
Germline Whole-Gene Deletion of FH Diagnosed from Tumor Profiling

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
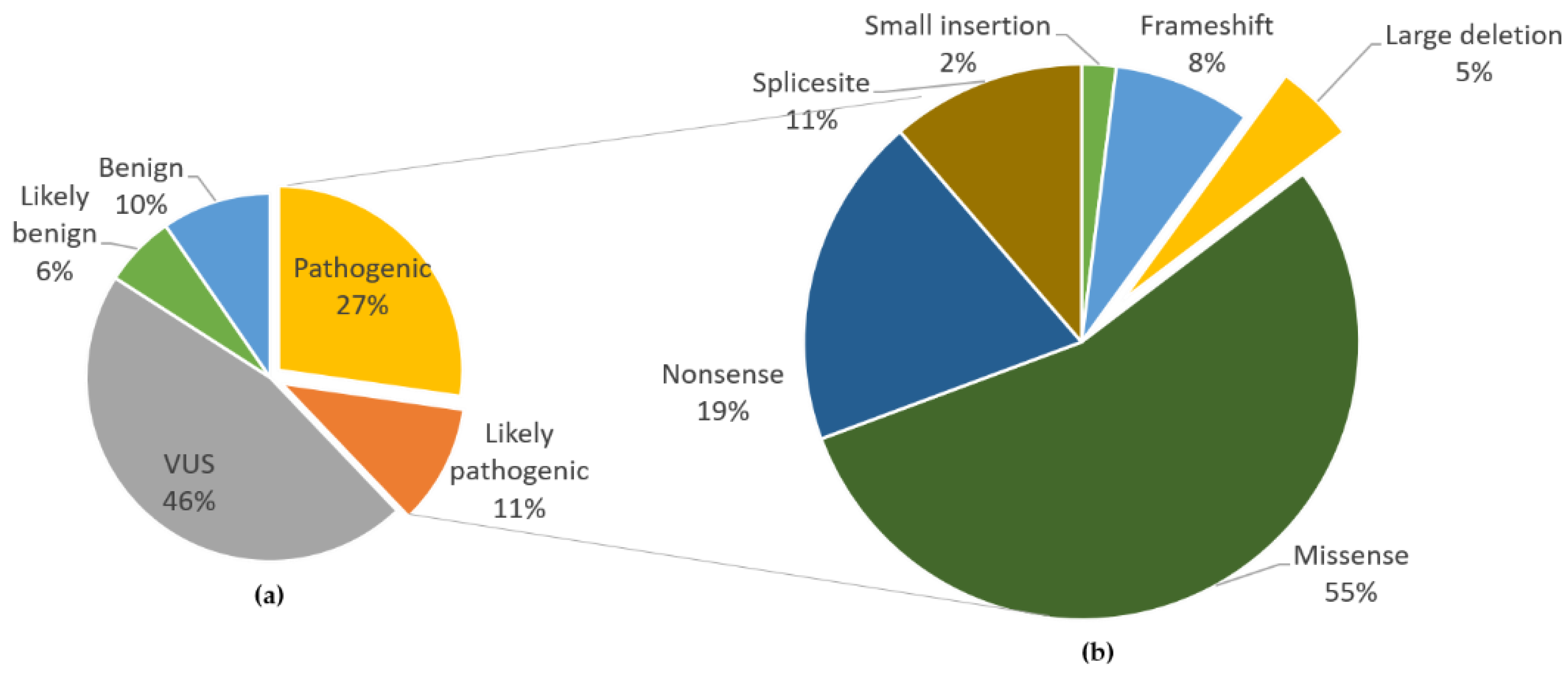
1. Introduction
2. Case Presentation
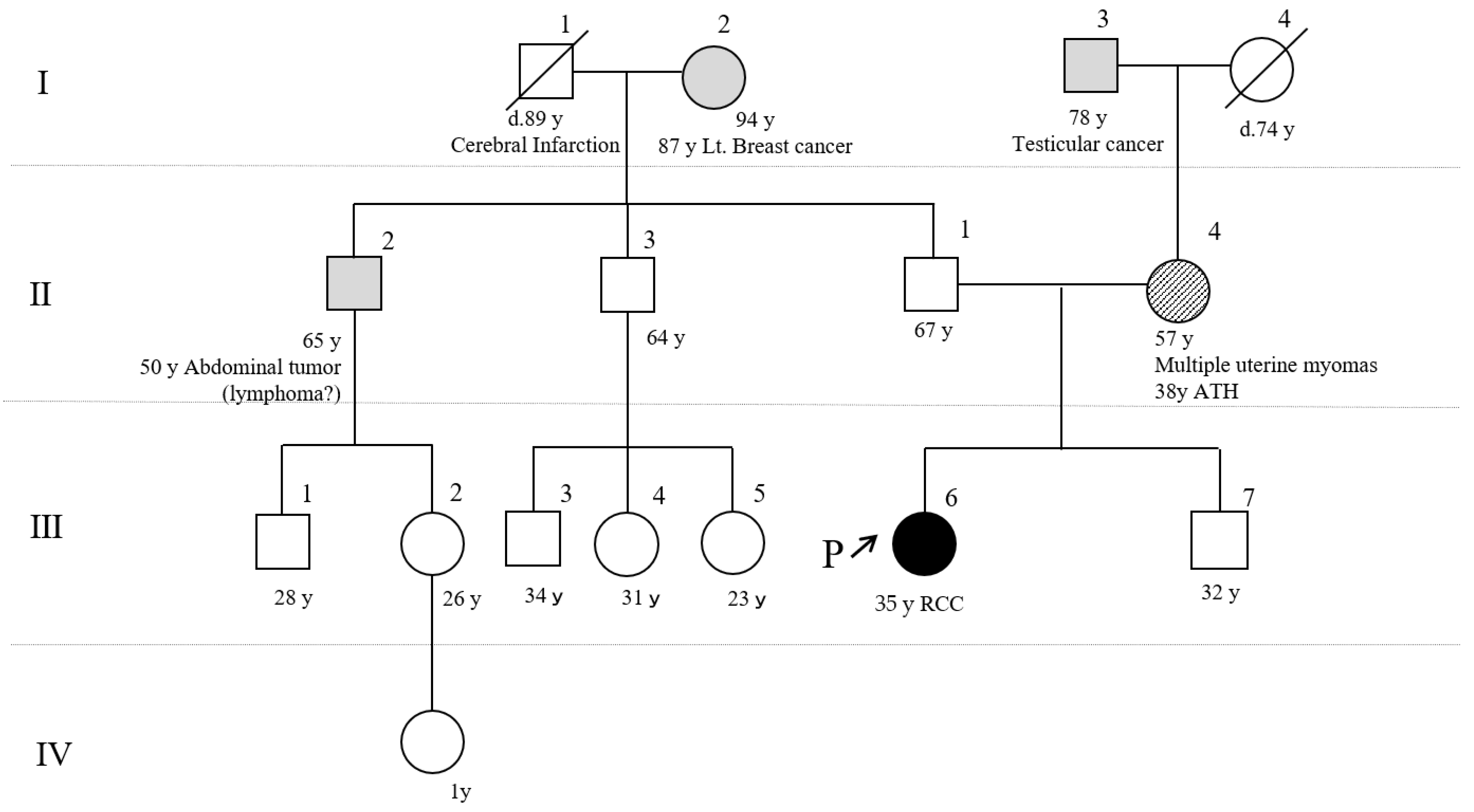
2.1. Patient’s Information and Clinical Course
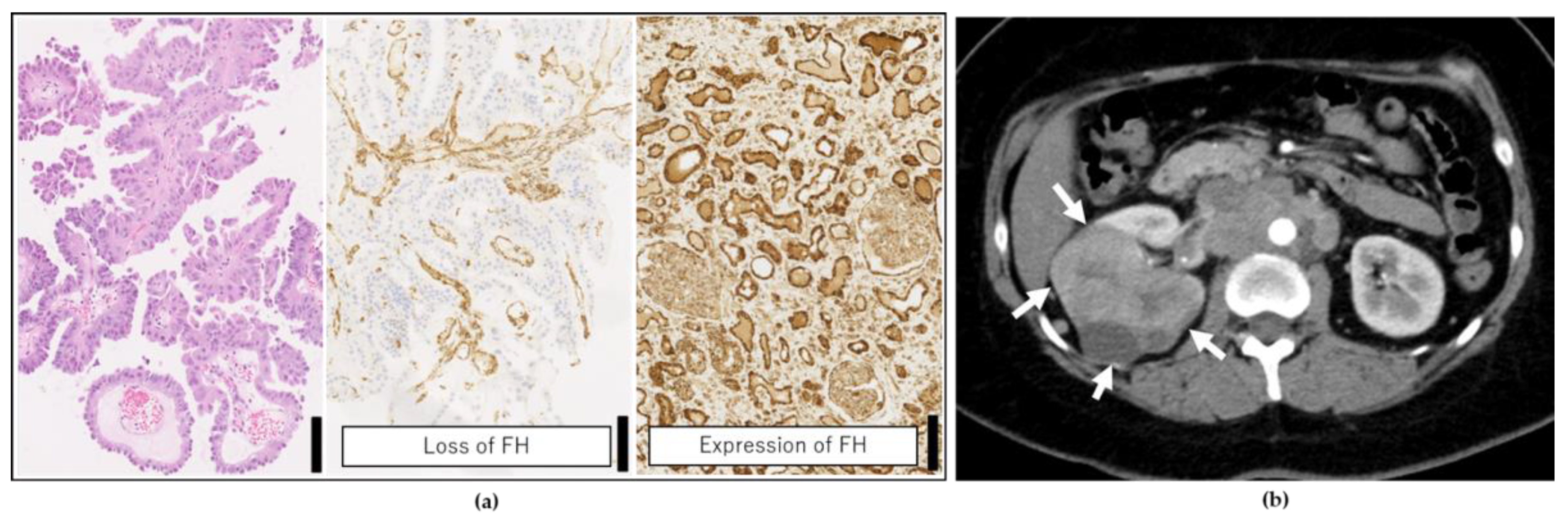
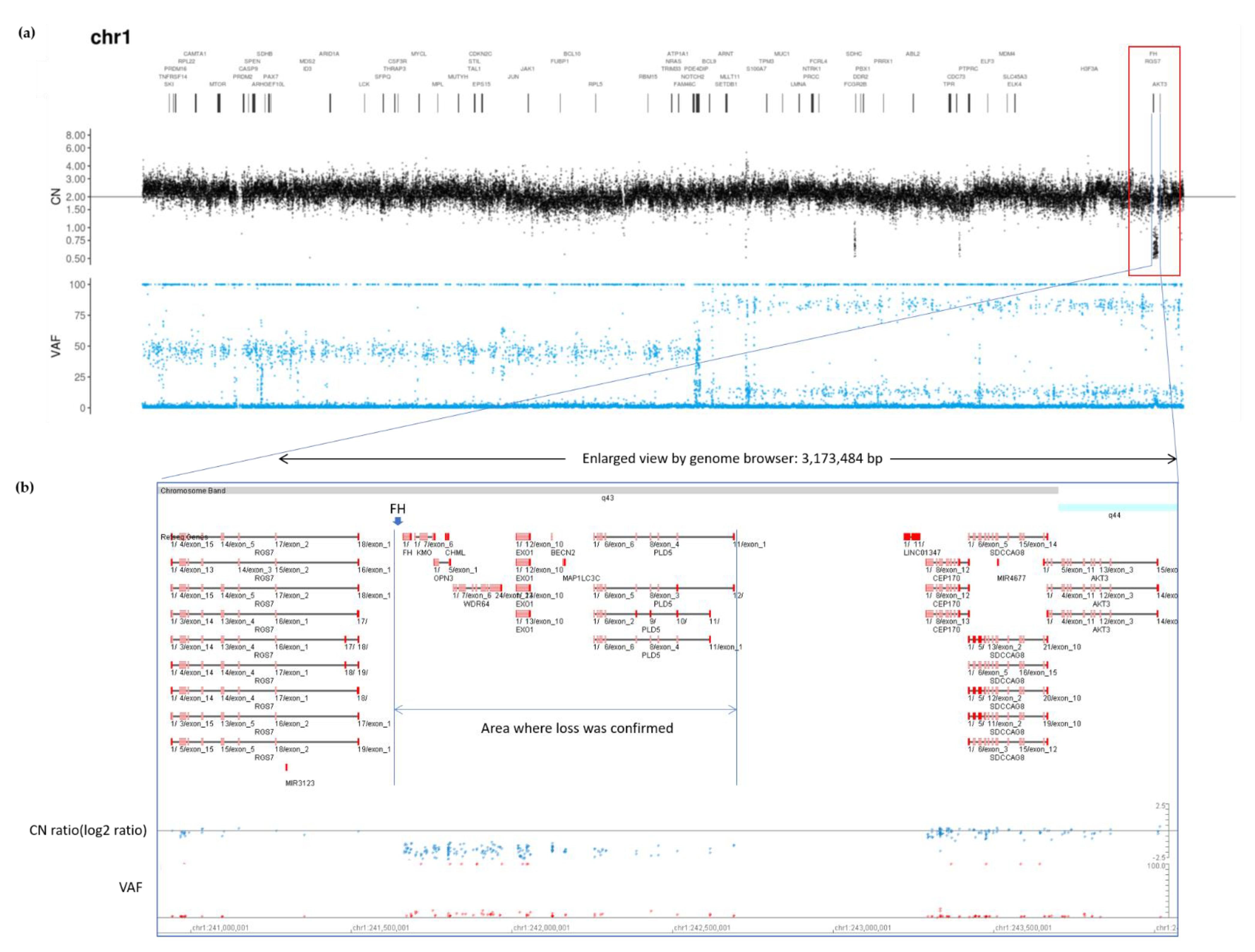
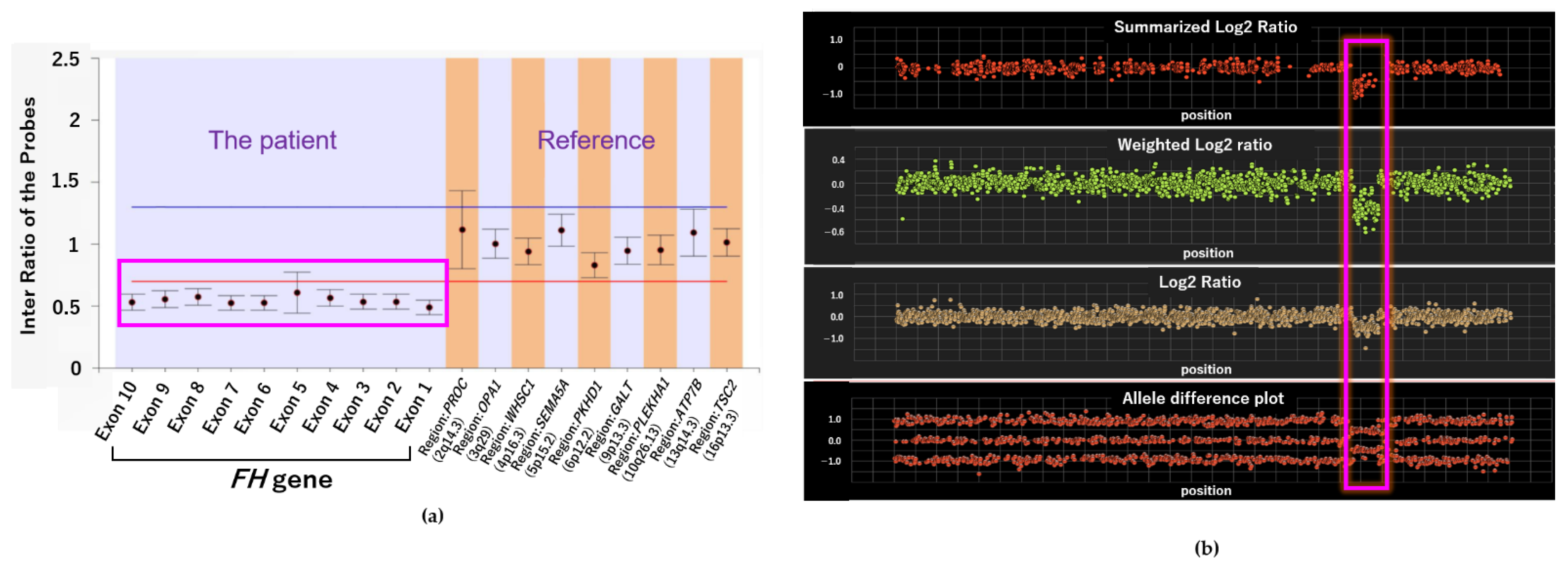
2.2. Genetic Testing Results
2.3. Materials and Methods
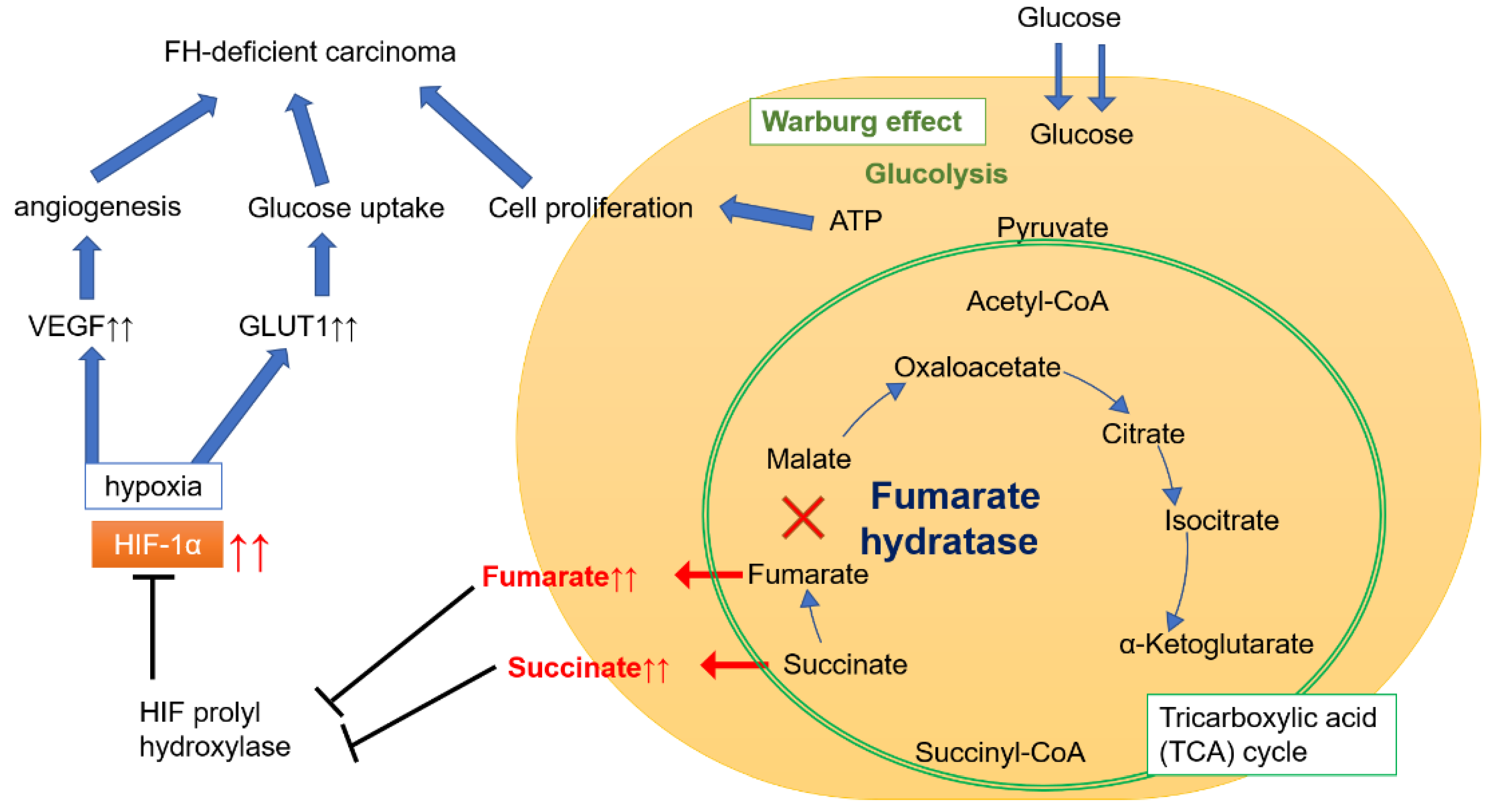
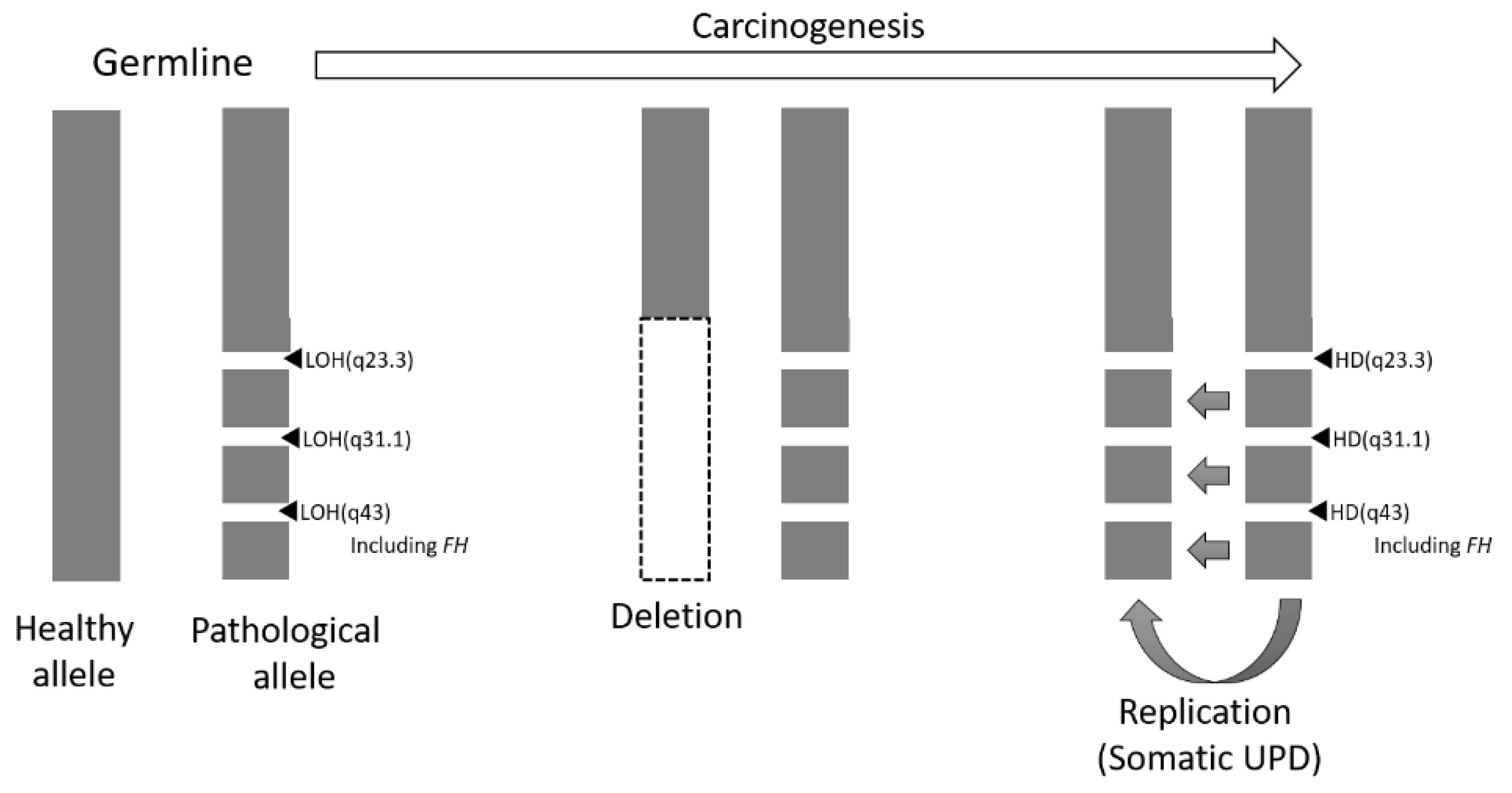
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Toro, J.R.; Nickerson, M.L.; Wei, M.-H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; et al. Mutations in the Fumarate Hydratase Gene Cause Hereditary Leiomyomatosis and Renal Cell Cancer in Families in North America. Am. J. Hum. Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Smit, D.L.; Mensenkamp, A.R.; Badeloe, S.; Breuning, M.H.; Simon, M.E.; van Spaendonck, K.Y.; Aalfs, C.M.; Post, J.G.; Shanley, S.; Krapels, I.P.; et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin. Genet. 2011, 79, 49–59. [Google Scholar] [CrossRef]
