
Effects of SGLT2 Inhibitors beyond Glycemic Control—Focus on Myocardial SGLT1

Abstract
:1. Introduction
2. The Rationale behind Pharmacological SGLT2 and SGLT1 Inhibition
3. Cardiorenal Benefits of Pharmacological SGLT2 and Dual SGLT1/2 Inhibition in Patients with and without Type 2 Diabetes Mellitus
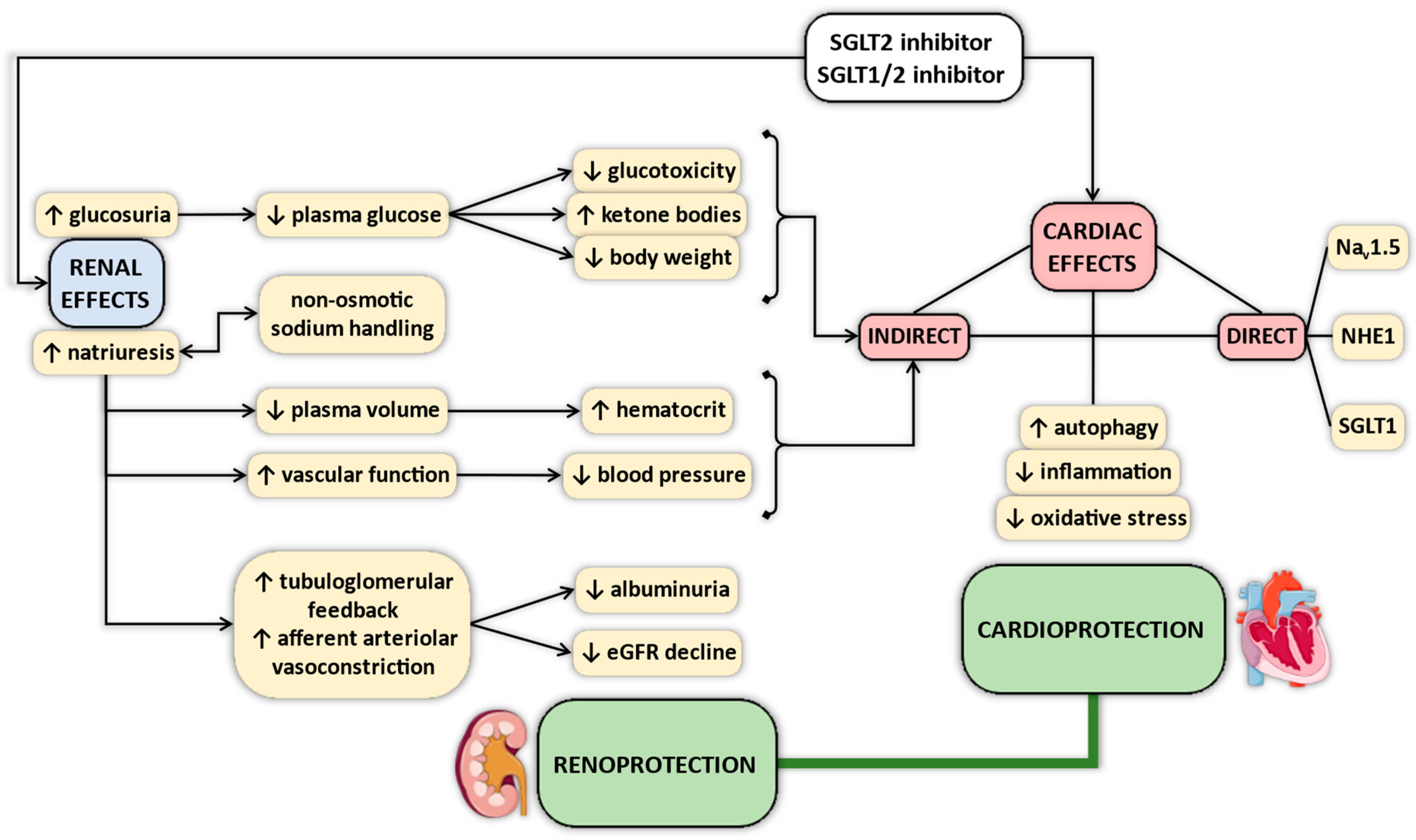
4. Proposed Mechanisms of Cardiovascular Protective Effects of SGLT2 Inhibitors—Why Myocardial SGLT1 Matters
5. Changes in Expression of SGLT1 in Various Myocardial Disease States
6. Localization of Myocardial SGLT1
7. Role of Myocardial SGLT1 in Glucose Uptake
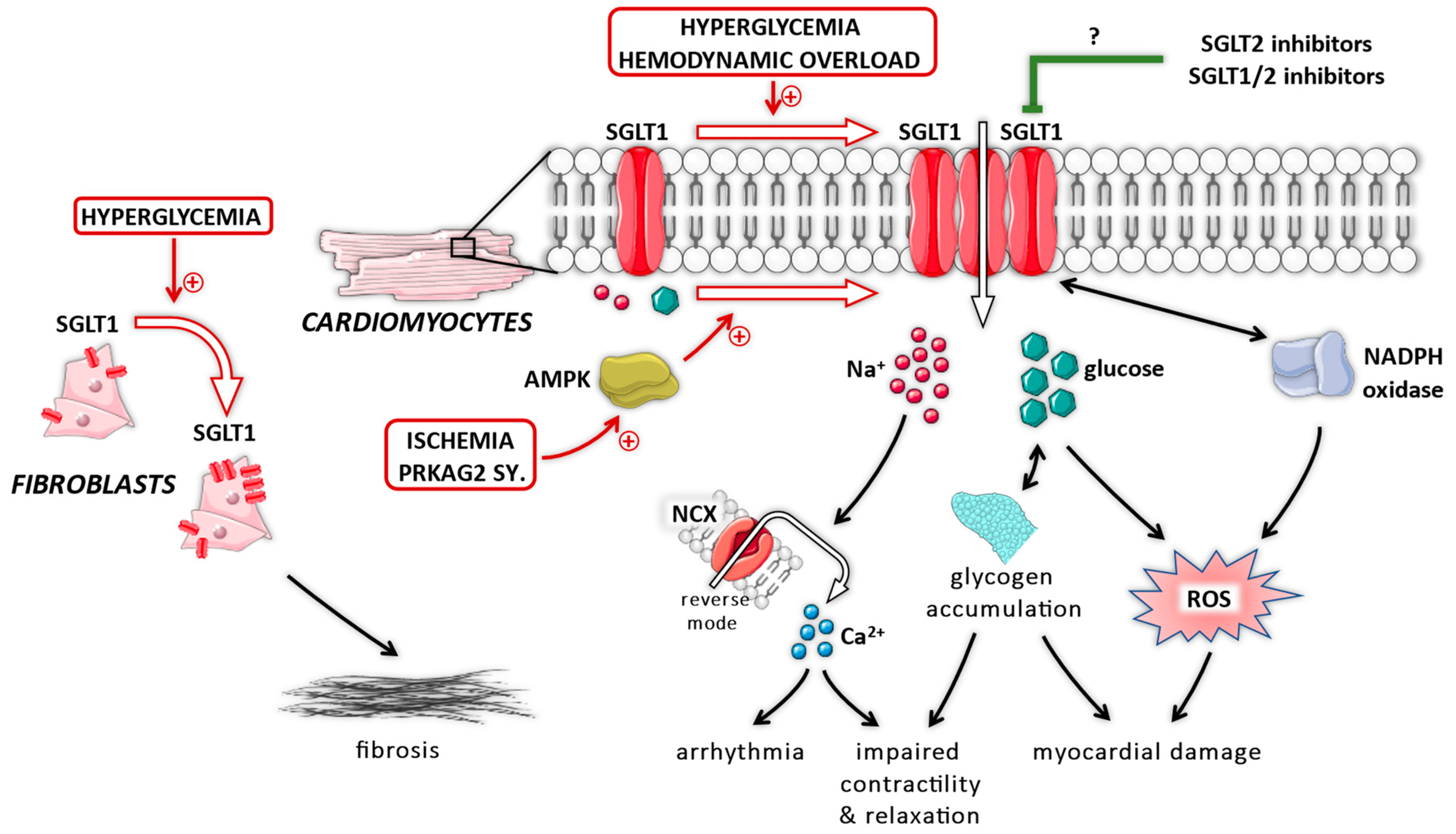
8. Role of Myocardial SGLT1 under Diabetic Conditions
9. Role of Myocardial SGLT1 under Non-Diabetic Conditions
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Lee, W.S.; You, G.; Brown, D.; Hediger, M.A. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Invest. 1994, 93, 397–404. [Google Scholar] [CrossRef]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol.-Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef] [Green Version]
- Komoroski, B.; Vachharajani, N.; Boulton, D.; Kornhauser, D.; Geraldes, M.; Li, L.; Pfister, M. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin. Pharmacol. Ther. 2009, 85, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cefalo, C.M.A.; Cinti, F.; Moffa, S.; Impronta, F.; Sorice, G.P.; Mezza, T.; Pontecorvi, A.; Giaccari, A. Sotagliflozin, the first dual SGLT inhibitor: Current outlook and perspectives. Cardiovasc. Diabetol. 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstock, J.; Cefalu, W.T.; Lapuerta, P.; Zambrowicz, B.; Ogbaa, I.; Banks, P.; Sands, A. Greater dose-ranging effects on A1C levels than on glucosuria with LX4211, a dual inhibitor of SGLT1 and SGLT2, in patients with type 2 diabetes on metformin monotherapy. Diabetes Care 2015, 38, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trico, D.; Baldi, S.; Tulipani, A.; Frascerra, S.; Macedo, M.P.; Mari, A.; Ferrannini, E.; Natali, A. Mechanisms through which a small protein and lipid preload improves glucose tolerance. Diabetologia 2015, 58, 2503–2512. [Google Scholar] [CrossRef]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; Charbonnel, B.; Cherney, D.Z.I.; Dagogo-Jack, S.; Pratley, R.; Greenberg, M.; Wang, S.; Huyck, S.; et al. Association of SGLT2 Inhibitors with Cardiovascular and Kidney Outcomes in Patients with Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Packer, M. Critical examination of mechanisms underlying the reduction in heart failure events with SGLT2 inhibitors: Identification of a molecular link between their actions to stimulate erythrocytosis and to alleviate cellular stress. Cardiovasc. Res. 2021, 117, 74–84. