
Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort?



Abstract
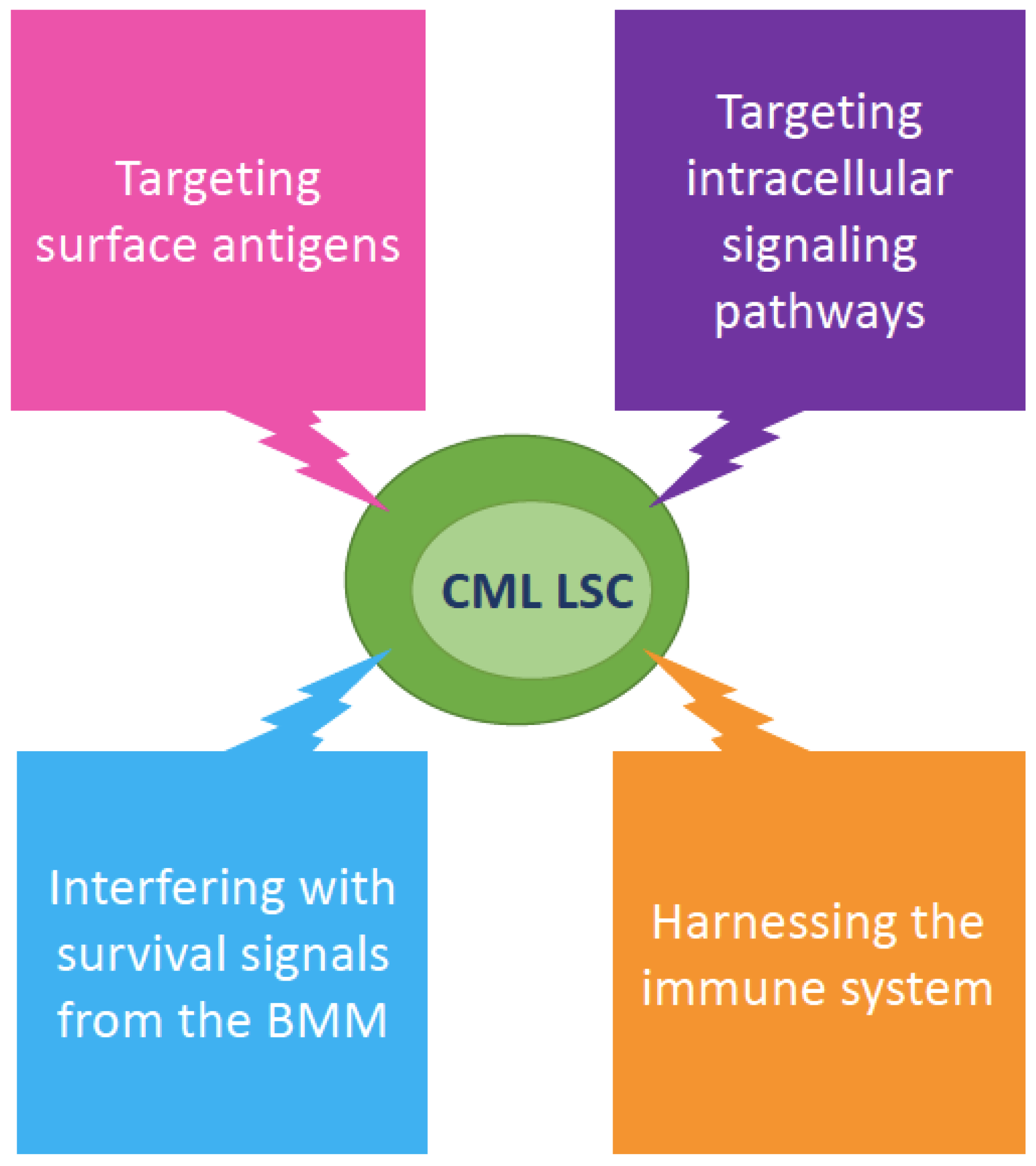
1. Introduction: Why Aren’t We Happy Yet with Clinical Results in CML?
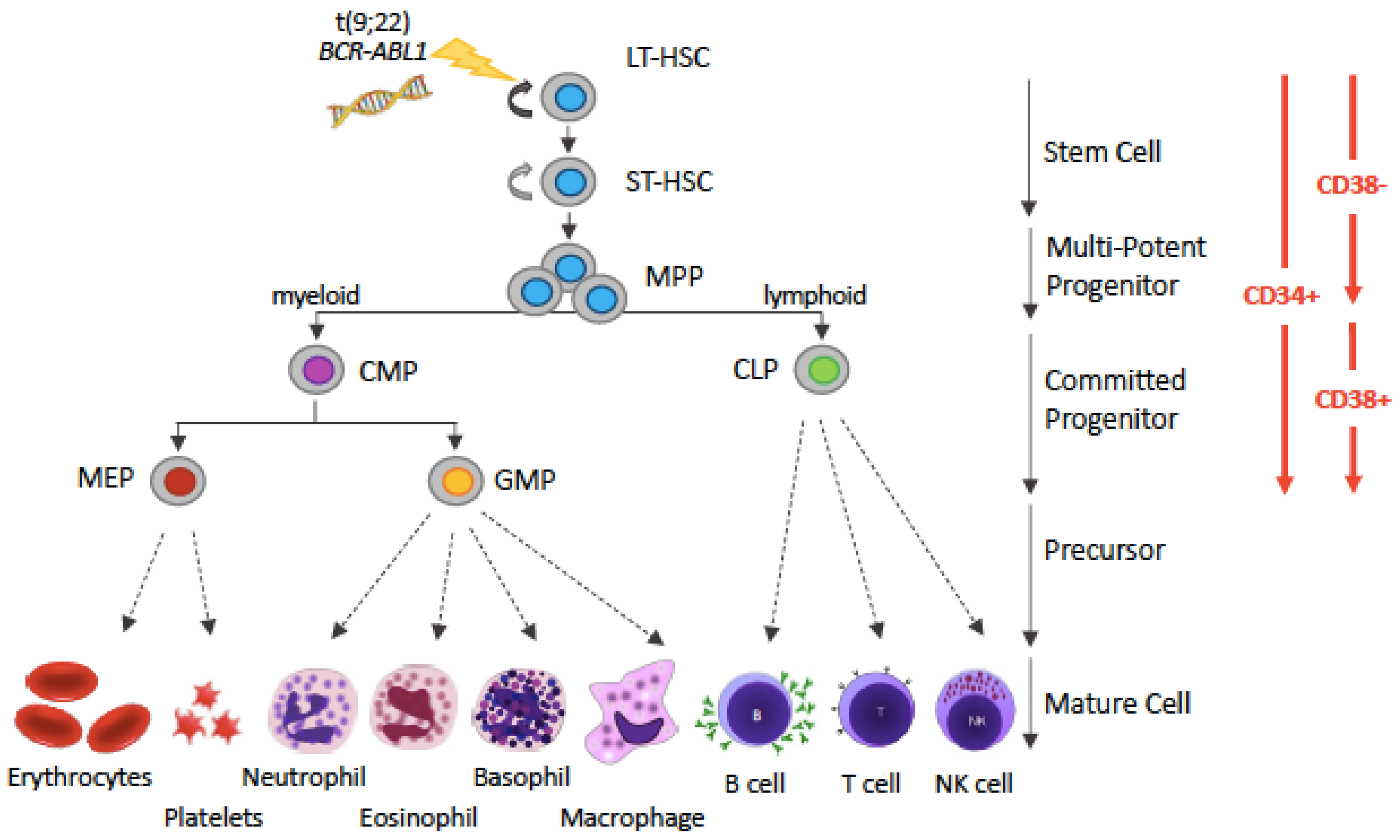
2. LSCs: The Ultimate Enemy in CML?