- Gomy, I.; Silva, W.A., Jr. Molecular Pathogenesis of Renal Cell Carcinoma: A Review. In Emerging Research and Treatments in Renal Cell Carcinoma; Amato, R.J., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0022-5. [Google Scholar] [CrossRef]
- Linehan, W.M.; Rouault, T.A. Molecular pathways: Fumarate hydratase-deficient kidney cancer—Targeting the Warburg effect in cancer. Clin. Cancer Res. 2013, 19, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Donoghue, M.; Talukdar, S.; Bandlamudi, C.; Srinivasan, P.; Vivek, M.; Jezdic, S.; Hanson, H.; Snape, K.; Kulkarni, A.; et al. Germline-focussed analysis of tumour-only sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2019, 30, 1221–1231. [Google Scholar] [CrossRef]
- Li, M.M.; Chao, E.; Esplin, E.D.; Miller, D.T.; Nathanson, K.L.; Plon, S.E.; Scheuner, M.T.; Stewart, D.R. Points to consider for reporting of germline variation in patients undergoing tumor testing: A statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Tanishima, S.; Fujii, K.; Mori, R.; Okada, C.; Yanagita, E.; Shibata, Y.; Matsuoka, R.; Amano, T.; Yamada, T.; et al. Clinical impact of a cancer genomic profiling test using an in-house comprehensive targeted sequencing system. Cancer Sci. 2020, 111, 3926–3937. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef]
- Yoshihama, T.; Hirasawa, A.; Sugano, K.; Yoshida, T.; Ushiama, M.; Ueki, A.; Akahane, T.; Nanki, Y.; Sakai, K.; Makabe, T.; et al. Germline multigene panel testing revealed a BRCA2 pathogenic variant in a patient with suspected Lynch syndrome. Int. Cancer Conf. J. 2021, 10, 6–10. [Google Scholar] [CrossRef]
- Sugano, K.; Nakajima, T.; Sekine, S.; Taniguchi, H.; Saito, S.; Takahashi, M.; Ushiama, M.; Sakamoto, H.; Yoshida, T. Germline PMS2 mutation screened by mismatch repair protein immunohistochemistry of colorectal cancer in Japan. Cancer Sci. 2016, 107, 1677–1686. [Google Scholar] [CrossRef]