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J. Sodium-glucose Cotransporter 2 Inhibitors in Heart Failure: Potential Mechanisms of Action, Adverse Effects and Future Developments. Eur. Cardiol. 2019, 14, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Garcia-Ropero, A.; Vargas-Delgado, A.P.; Santos-Gallego, C.G.; Badimon, J.J. Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance. Int. J. Mol. Sci. 2019, 20, 3289. [Google Scholar] [CrossRef] [Green Version]
- Sayour, A.A.; Celeng, C.; Olah, A.; Ruppert, M.; Merkely, B.; Radovits, T. Sodium-glucose cotransporter 2 inhibitors reduce myocardial infarct size in preclinical animal models of myocardial ischaemia-reperfusion injury: A meta-analysis. Diabetologia 2021, 64, 737–748. [Google Scholar] [CrossRef]
- Bjornstad, P.; Greasley, P.J.; Wheeler, D.C.; Chertow, G.M.; Langkilde, A.M.; Heerspink, H.J.L.; van Raalte, D.H. The potential roles of osmotic and non-osmotic sodium handling in mediating effects of SGLT2 inhibitors on heart failure. J. Card. Fail. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.E.; McGuire, D.K.; Pitt, B.; Scirica, B.M.; Austin, B.; et al. Dapagliflozin Effects on Biomarkers, Symptoms, and Functional Status in Patients with Heart Failure With Reduced Ejection Fraction: The DEFINE-HF Trial. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Sattar, N.; Brueckmann, M.; Jamal, W.; Cotton, D.; et al. Empagliflozin in Patients With Heart Failure, Reduced Ejection Fraction, and Volume Overload: EMPEROR-Reduced Trial. J. Am. Coll. Cardiol. 2021, 77, 1381–1392. [Google Scholar] [CrossRef]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur. Heart J. 2021. [Google Scholar] [CrossRef]
- Zhou, L.; Cryan, E.V.; D’Andrea, M.R.; Belkowski, S.; Conway, B.R.; Demarest, K.T. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J. Cell Biochem. 2003, 90, 339–346. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef] [Green Version]
- Van Steenbergen, A.; Balteau, M.; Ginion, A.; Ferte, L.; Battault, S.; Ravenstein, C.M.; Balligand, J.L.; Daskalopoulos, E.P.; Gilon, P.; Despa, F.; et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 2017, 7, 41166. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- von Lewinski, D.; Gasser, R.; Rainer, P.P.; Huber, M.S.; Wilhelm, B.; Roessl, U.; Haas, T.; Wasler, A.; Grimm, M.; Bisping, E.; et al. Functional effects of glucose transporters in human ventricular myocardium. Eur. J. Heart Fail. 2010, 12, 106–113. [Google Scholar] [CrossRef]
