
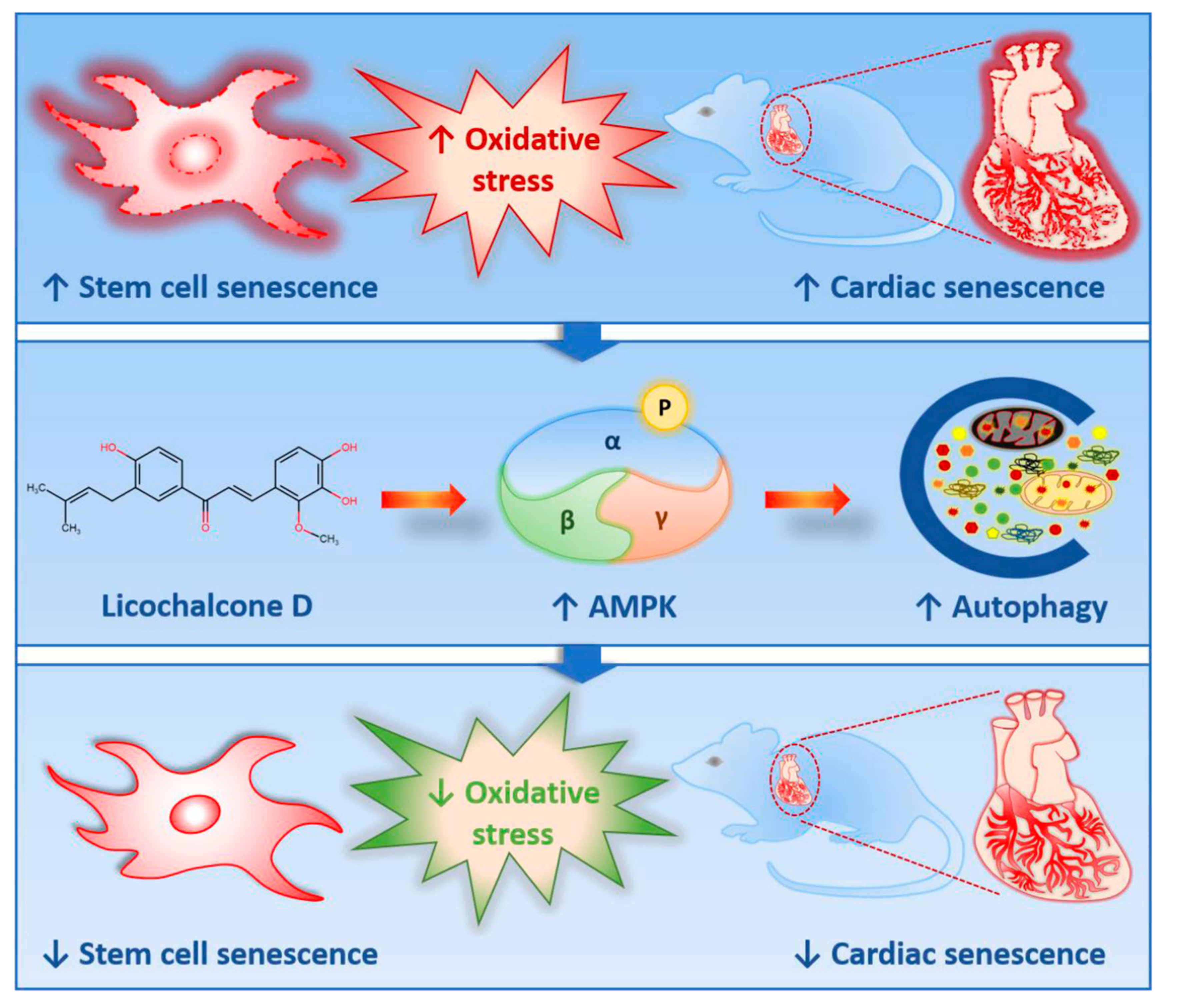
Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation

 ,
,  ,
,  ,
,  , and
, and 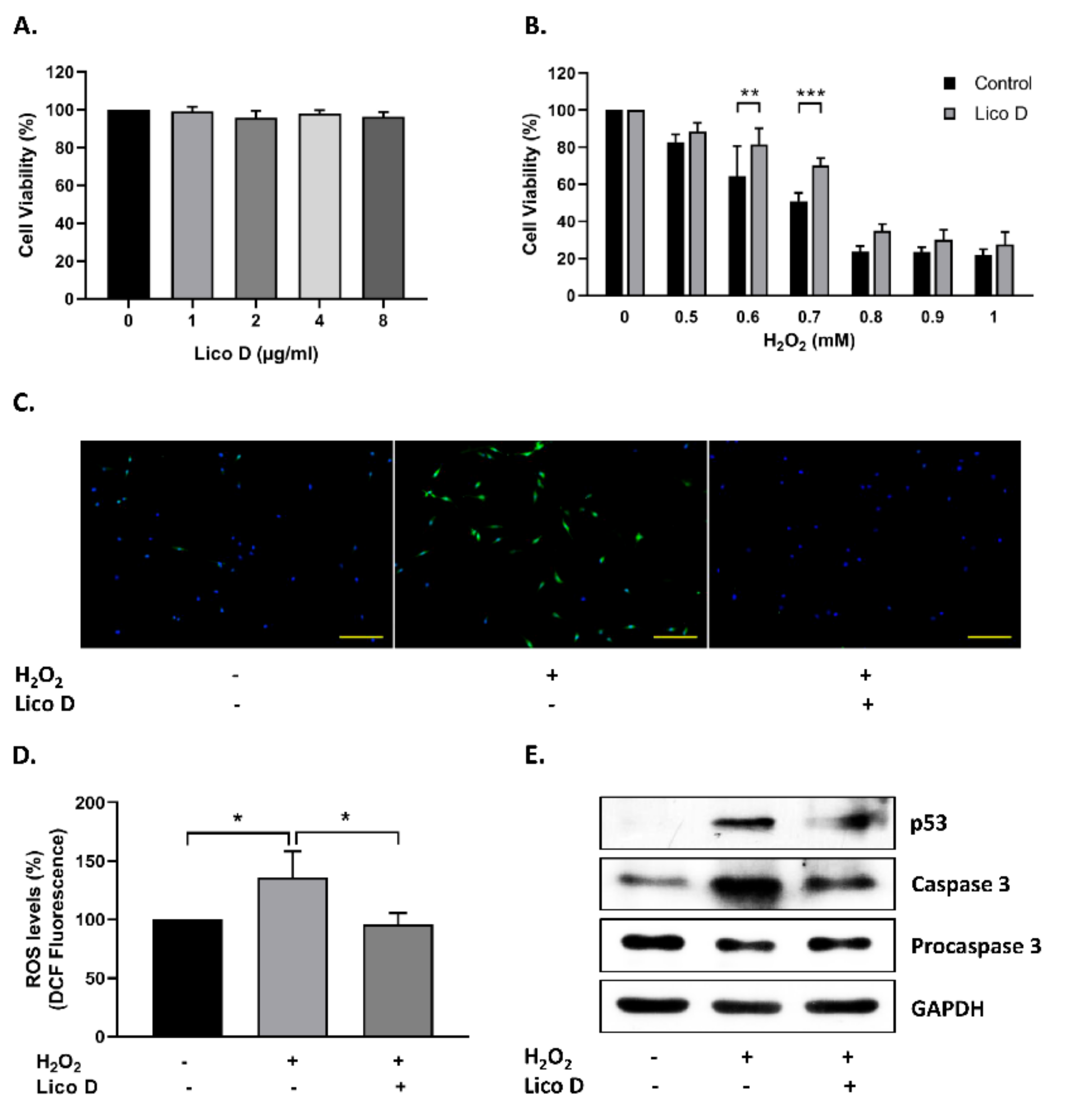{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
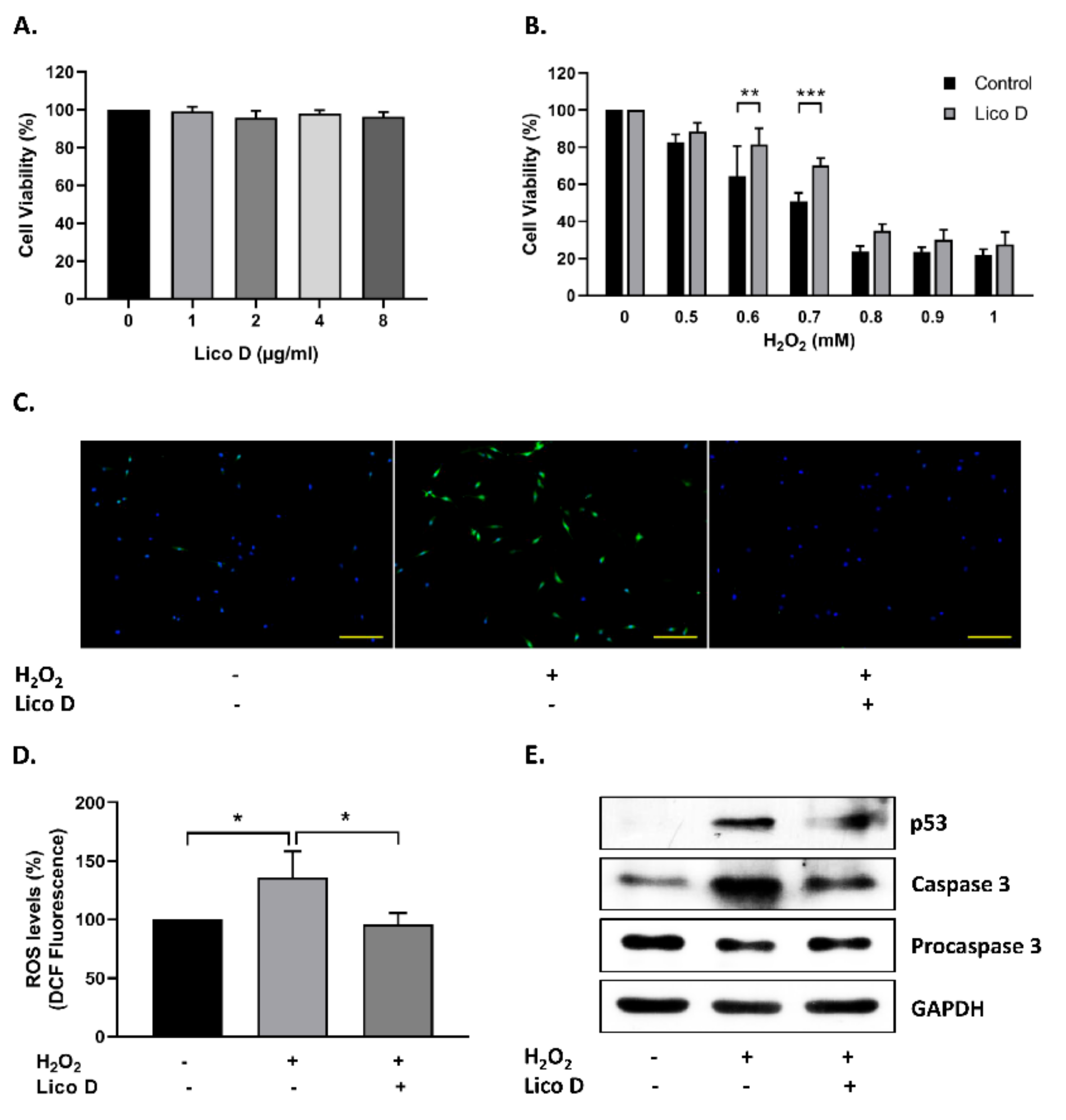
2.1. Lico D Protects the hBM-MSCs against Oxidative Stress
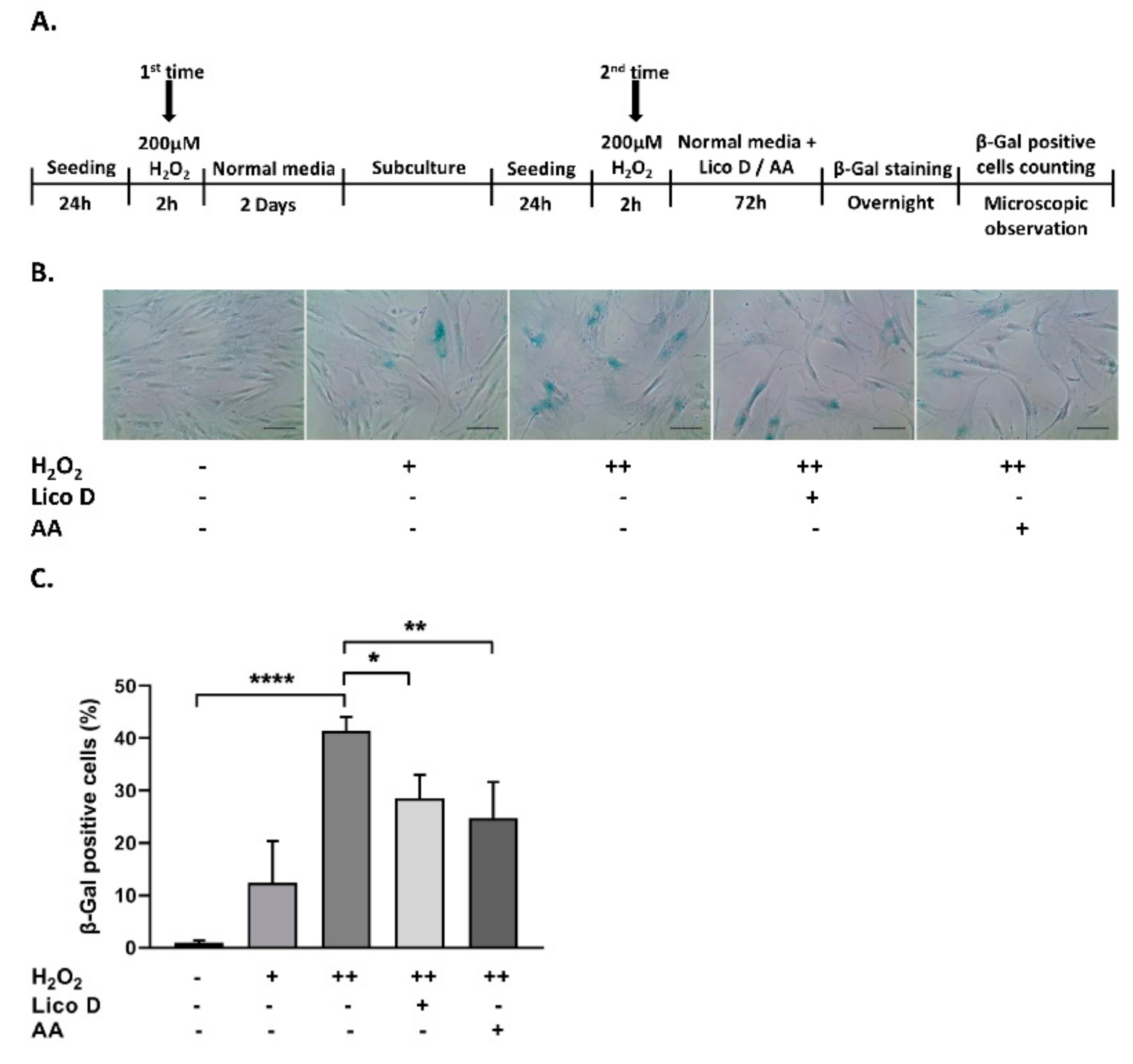
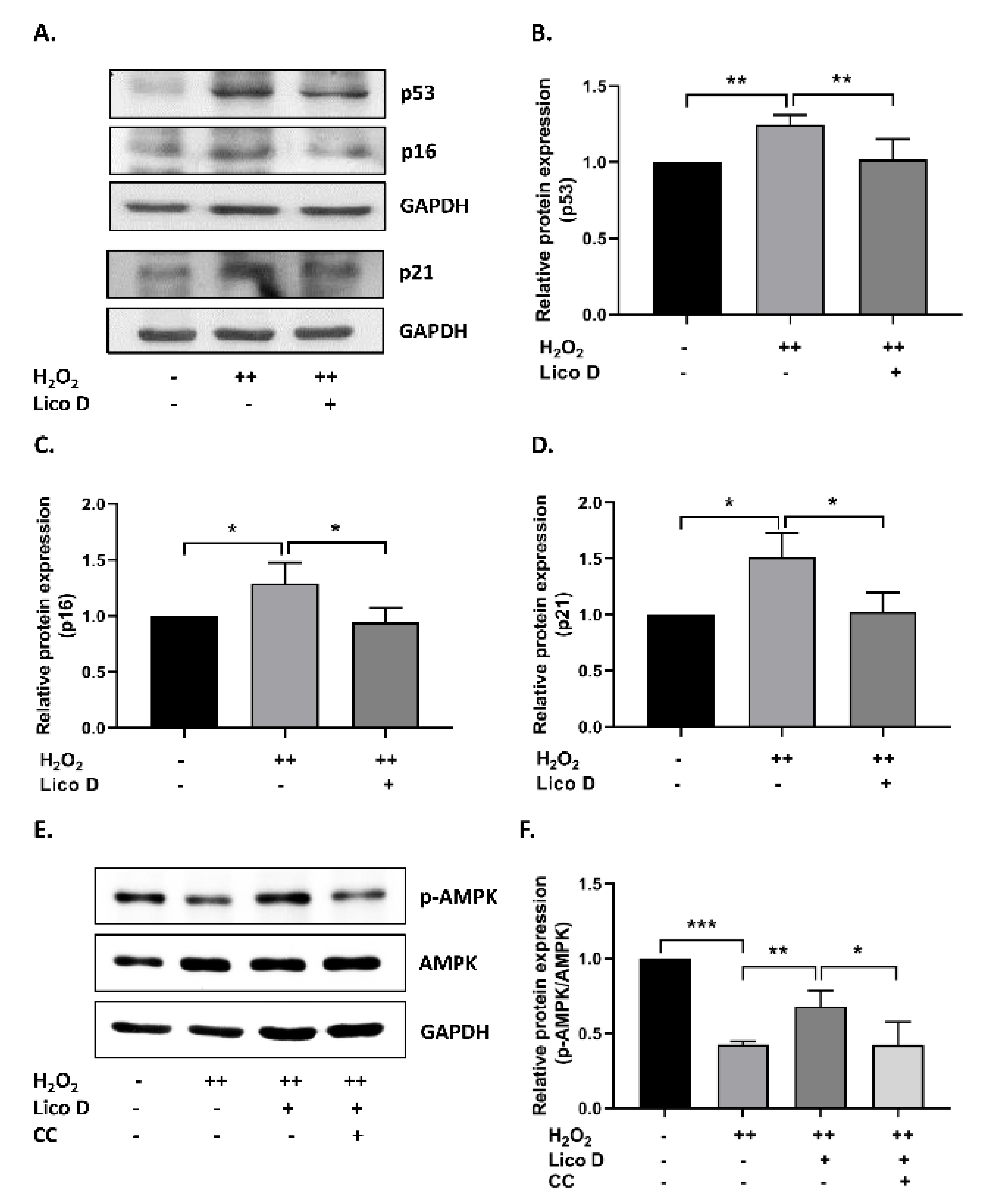
2.2. Lico D Reduces the Oxidative Stress Induced Senescence in hBM-MSCs
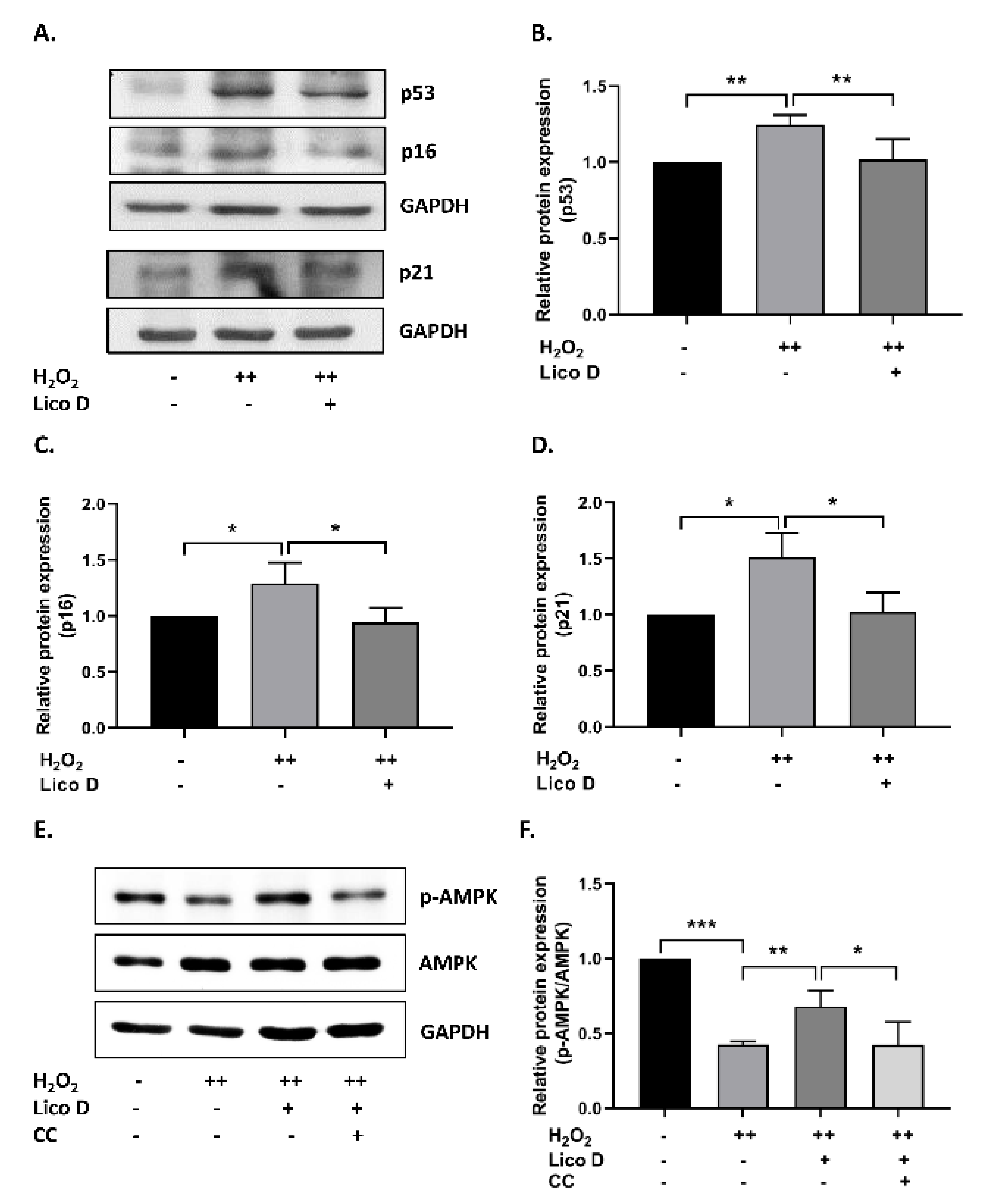
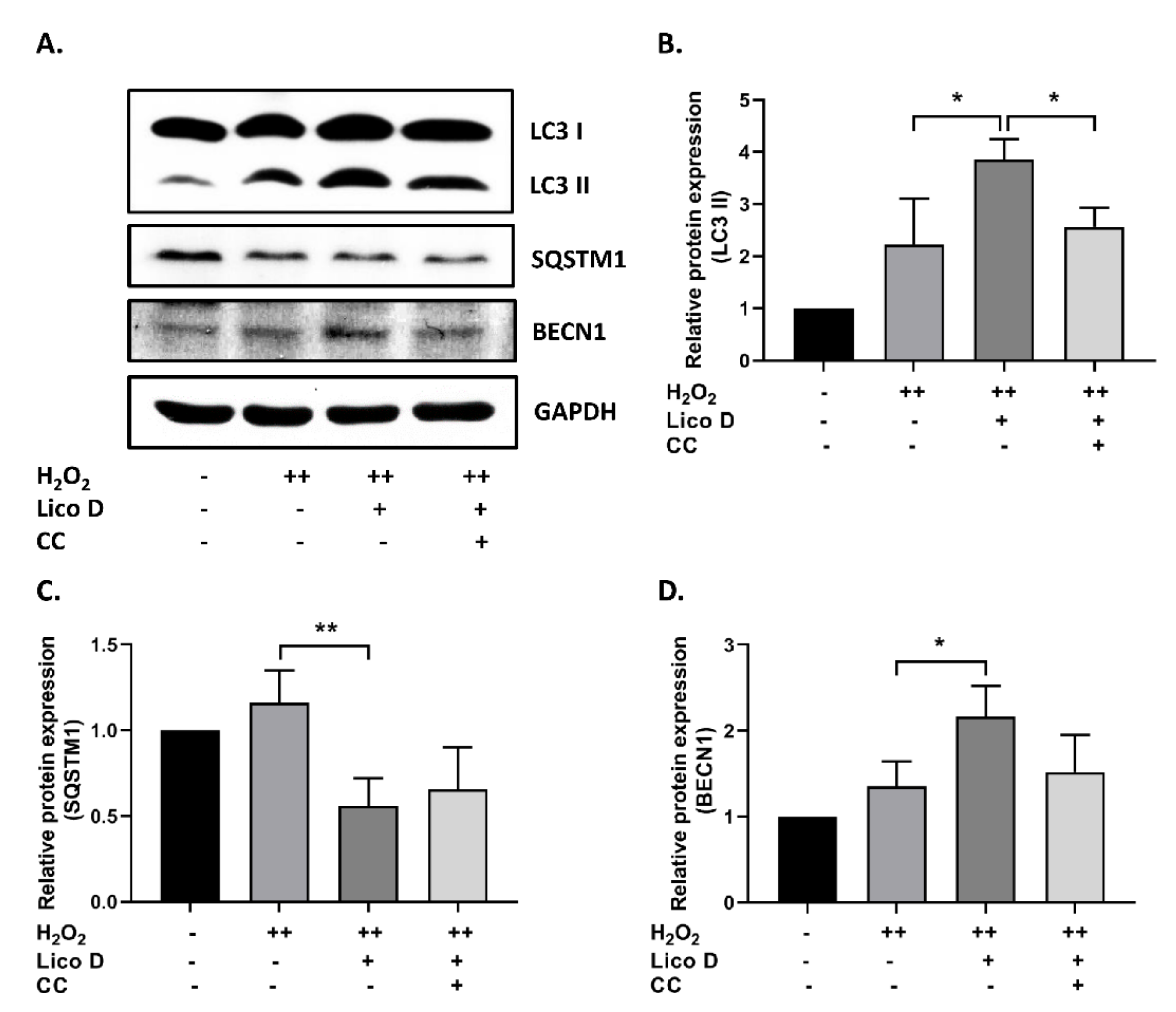
2.3. Lico D Reduces the Oxidative Stress Induced Senescence via Activation of AMPK and Autophagy
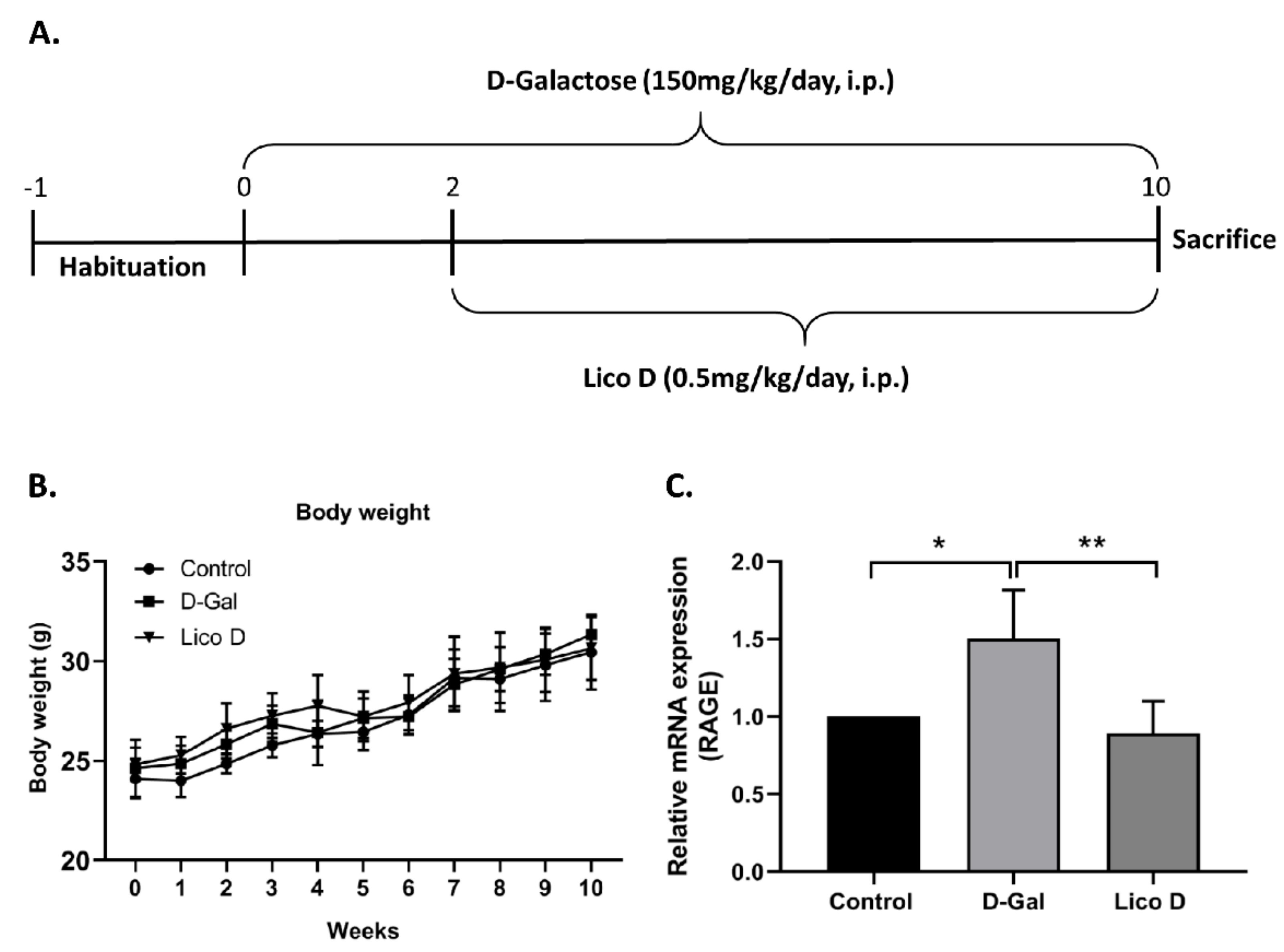
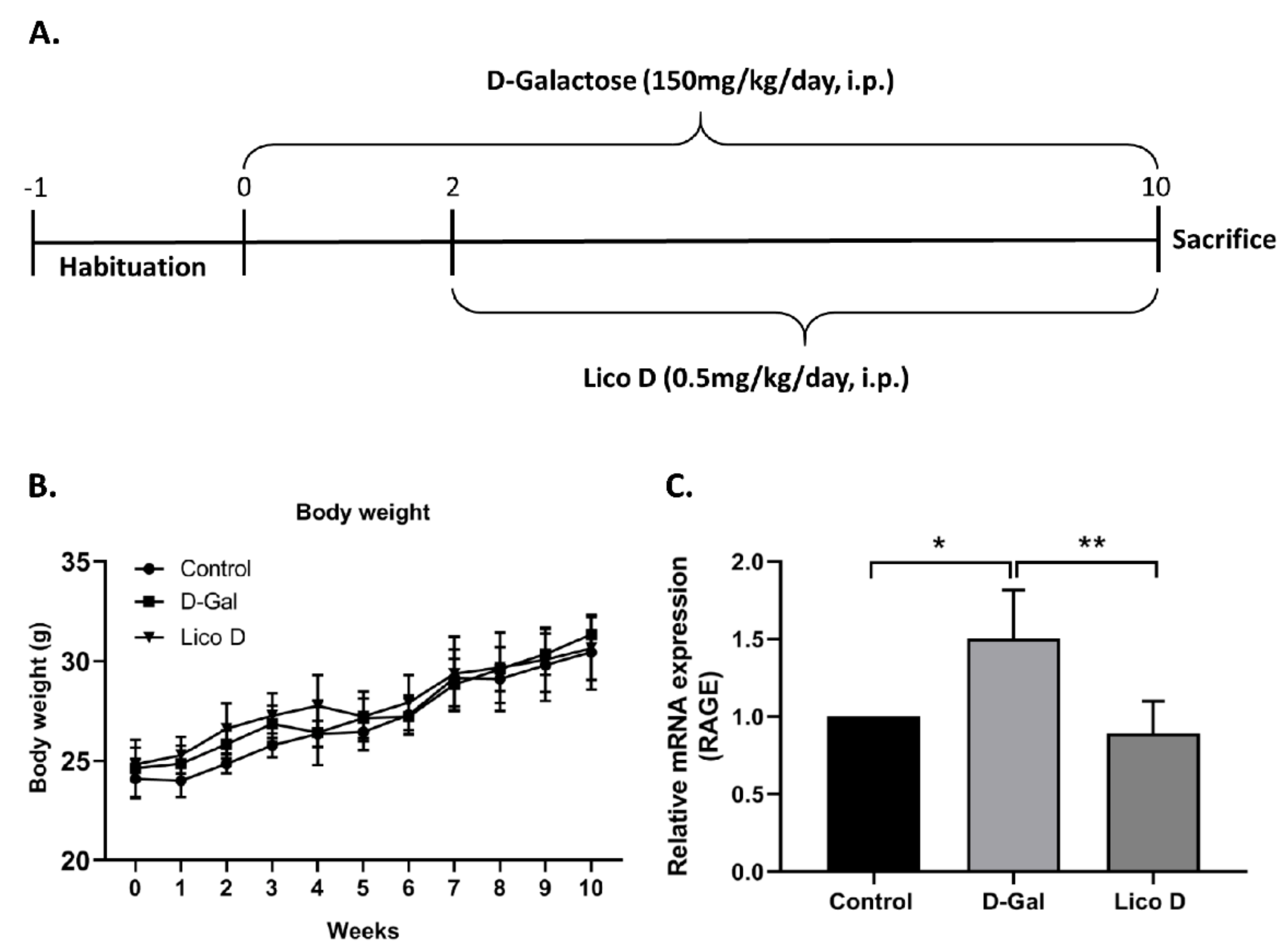
2.4. Effect of Lico D on Body Weight in D-Gal Induced Aging Mice
2.5. Lico D Reduces RAGE Expression in D-Gal Induced Aging Mice
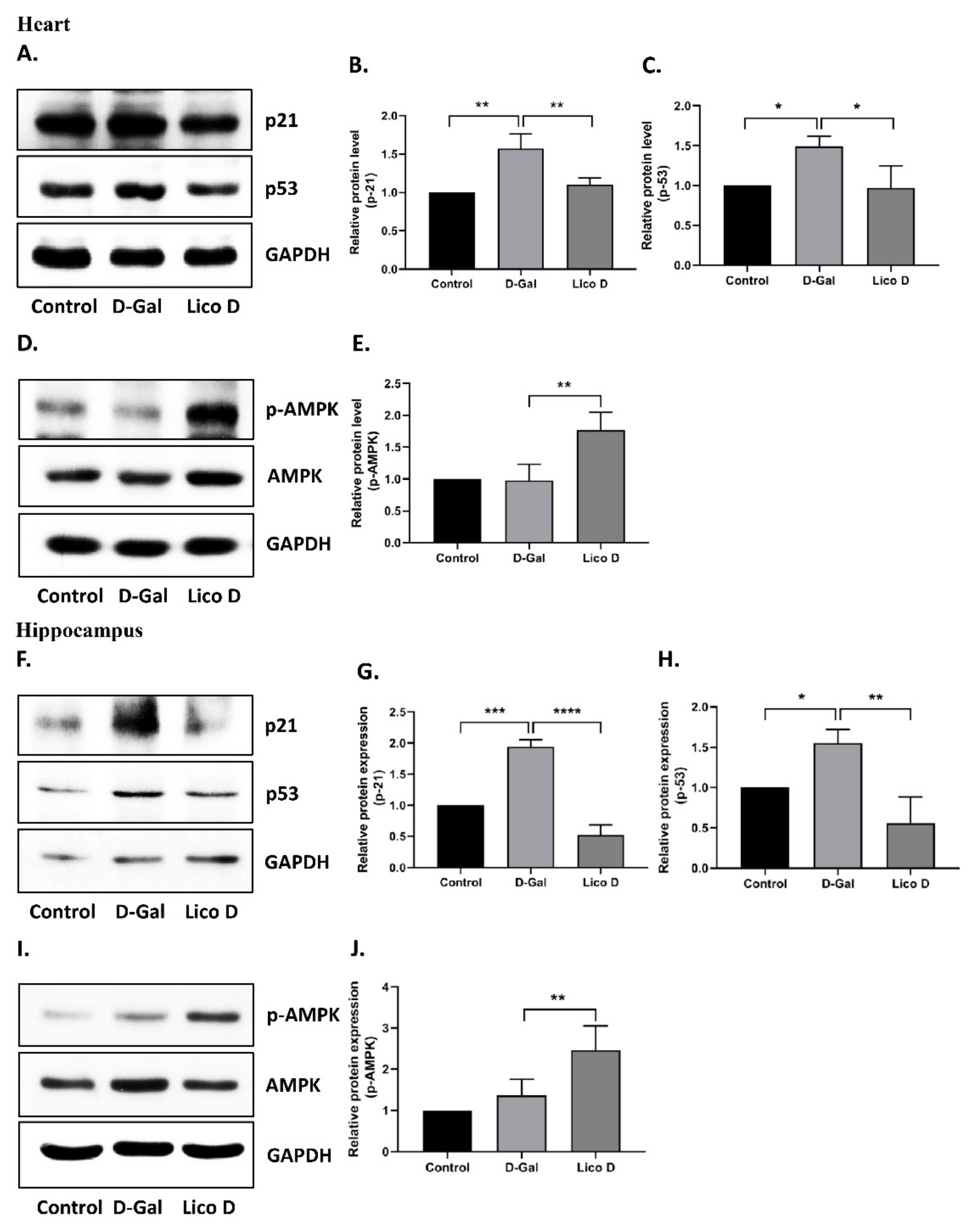
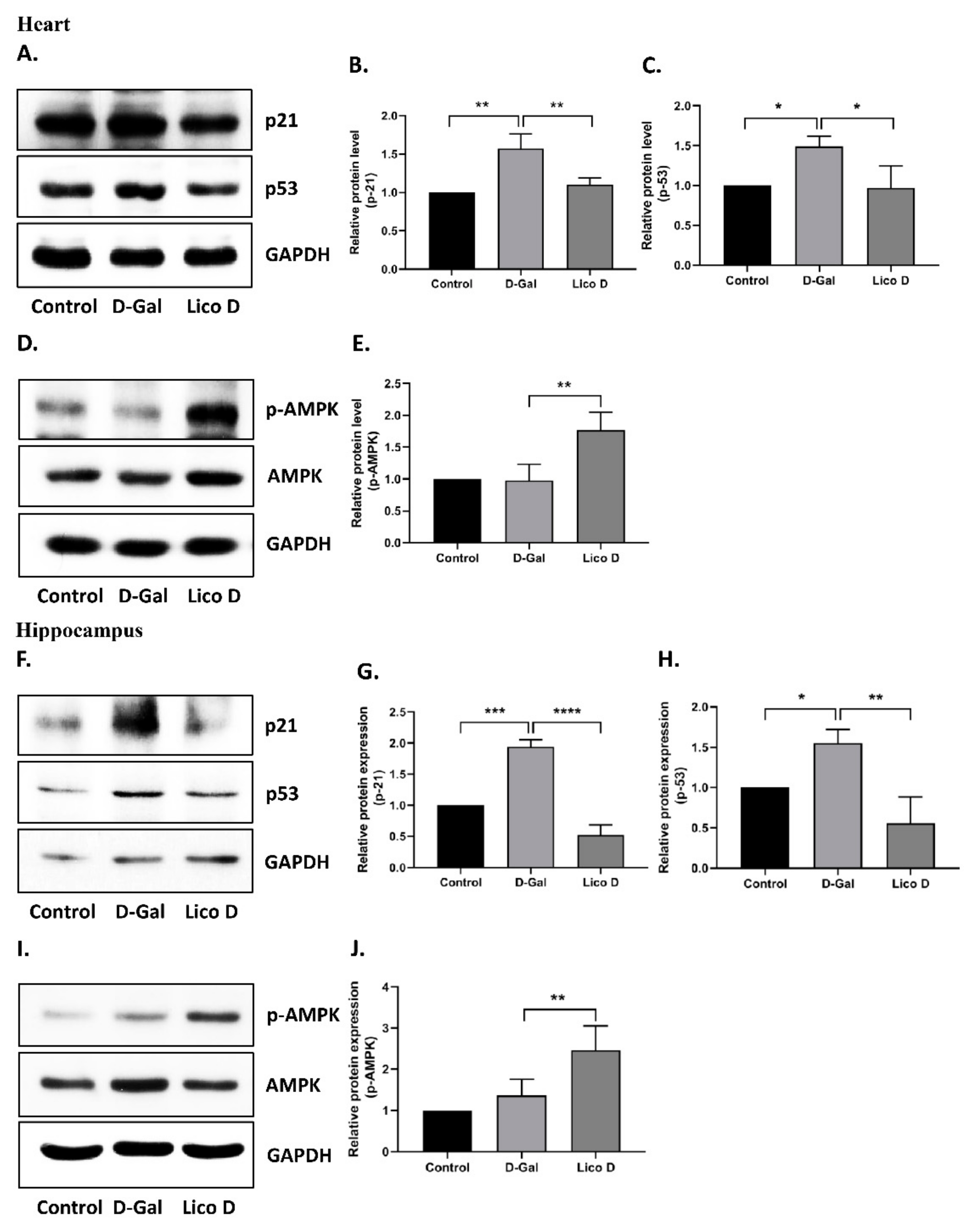
2.6. AMPK Activation by Lico D Ameliorates Heart and Hippocampus Senescence in D-Gal Induced Aging Mice
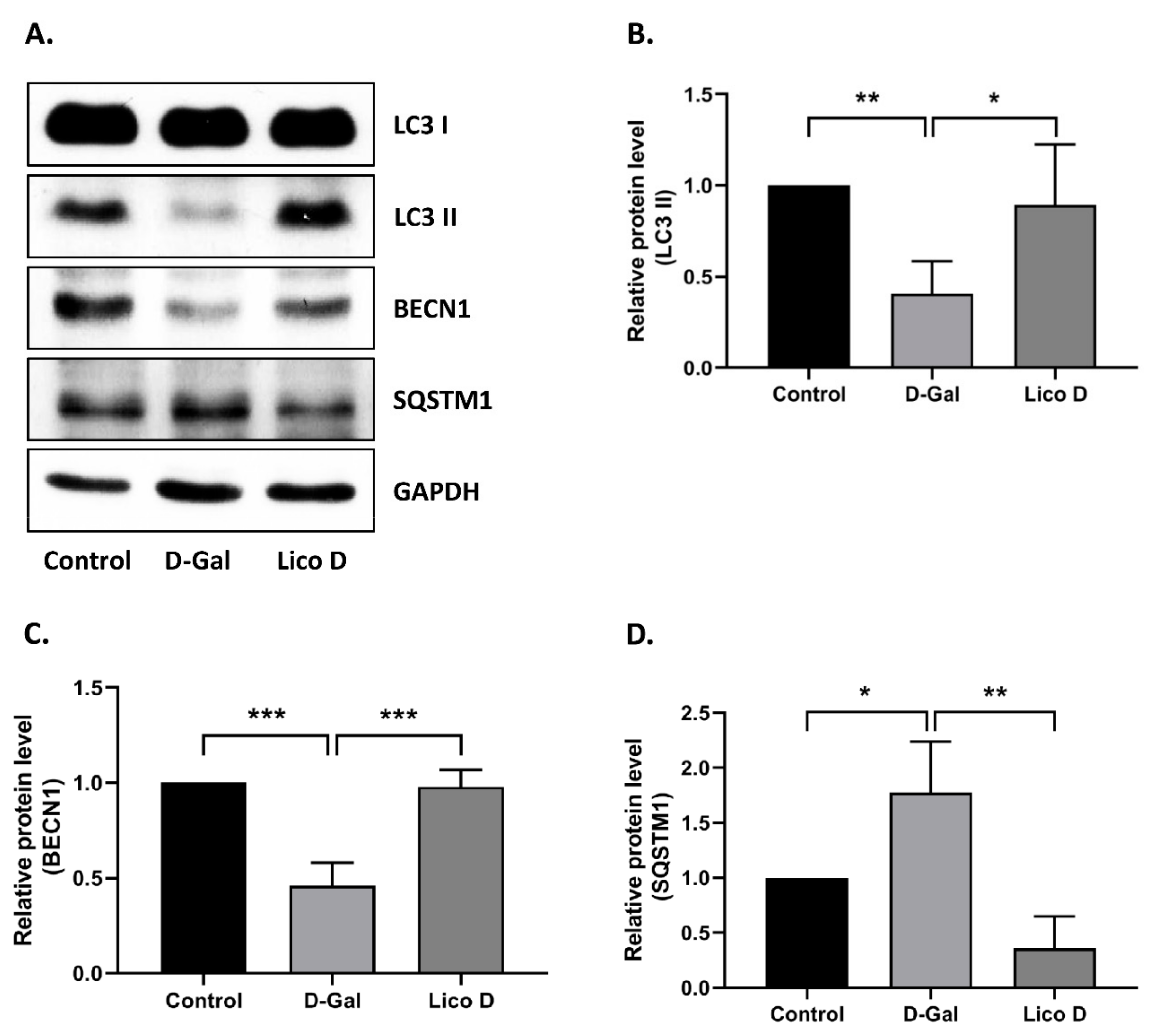
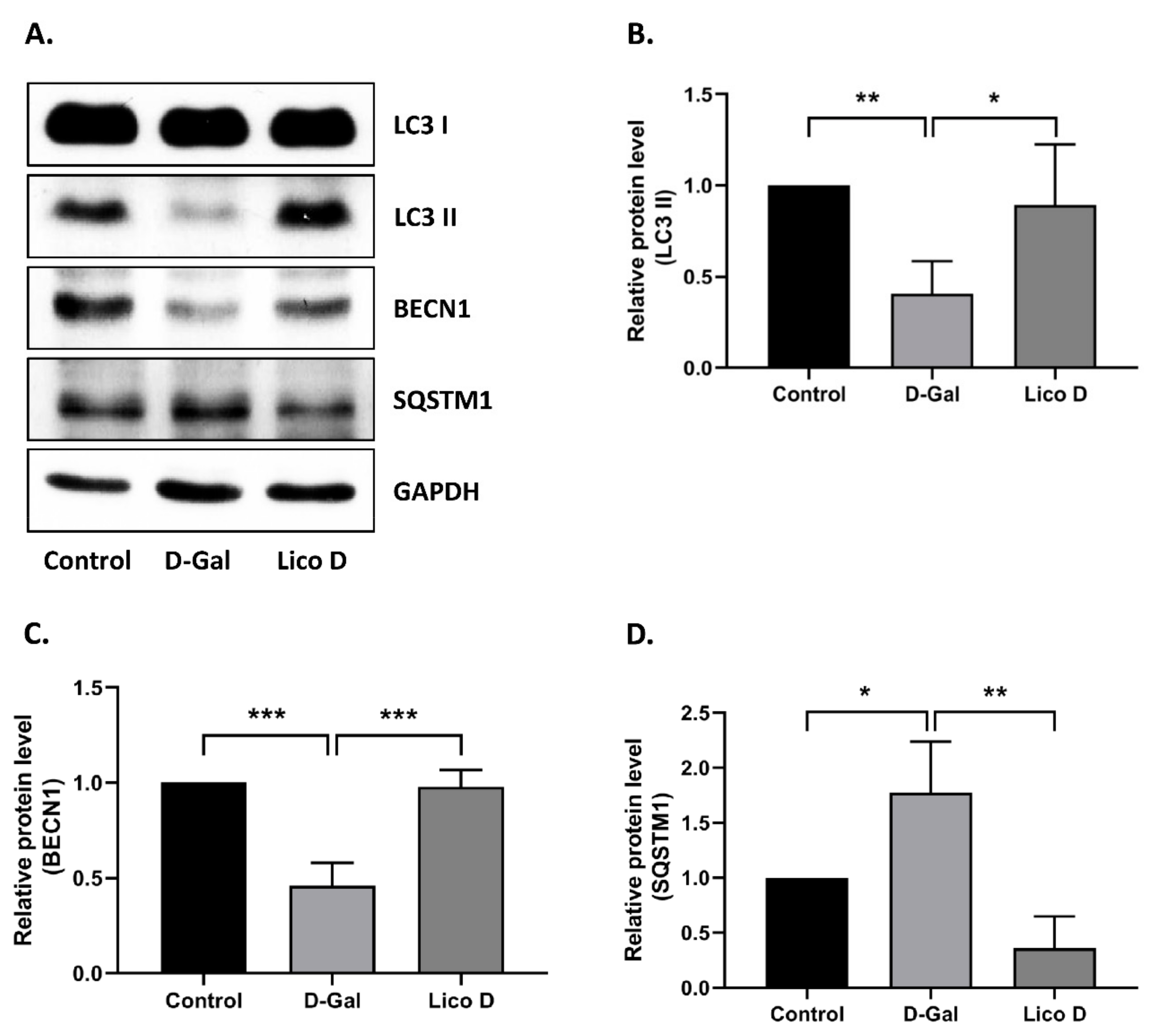
2.7. Lico D Activates Autophagy in D-Gal Induced Aging Mice Heart Tissue
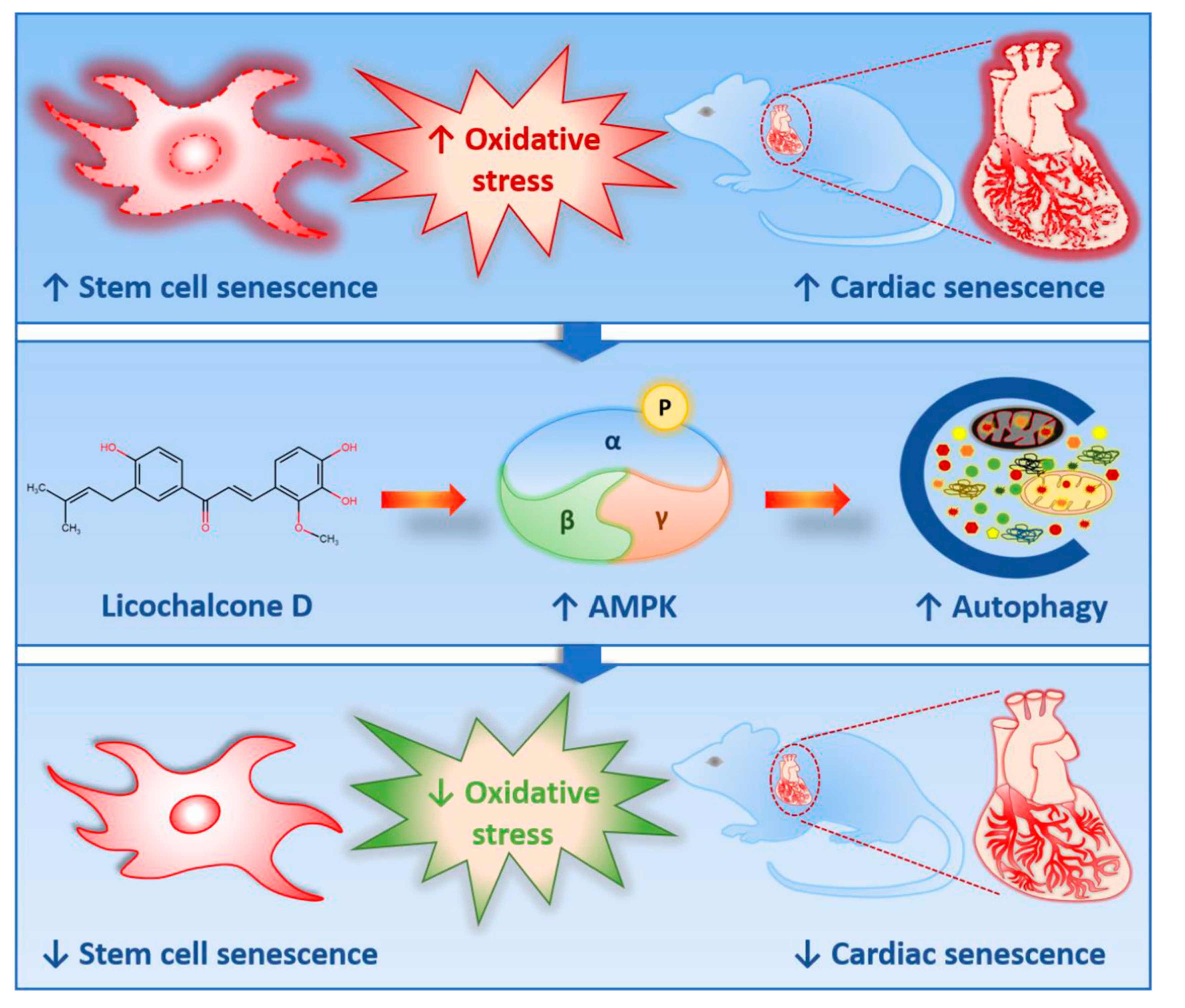
3. Discussion
3.1. Lico D Ameliorated the Oxidative Stress Induced Premature Senescence via AMPK Activation