- Bayley, J.P.; Launonen, V.; Tomlinson, I.P. The FH mutation database: An online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med. Genet. 2008, 9, 20. [Google Scholar] [CrossRef]
- Bayley, J.P. Global Variome Shared LOVD FH (Fumarate Hydratase). Available online: https://databases.lovd.nl/shared/genes/FH (accessed on 9 February 2021).
- Ahvenainen, T.; Lehtonen, H.J.; Lehtonen, R.; Vahteristo, P.; Aittomaki, K.; Baynam, G.; Dommering, C.; Eng, C.; Gruber, S.B.; Gronberg, H.; et al. Mutation screening of fumarate hydratase by multiplex ligation-dependent probe amplification: Detection of exonic deletion in a patient with leiomyomatosis and renal cell cancer. Cancer Genet. Cytogenet. 2008, 183, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Gardie, B.; Remenieras, A.; Kattygnarath, D.; Bombled, J.; Lefevre, S.; Perrier-Trudova, V.; Rustin, P.; Barrois, M.; Slama, A.; Avril, M.F.; et al. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J. Med. Genet. 2011, 48, 226–234. [Google Scholar] [CrossRef]
- Furuya, M.; Iribe, Y.; Nagashima, Y.; Kambe, N.; Ohe, C.; Kinoshita, H.; Sato, C.; Kishida, T.; Okubo, Y.; Numakura, K.; et al. Clinicopathological and molecular features of hereditary leiomyomatosis and renal cell cancer-associated renal cell carcinomas. J. Clin. Pathol. 2020, 73, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Kamai, T.; Abe, H.; Arai, K.; Murakami, S.; Sakamoto, S.; Kaji, Y.; Yoshida, K.I. Radical nephrectomy and regional lymph node dissection for locally advanced type 2 papillary renal cell carcinoma in an at-risk individual from a family with hereditary leiomyomatosis and renal cell cancer: A case report. BMC Cancer 2016, 16, 232. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N.; Tsutsui, M.; Iguchi, M.; Nobuoka, E.; Uehara, T.; Sonobe, Y.; Morinaga, Y.; Shibuya, S.; Oda, W.; Yanai, H.; et al. Fumarate hydratase-deficient renal cell carcinoma: A clinicopathological study of seven cases including hereditary and sporadic forms. Ann. Diagn. Pathol. 2020, 49, 151599. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, M.; Chihara, Y.; Lou, Y.; Torimoto, K.; Kagebayashi, Y.; Tamura, K.; Shuin, T.; Fujimoto, K.; Kuniyasu, H.; Samma, S. Novel missense mutation in the FH gene in familial renal cell cancer patients lacking cutaneous leiomyomas. BMC Res. Notes 2014, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Udaka, N.; Hasumi, H.; Nakaigawa, N.; Nagashima, Y.; Tanaka, R.; Kato, I.; Yao, M.; Furuya, M. Histopathological analysis of aggressive renal cell carcinoma harboring a unique germline mutation in fumarate hydratase. Pathol. Int. 2018. [Google Scholar] [CrossRef]
- Yonamine, T.; Kaname, T.; Chinen, Y.; Tamashiro, K.; Kosuge, N.; Saito, S. Hereditary leiomyomatosis and renal cell cancer (HLRCC): A case report. Urol. Case Rep. 2020, 30, 101141. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.A.; Barclay, E.; Rowan, A.J.; Tyrer, J.P.; Calonje, E.; Manek, S.; Kelsell, D.; Leigh, I.; Olpin, S.; Tomlinson, I.P. Clinical features of multiple cutaneous and uterine leiomyomatosis: An underdiagnosed tumor syndrome. Arch Dermatol. 