- Sayour, A.A.; Olah, A.; Ruppert, M.; Barta, B.A.; Horvath, E.M.; Benke, K.; Polos, M.; Hartyanszky, I.; Merkely, B.; Radovits, T. Characterization of left ventricular myocardial sodium-glucose cotransporter 1 expression in patients with end-stage heart failure. Cardiovasc. Diabetol. 2020, 19. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuurbier, C.J.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Coronel, R. SGLT2 inhibitor empagliflozin inhibits the cardiac Na+/H+ exchanger 1: Persistent inhibition under various experimental conditions. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Uthman, L.; Nederlof, R.; Eerbeek, O.; Baartscheer, A.; Schumacher, C.; Buchholtz, N.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc. Res. 2019, 115, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Off-target effects of SGLT2 blockers: Empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, Q.; Qiu, Y.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.; Omoto, A.C.M.; Hall, M.E.; Li, J.; et al. Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure. J. Am. Heart Assoc. 2021, 10, e018298. [Google Scholar] [CrossRef] [PubMed]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Braunwald, E. Clinical Benefit of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Seidelmann, S.B.; Feofanova, E.; Yu, B.; Franceschini, N.; Claggett, B.; Kuokkanen, M.; Puolijoki, H.; Ebeling, T.; Perola, M.; Salomaa, V.; et al. Genetic Variants in SGLT1, Glucose Tolerance, and Cardiometabolic Risk. J. Am. Coll. Cardiol. 2018, 72, 1763–1773. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Mason, A.M.; Marz, W.; Kleber, M.E.; Niessner, A.; Bluher, M.; Speer, T.; Laufs, U. Genetic variation in sodium-glucose cotransporter 2 and heart failure. Clin. Pharmacol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ Concentration ([Na+]i) Is Elevated in Diabetic Hearts Due to Enhanced Na+-Glucose Cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef]
- Kanwal, A.; Nizami, H.L.; Mallapudi, S.; Putcha, U.K.; Mohan, G.K.; Banerjee, S.K. Inhibition of SGLT1 abrogates preconditioning-induced cardioprotection against ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 2016, 472, 392–398. [Google Scholar] [CrossRef]