3.2. Lico D Restored the Impaired Autophagy via AMPK Activation under Oxidative Stress
3.3. Lico D Reduced the RAGE Expression in Hippocampus
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. hBM-MSCs Cell Culture
4.3. Cell Viability Assay
4.4. Detection of Intracellular ROS
4.5. Oxidative Stress-Induced Senescence
4.6. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
4.7. Immunoblotting Analysis
4.8. Animals and Administration of Drugs
4.9. RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Torres, B.; Estepa-Fernández, A.; Rovira, M.; Orzáez, M.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. The chemistry of senescence. Nat. Rev. Chem. 2019, 3, 426–441. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [Green Version]
- Crowe, E.P.; Tuzer, F.; Gregory, B.D.; Donahue, G.; Gosai, S.J.; Cohen, J.; Leung, Y.Y.; Yetkin, E.; Nativio, R.; Wang, L.S.; et al. Changes in the Transcriptome of Human Astrocytes Accompanying Oxidative Stress-Induced Senescence. Front Aging Neurosci 2016, 8, 208. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 1998, 332 (Pt 1), 43–50. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Yang, P.; Zhao, R.; Bai, Y.; Guo, Z. Matrine Attenuates D-Galactose-Induced Aging-Related Behavior in Mice via Inhibition of Cellular Senescence and Oxidative Stress. Oxid Med Cell Longev 2018, 2018, 7108604. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Sun, Y.; Li, H.; Han, D.; Bai, Y.; Zhao, R.; Guo, Z. Anti-Ageing Effect of Physalis alkekengi Ethyl Acetate Layer on a d-galactose-Induced Mouse Model through the Reduction of Cellular Senescence and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zeng, J.; Su, M.; He, Y.; Zhu, B. Acetylshikonin from Zicao attenuates cognitive impairment and hippocampus senescence in d-galactose-induced aging mouse model via upregulating the expression of SIRT1. Brain Res. Bull. 2018, 137, 311–318. [Google Scholar] [CrossRef]
- Nam, S.M.; Hwang, H.; Seo, M.; Chang, B.J.; Kim, H.J.; Choi, S.H.; Rhim, H.; Kim, H.C.; Cho, I.H.; Nah, S.Y. Gintonin Attenuates D-Galactose-Induced Hippocampal Senescence by Improving Long-Term Hippocampal Potentiation, Neurogenesis, and Cognitive Functions. Gerontology 2018, 64, 562–575. [Google Scholar] [CrossRef]
- Huang, D.; Yin, L.; Liu, X.; Lv, B.; Xie, Z.; Wang, X.; Yu, B.; Zhang, Y. Geraniin protects bone marrow-derived mesenchymal stem cells against hydrogen peroxide-induced cellular oxidative stress in vitro. Int. J. Mol. Med. 2018, 41, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Bo-Htay, C.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Effects of d-galactose-induced ageing on the heart and its potential interventions. J. Cell Mol. Med. 2018, 22, 1392–1410. [Google Scholar] [CrossRef] [Green Version]