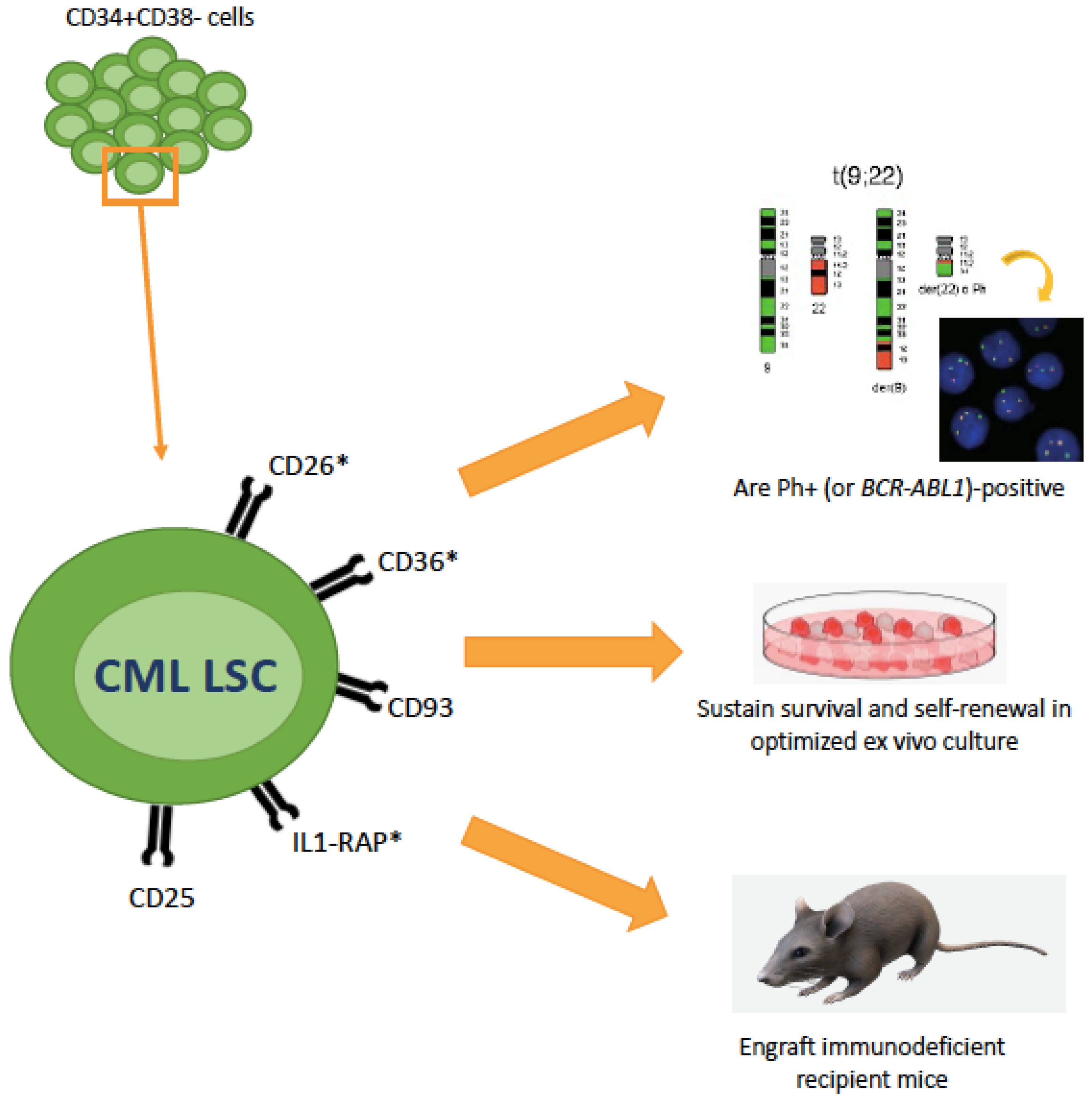
3. Aberrant Surface Markers in CML LSCs and Their Diagnostic, Predictive and Therapeutic Role
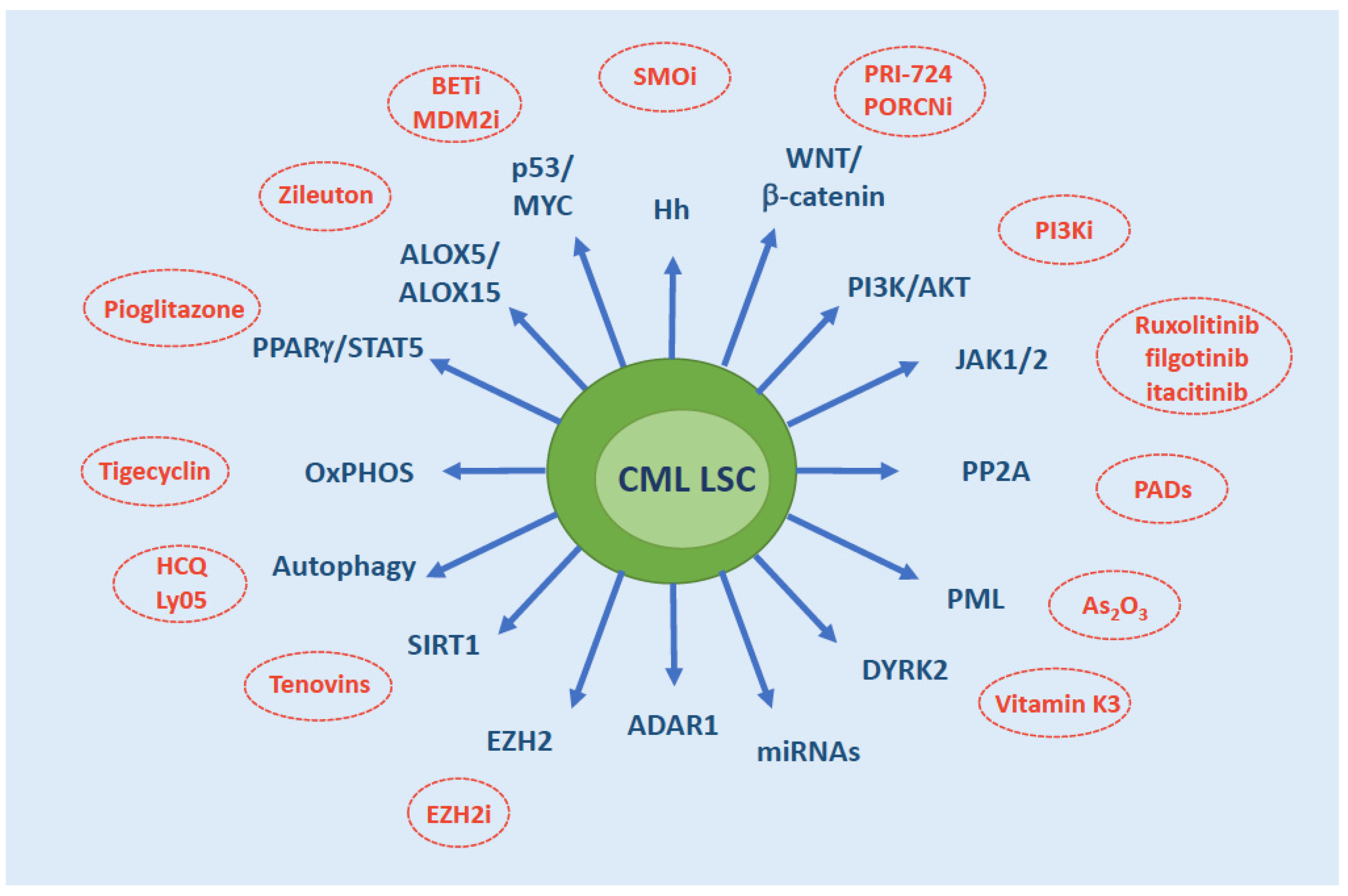
4. Cell-Intrinsic Survival Pathways
- -
- Sonic Hedgehog (Hh) pathway: Hh is a master regulator of self-renewal of normal SCs as well as LSCs. The binding of Hh ligand to the Patched (PTCH) receptor relieves the inhibition of the G protein–coupled receptor Smoothened (SMO) that, in turn, activates the transcription factor GLI1. GLI1 then translocates into the nucleus, where it modulates the transcription of several target genes implicated in cell cycle regulation and apoptosis (CCND1, MYC, etc.) and promote MDM2-dependent degradation of p53. The pathway is hyperactivated in CML LSCs through the upregulation of SMO [56,57].