- Sawa, Y.; Saito, M.; Ishida, N.; Ibi, M.; Matsushita, N.; Morino, Y.; Taira, E.; Hirose, M. Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 2020, 142, 16–25. [Google Scholar] [CrossRef]
- Sanchez-Mas, J.; Saura-Guillen, E.; Asensio-Lopez, M.C.; Soriano-Filiu, A.; Carmen Sanchez-Perez, M.; Hernandez-Martinez, A.M.; Lax, A.; Pascual-Figal, D. Temporal characterization of cardiac expression of glucose transporters SGLT and GLUT in an experimental model of myocardial infarction. Diabetes Metab. 2019, 45, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, N.; Ishida, N.; Ibi, M.; Saito, M.; Sanbe, A.; Shimojo, H.; Suzuki, S.; Koepsell, H.; Takeishi, Y.; Morino, Y.; et al. Chronic Pressure Overload Induces Cardiac Hypertrophy and Fibrosis via Increases in SGLT1 and IL-18 Gene Expression in Mice. Int. Heart J. 2018, 59, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Sayour, A.A.; Ruppert, M.; Oláh, A.; Benke, K.; Barta, B.A.; Zsáry, E.; Ke, H.; Horváth, E.M.; Merkely, B.; Radovits, T. Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure. Antioxidants 2021, 10, 1190. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, Q.; Zhang, Z.; Lu, D.; Meng, Z.; Wu, W. Inhibition of SGLT1 protects against glycemic variability-induced cardiac damage and pyroptosis of cardiomyocytes in diabetic mice. Life Sci. 2021, 271, 119116. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chai, Q.; Zhang, Z. Glucose fluctuation accelerates cardiac injury of diabetic mice via sodium-dependent glucose cotransporter 1 (SGLT1). Arch. Biochem. Biophys. 2021, 709, 108968. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Guan, L.; Meng, L.; Uzui, H.; Guo, H. SGLT1 Knockdown Attenuates Cardiac Fibroblast Activation in Diabetic Cardiac Fibrosis. Front. Pharmacol. 2021, 12, 700366. [Google Scholar] [CrossRef]
- Yoshii, A.; Nagoshi, T.; Kashiwagi, Y.; Kimura, H.; Tanaka, Y.; Oi, Y.; Ito, K.; Yoshino, T.; Tanaka, T.D.; Yoshimura, M. Cardiac ischemia-reperfusion injury under insulin-resistant conditions: SGLT1 but not SGLT2 plays a compensatory protective role in diet-induced obesity. Cardiovasc. Diabetol. 2019, 18. [Google Scholar] [CrossRef]
- Connelly, K.A.; Zhang, Y.; Desjardins, J.F.; Thai, K.; Gilbert, R.E. Dual inhibition of sodium-glucose linked cotransporters 1 and 2 exacerbates cardiac dysfunction following experimental myocardial infarction. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, Y.; Nagoshi, T.; Yoshino, T.; Tanaka, T.D.; Ito, K.; Harada, T.; Takahashi, H.; Ikegami, M.; Anzawa, R.; Yoshimura, M. Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice. PLoS ONE 2015, 10, e0130605. [Google Scholar]