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of D-galactose-induced brain aging and its potential used for therapeutic interventions. Exp. Gerontol. 2018, 101, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Azman, K.F.; Zakaria, R. D-Galactose-induced accelerated aging model: An overview. Biogerontology 2019, 20, 763–782. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.F.; Ouyang, S.H.; Tu, L.F.; Wang, X.; Yuan, W.L.; Wang, G.E.; Wu, Y.P.; Duan, W.J.; Yu, H.M.; Fang, Z.Z.; et al. Caffeine Protects Skin from Oxidative Stress-Induced Senescence through the Activation of Autophagy. Theranostics 2018, 8, 5713–5730. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar] [CrossRef] [Green Version]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912–915. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, K.; Tamura, Y.; Yamamoto, M.; Inoue, Y.; Takagaki, R.; Takahashi, K.; Demizu, S.; Kajiyama, K.; Hiraga, Y.; Kinoshita, T. Identification of antimicrobial and antioxidant constituents from licorice of Russian and Xinjiang origin. Chem. Pharm. Bull. 1989, 37, 2528–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiyama, K.; Demizu, S.; Hiraga, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tamura, Y.; Okada, K.; Kinoshita, T. Two prenylated retrochalcones from Glycyrrhiza inflata. Phytochemistry 1992, 31, 3229–3232. [Google Scholar] [CrossRef]
