase-3 Is a Pivotal Regulator of Microvascular Rarefaction and Renal Fibrosis after Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2018, 29, 1900–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, B.; Li, Y.; Sun, S.; Yang, Y.; Lv, Y.; Wang, L.; Song, M.; Chen, M.; Wu, C.; Pan, H.; et al. Ganoderic acid A attenuates lipopolysaccharide-induced lung injury in mice. Biosci. Rep. 2019, 39, BSR20190301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lixin, X.; Lijun, Y.; Songping, H. Ganoderic acid A against cyclophosphamide-induced hepatic toxicity in mice. J. Biochem. Mol. Toxicol. 2019, 33, e22271. [Google Scholar] [CrossRef]
- Shi, J.; Wang, H.; Liu, J.; Zhang, Y.; Luo, J.; Li, Y.; Yang, C.; Jiang, J. Ganoderic acid B attenuates LPS-induced lung injury. Int. Immunopharmacol. 2020, 88, 106990. [Google Scholar] [CrossRef]
- Meng, J.; Sai-Zhen, W.; He, J.Z.; Zhu, S.; Huang, B.Y.; Wang, S.Y.; Li, M.; Zhou, H.; Lin, S.Q.; Yang, B.X. Ganoderic acid A is the effective ingredient of Ganoderma triterpenes in retarding renal cyst development in polycystic kidney disease. Acta Pharmacol. Sin. 2020, 41, 782–790. [Google Scholar] [CrossRef]
- Zhao, R.L.; He, Y.M. Network pharmacology analysis of the anti-cancer pharmacological mechanisms of Ganoderma lucidum extract with experimental support using Hepa1-6-bearing C57 BL/6 mice. J. Ethnopharmacol. 2018, 210, 287–295. [Google Scholar] [CrossRef]
- Lin, D.M.; Luo, H.J.; Lin, Z.X.; Lin, S.Q. Rapid separation of ganoderic acid from the extraction by-products of Ganoderma lucidum. Fujian Med. J. 2018, 40, 135–138. [Google Scholar]
- Geng, X.Q.; Ma, A.; He, J.Z.; Wang, L.; Jia, Y.L.; Shao, G.Y.; Li, M.; Zhou, H.; Lin, S.Q.; Ran, J.H.; et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-beta/Smad and MAPK signaling pathways. Acta Pharmacol. Sin. 2020, 41, 670–677. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, G.; He, J.; Meng, J.; Ma, A.; Geng, X.; Zhang, S.; Qiu, Z.; Lin, D.; Li, M.; Zhou, H.; et al. Ganoderic Acids Prevent Renal Ischemia Reperfusion Injury by Inhibiting Inflammation and Apoptosis. Int. J. Mol. Sci. 2021, 22, 10229. https://doi.org/10.3390/ijms221910229
Shao G, He J, Meng J, Ma A, Geng X, Zhang S, Qiu Z, Lin D, Li M, Zhou H, et al. Ganoderic Acids Prevent Renal Ischemia Reperfusion Injury by Inhibiting Inflammation and Apoptosis. International Journal of Molecular Sciences. 2021; 22(19):10229. https://doi.org/10.3390/ijms221910229
Chicago/Turabian StyleShao, Guangying, Jinzhao He, Jia Meng, Ang Ma, Xiaoqiang Geng, Shun Zhang, Zhiwei Qiu, Dongmei Lin, Min Li, Hong Zhou, and et al. 2021. "Ganoderic Acids Prevent Renal Ischemia Reperfusion Injury by Inhibiting Inflammation and Apoptosis" International Journal of Molecular Sciences 22, no. 19: 10229. https://doi.org/10.3390/ijms221910229
APA StyleShao, G., He, J., Meng, J., Ma, A., Geng, X., Zhang, S., Qiu, Z., Lin, D., Li, M., Zhou, H., Lin, S., & Yang, B. (2021). Ganoderic Acids Prevent Renal Ischemia Reperfusion Injury by Inhibiting Inflammation and Apoptosis. International Journal of Molecular Sciences, 22(19), 10229. https://doi.org/10.3390/ijms221910229