- Banerjee, S.K.; Wang, D.W.; Alzamora, R.; Huang, X.N.; Pastor-Soler, N.M.; Hallows, K.R.; McGaffin, K.R.; Ahmad, F. SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J. Mol. Cell Cardiol. 2010, 49, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Lee, S.J.; Wang, D.; Huang, X.Y.; Ahmad, F. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J. Am. Heart Assoc. 2014, 3, e000899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.J.; Park, S.H.; Lee, Y.J. Signaling cascade of ANG II-induced inhibition of alpha-MG uptake in renal proximal tubule cells. Am. J. Physiol.-Ren. Physiol. 2004, 286, F634–F642. [Google Scholar] [CrossRef] [PubMed]
- Jae Han, H.; Yeong Park, J.; Jung Lee, Y.; Taub, M. Epidermal growth factor inhibits 14C-alpha-methyl-D-glucopyranoside uptake in renal proximal tubule cells: Involvement of PLC/PKC, p44/42 MAPK, and cPLA2. J. Cell Physiol. 2004, 199, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Elfeber, K.; Stumpel, F.; Gorboulev, V.; Mattig, S.; Deussen, A.; Kaissling, B.; Koepsell, H. Na(+)-D-glucose cotransporter in muscle capillaries increases glucose permeability. Biochem. Biophys. Res. Commun. 2004, 314, 301–305. [Google Scholar] [CrossRef]
- Meng, L.; Uzui, H.; Guo, H.; Tada, H. Role of SGLT1 in high glucose level-induced MMP-2 expression in human cardiac fibroblasts. Mol. Med. Rep. 2018, 17, 6887–6892. [Google Scholar] [CrossRef] [Green Version]
- Vrhovac, I.; Balen Eror, D.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radovic, N.; Jadrijevic, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na(+)-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch.-Eur. J. Physiol. 2015, 467, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Sala-Rabanal, M.; Hirayama, B.A.; Ghezzi, C.; Liu, J.; Huang, S.C.; Kepe, V.; Koepsell, H.; Yu, A.; Powell, D.R.; Thorens, B.; et al. Revisiting the physiological roles of SGLTs and GLUTs using positron emission tomography in mice. J. Physiol. 2016, 594, 4425–4438. [Google Scholar] [CrossRef] [PubMed]
- Ferte, L.; Marino, A.; Battault, S.; Bultot, L.; Van Steenbergen, A.; Bol, A.; Cumps, J.; Ginion, A.; Koepsell, H.; Dumoutier, L.; et al. New insight in understanding the contribution of SGLT1 in cardiac glucose uptake: Evidence for a truncated form in mice and humans. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H838–H853. [Google Scholar] [CrossRef] [PubMed]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Sayour, A.A.; Korkmaz-Icoz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Olah, A.; Benke, K.; Brune, M.; Benko, R.; Horvath, E.M.; et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Harada, S.; Tokuyama, S. Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res. 2012, 1489, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Ogihara, S.; Harada, S.; Tokuyama, S. Activation of cerebral sodium-glucose transporter type 1 function mediated by post-ischemic hyperglycemia exacerbates the development of cerebral ischemia. Neuroscience 2015, 310, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Nespoux, J.; Patel, R.; Hudkins, K.L.; Huang, W.; Freeman, B.; Kim, Y.C.; Koepsell, H.; Alpers, C.E.; Vallon, V. Gene deletion of the Na(+)-glucose cotransporter SGLT1 ameliorates kidney recovery in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol.-Ren. Physiol. 2019, 316, F1201–F1210. [Google Scholar] [CrossRef]
- Nespoux, J.; Patel, R.; Zhang, H.; Huang, W.; Freeman, B.; Sanders, P.W.; Kim, Y.C.; Vallon, V. Gene knockout of the Na(+)-glucose cotransporter SGLT2 in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol.-Ren. Physiol. 2020, 318, F1100–F1112. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trial Name (Acronym) | Population | Type of SGLT2 Inhibitor (vs. Placebo) | Selectivity for SGLT2 over SGLT1 | Primary Outcome | Hazard Ratio (and 95% CI) for Primary Outcome |
|---|---|---|---|---|---|
| EMPA-REG OUTCOME (ref: [2]) | T2DM + established CVD | empagliflozin | ~2700 | CV death, nonfatal myocardial infarction, and nonfatal stroke | 0.86 (0.74, 0.99) * |
| CANVAS Program (ref: [3]) | T2DM ± CVD (66% established CVD) | canagliflozin | ~260 | CV death, nonfatal myocardial infarction, and nonfatal stroke | 0.86 (0.75, 0.97) * |
| DECLARE-TIMI 58 (ref: [4]) | T2DM ± CVD (41% established CVD) | dapagliflozin | ~1200 | CV death, nonfatal myocardial infarction, and nonfatal ischemic stroke | 0.93 (0.84, 1.03) |
| CREDENCE (ref: [5]) | T2DM + albuminuric CKD (50% established CVD) | canagliflozin | ~260 | renal composite | 0.70 (0.59, 0.82) *** |
| VERTIS CV (ref: [6]) | T2DM + established CVD | ertugliflozin | ~2200 | CV death, nonfatal myocardial infarction, and nonfatal stroke | 0.97 (0.85, 1.11) |
| SCORED (ref: [7]) | T2DM + CKD (89% established CVD) | sotagliflozin | ~20 | CV death, hospitalization for HF, and urgent visits for HF | 0.74 (0.63, 0.88) *** |
| Trial Name (Acronym) | Population | Type of SGLT2 Inhibitor (vs. Placebo) | Selectivity for SGLT2 over SGLT1 | Primary Outcome | Hazard Ratio (and 95% CI) for Primary Outcome |