- Maria Pia, G.D.; Sara, F.; Mario, F.; Lorenza, S. Biological Effects of Licochalcones. Mini Rev. Med. Chem. 2019, 19, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-Z.; Zhao, F.-F.; Gao, L.; Du, G.-H.; Zhang, X.; Qin, X.-M. Licorice extract attenuates brain aging of d-galactose induced rats through inhibition of oxidative stress and attenuation of neuronal apoptosis. RSC Adv. 2017, 7, 47758–47766. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Yan, X.; Hao, W.; Ma, X.; Ren, H.; Ren, B.; Li, D.; Dong, Z.; Zheng, Q. Licochalcone D induces apoptosis and inhibits migration and invasion in human melanoma A375 cells. Oncol. Rep. 2018, 39, 2160–2170. [Google Scholar]
- Seo, J.H.; Choi, H.W.; Oh, H.N.; Lee, M.H.; Kim, E.; Yoon, G.; Cho, S.S.; Park, S.M.; Cho, Y.S.; Chae, J.I.; et al. Licochalcone D directly targets JAK2 to induced apoptosis in human oral squamous cell carcinoma. J. Cell Physiol. 2019, 234, 1780–1793. [Google Scholar] [CrossRef]
- Oh, H.N.; Lee, M.H.; Kim, E.; Kwak, A.W.; Yoon, G.; Cho, S.S.; Liu, K.; Chae, J.I.; Shim, J.H. Licochalcone D Induces ROS-Dependent Apoptosis in Gefitinib-Sensitive or Resistant Lung Cancer Cells by Targeting EGFR and MET. Biomolecules 2020, 10, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Haraguchi, H.; Ishikawa, H.; Mizutani, K.; Tamura, Y.; Kinoshita, T. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg. Med. Chem. 1998, 6, 339–347. [Google Scholar] [CrossRef]
- Yuan, X.; Niu, H.T.; Wang, P.L.; Lu, J.; Zhao, H.; Liu, S.H.; Zheng, Q.S.; Li, C.G. Cardioprotective Effect of Licochalcone D against Myocardial Ischemia/Reperfusion Injury in Langendorff-Perfused Rat Hearts. PLoS ONE 2015, 10, e0128375. [Google Scholar] [CrossRef]
- Chen, J.H.; Ozanne, S.E.; Hales, C.N. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2007, 371, 179–189. [Google Scholar] [PubMed]
- Chen, J.H.; Stoeber, K.; Kingsbury, S.; Ozanne, S.E.; Williams, G.H.; Hales, C.N. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J. Biol. Chem. 2004, 279, 49439–49446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zhang, H.; Wang, B.; Zhang, Y.; Zheng, X.; Shao, B.; Zhuge, Q.; Jin, K. Key Signaling Pathways in Aging and Potential Interventions for Healthy Aging. Cells 2021, 10, 660. [Google Scholar] [CrossRef]
- Lin, J.Y.; Kuo, W.W.; Baskaran, R.; Kuo, C.H.; Chen, Y.A.; Chen, W.S.; Ho, T.J.; Day, C.H.; Mahalakshmi, B.; Huang, C.Y. Swimming exercise stimulates IGF1/ PI3K/Akt and AMPK/SIRT1/PGC1α survival signaling to suppress apoptosis and inflammation in aging hippocampus. Aging 2020, 12, 6852–6864. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16r–28r. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Pinkas, A.; Aschner, M. Advanced Glycation End-Products and Their Receptors: Related Pathologies, Recent Therapeutic Strategies, and a Potential Model for Future Neurodegeneration Studies. Chem. Res. Toxicol. 2016, 29, 707–714. [Google Scholar] [CrossRef]
- Ma, Y.; Ma, B.; Shang, Y.; Yin, Q.; Wang, D.; Xu, S.; Hong, Y.; Hou, X.; Liu, X. Flavonoid-Rich Ethanol Extract from the Leaves of Diospyros kaki Attenuates D-Galactose-Induced Oxidative Stress and Neuroinflammation-Mediated Brain Aging in Mice. Oxid. Med. Cell Longev. 2018, 2018, 8938207. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal. Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Ye, Q.; Liu, C.M.; Shan, Q.; Wang, Y.J. Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-κB pathway activation. Cereb. Cortex. 2010, 20, 2540–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Tang, Y.; Jin, X.; Chen, C.; Lu, Y.; Liu, L.; Shen, C. Metformin Inhibits Advanced Glycation End Products-Induced Inflammatory Response in Murine Macrophages Partly through AMPK Activation and RAGE/NFκB Pathway Suppression. J. Diabetes Res. 2016, 2016, 4847812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, J.; Funakoshi-Tago, M.; Mashino, T.; Tago, K.; Inoue, H.; Sonoda, Y.; Kasahara, T. Glycyrrhiza inflata-derived chalcones, Licochalcone A, Licochalcone B and Licochalcone D, inhibit phosphorylation of NF-kappaB p65 in LPS signaling pathway. Int. Immunopharmacol. 2009, 9, 499–507. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharajan, N.; Ganesan, C.D.; Moon, C.; Jang, C.-H.; Oh, W.-K.; Cho, G.-W. Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation. Int. J. Mol. Sci. 2021, 22, 7324. https://doi.org/10.3390/ijms22147324
Maharajan N, Ganesan CD, Moon C, Jang C-H, Oh W-K, Cho G-W. Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation. International Journal of Molecular Sciences. 2021; 22(14):7324. https://doi.org/10.3390/ijms22147324
Chicago/Turabian StyleMaharajan, Nagarajan, Chitra Devi Ganesan, Changjong Moon, Chul-Ho Jang, Won-Keun Oh, and Gwang-Won Cho. 2021. "Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation" International Journal of Molecular Sciences 22, no. 14: 7324. https://doi.org/10.3390/ijms22147324
APA StyleMaharajan, N., Ganesan, C. D., Moon, C., Jang, C.-H., Oh, W.-K., & Cho, G.-W. (2021). Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation. International Journal of Molecular Sciences, 22(14), 7324. https://doi.org/10.3390/ijms22147324