2005, 141, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Ferlicot, S.; Guillaud-Bataille, M.; Le Teuff, G.; Genestie, C.; Deveaux, S.; Slama, A.; Poulalhon, N.; Escudier, B.; Albiges, L.; et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin. Genet. 2017, 92, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Hol, J.A.; Jongmans, M.C.J.; Littooij, A.S.; de Krijger, R.R.; Kuiper, R.P.; van Harssel, J.J.T.; Mensenkamp, A.; Simons, M.; Tytgat, G.A.M.; van den Heuvel-Eibrink, M.M.; et al. Renal cell carcinoma in young FH mutation carriers: Case series and review of the literature. Fam. Cancer 2020, 19, 55–63. [Google Scholar] [CrossRef]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomäki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueki, A.; Sugano, K.; Misu, K.; Aimono, E.; Nakamura, K.; Tanishima, S.; Tanaka, N.; Mikami, S.; Hirasawa, A.; Ando, M.; et al. Germline Whole-Gene Deletion of FH Diagnosed from Tumor Profiling. Int. J. Mol. Sci. 2021, 22, 7962. https://doi.org/10.3390/ijms22157962
Ueki A, Sugano K, Misu K, Aimono E, Nakamura K, Tanishima S, Tanaka N, Mikami S, Hirasawa A, Ando M, et al. Germline Whole-Gene Deletion of FH Diagnosed from Tumor Profiling. International Journal of Molecular Sciences. 2021; 22(15):7962. https://doi.org/10.3390/ijms22157962
Chicago/Turabian StyleUeki, Arisa, Kokichi Sugano, Kumiko Misu, Eriko Aimono, Kohei Nakamura, Shigeki Tanishima, Nobuyuki Tanaka, Shuji Mikami, Akira Hirasawa, Miho Ando, and et al. 2021. "Germline Whole-Gene Deletion of FH Diagnosed from Tumor Profiling" International Journal of Molecular Sciences 22, no. 15: 7962. https://doi.org/10.3390/ijms22157962
APA StyleUeki, A., Sugano, K., Misu, K., Aimono, E., Nakamura, K., Tanishima, S., Tanaka, N., Mikami, S., Hirasawa, A., Ando, M., Yoshida, T., Oya, M., Nishihara, H., & Kosaki, K. (2021). Germline Whole-Gene Deletion of FH Diagnosed from Tumor Profiling. International Journal of Molecular Sciences, 22(15), 7962. https://doi.org/10.3390/ijms22157962