|---|---|---|---|---|---|
| DAPA-HF (ref: [17]) | HFrEF ± T2DM | dapagliflozin | ~1200 | worsening HF and CV death | 0.74 (0.65, 0.85) *** |
| EMPEROR- Reduced (ref: [18]) | HFrEF ± T2DM | empagliflozin | ~2700 | hospitalization for worsening HF and CV death | 0.75 (0.65, 0.86) *** |
| SOLOIST-WHF (ref: [19]) | recent hospitalization for HF + T2DM | sotagliflozin | ~20 | hospitalizations and urgent visits for HF, and CV death | 0.67 (0.52, 0.85) *** |
| EMPEROR- Preserved (ref: [20]) | HFpEF ± T2DM | empagliflozin | ~2700 | hospitalization for HF and CV death | 0.79 (0.69, 0.90) *** |
| SGLT1 Expression | ||||||
|---|---|---|---|---|---|---|
| Condition | Subtype | Ref: [41] | Ref: [39] | Ref: [51] | Ref: [54] | Ref: [40] |
| HF | hypertrophic CM | ~ | ↑ | (↑) | ||
| HF | ischemic CM | ↑ | ↑ | ↑ | (↑) | ~ |
| HF | dilated CM | ↑ | ~ | (↑) | ~ | |
| HF | metabolic syndrome/T2DM | ↑ | ↑ | ↑ | ||
| HF | post-LVAD | ↑ | ||||
| Condition | SGLT1 Expression | Ref: | SGLT1 Expression | Ref: |
|---|---|---|---|---|
| acute myocardial IRI | ↑ | [55,56] | ~ | [65,67] |
| permanent LAD ligation | ↑ | [51,57,58] | ~ | [64] |
| hemodynamic overload-induced HF | ↑ | [59,60] | ||
| metabolic syndrome/T2DM | ↑ | [51,54,61,62,63,64] | ~ | [65] |
| T1DM | ↓ | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayour, A.A.; Ruppert, M.; Oláh, A.; Benke, K.; Barta, B.A.; Zsáry, E.; Merkely, B.; Radovits, T. Effects of SGLT2 Inhibitors beyond Glycemic Control—Focus on Myocardial SGLT1. Int. J. Mol. Sci. 2021, 22, 9852. https://doi.org/10.3390/ijms22189852
Sayour AA, Ruppert M, Oláh A, Benke K, Barta BA, Zsáry E, Merkely B, Radovits T. Effects of SGLT2 Inhibitors beyond Glycemic Control—Focus on Myocardial SGLT1. International Journal of Molecular Sciences. 2021; 22(18):9852. https://doi.org/10.3390/ijms22189852
Chicago/Turabian StyleSayour, Alex Ali, Mihály Ruppert, Attila Oláh, Kálmán Benke, Bálint András Barta, Eszter Zsáry, Béla Merkely, and Tamás Radovits. 2021. "Effects of SGLT2 Inhibitors beyond Glycemic Control—Focus on Myocardial SGLT1" International Journal of Molecular Sciences 22, no. 18: 9852. https://doi.org/10.3390/ijms22189852
APA StyleSayour, A. A., Ruppert, M., Oláh, A., Benke, K., Barta, B. A., Zsáry, E., Merkely, B., & Radovits, T. (2021). Effects of SGLT2 Inhibitors beyond Glycemic Control—Focus on Myocardial SGLT1. International Journal of Molecular Sciences, 22(18), 9852. https://doi.org/10.3390/ijms22189852