- -
- Wnt/β-catenin: nuclear β-catenin, a co-activator of the T-cell factor/lymphoid enhancer-binding factor (Tcf/Lef) transcription factors, is required for self-renewal and survival of normal HSCs through the activation of key genes like NOTCH [58] and is, similarly, a key mediator of LSC survival [59]. Both canonical and noncanonical Wnt pathways result in relocation of β-catenin in the cytoplasm, where it is ultimately phosphorylated by Glycogen Sinthase Kinase 3β (GSK3β) and targeted for degradation by an Axin-mediated multimeric complex. Other pathways, like CD70/CD27 [60,61] have been shown to converge on Wnt, thus ultimately influencing β-catenin levels and activity. Noncanonical Wnt pathways, that convey signals in a β-catenin-independent fashion, have also been implicated in LSC survival. Via activation of phospholipase C (PLC), Wnt has indeed been shown to increase intracellular Ca2+, leading to the activation of the Ca2+-calmodulin-dependent protein phosphatase calcineurin and ultimately resulting in the activation of Nuclear Factor of Activated T-cells (NFAT) transcription factor [62]. Enhanced NFAT activity mediated by this noncanonical Wnt/Ca2+ signaling increases autocrine cytokine production that provides to LSCs compensatory prosurvival signals upon exposure to TKIs.
- -
- PI3K/AKT: the PI3K/AKT pathway mediates proliferation and is physiologically activated by binding of growth factors to receptor tyrosine kinases. BCR-ABL1-dependent activation of PI3K/AKT signaling contributes to CML LSCs maintenance, as it leads to AKT-mediated phosphorylation and cytosolic retention of FOXO, hence blocks the transcription of FOXO target genes involved in apoptosis [63]. Among others, transforming growth factor β (TGF-β) signaling influences FOXO nuclear translocation via PI3K/Akt signaling [63].
- -
- Janus kinases (JAK): in CML, JAK2 signaling has been shown to converge on STAT3 and STAT5 activation. STAT5 phosphorylation and translocation into the nucleus is well known to play a key role in CML pathogenesis [64,65]. STAT3 signaling has also been implicated in CML LSC survival [66,67,68]. Both JAK2 and JAK1 have been implicated in STAT3 activation.
- -
- Protein phosphatase 2A (PP2A): the tumor suppressor PP2A gene encodes a multimeric serine/threonine phosphatase that is inactivated via overexpression of its endogenous inhibitors SET and CIP2A. In CML, PP2A loss as a result of BCR-ABL1-dependent expression of SET has been implicated in quiescent LSC maintenance [64]. BCR-ABL1 expression (but not activity) was found to be essential for the recruitment of JAK2, activation of a JAK2/β-catenin survival/self-renewal pathway.
- -
- Promyelocytic leukemia (PML): the PML gene, involved in the t(15;17) chromosomal translocation of acute promyelocytic leukaemia, encodes a protein localizing to nuclear bodies that acts as a tumor suppressor, controlling apoptosis, cellular proliferation and senescence [69] and negatively regulating mTOR [70]. PML has been shown to play an indispensable role in maintaining LSC quiescence in CML [71].
- -
- Dual Specificity Tyrosine Phosphorylation Regulated Kinase 2 (DYRK2): the KLF4 transcription factor has been shown to sustain CML LSCs via downregulation of the dual-specificity kinase DYRK2, that mediates the stabilization of p53 and the proteasomal degradation of c-MYC. This results in reduced apoptosis and increased self-renewal [72]. The central role of p53 and c-MYC in LSC maintenance had already been elegantly demonstrated by a seminal proteomic study conducted by Tessa Holyoake’s group [73].
- -
- Micro RNAs (miRNAs): comparative miRNA expression profiling has highlighted several deregulated miRNAs in CML LSCs. For example, the BCR-ABL1 kinase-independent upregulation of miR-29a-3p and miR-660-5p was observed in the CD34+CD38- fraction, and via the downregulation of their respective targets Ten Eleven Translocation 2 (TET2) and Endothelial PAS Domain Protein 1 (EPAS1) was found to confer TKI resistance in vitro [74]. More recently, Pellicano et al. [75] have shown that BCR-ABL1 kinase-dependent upregulation of hsa-mir183 results in the downregulation of its direct target Early Growth Response 1 (EGR1) transcription factor, and, as a consequence, upregulation of the cell cycle regulator E2F1, that is known to control both cell proliferation and p53-dependent/independent apoptosis. The latter was found to play a pivotal role in controlling LSC (but not normal HSC) survival and proliferation, since E2F1 inhibition led to a decrease in colony-forming potential, cell-cycle arrest, and induction of p53-mediated apoptosis. Even more recently, it has been reported that miR-196a-5p is also upregulated in the CD26+CD34+CD38– fraction as compared to the CD26–CD34+CD38–, although the roles of this microRNA in CML LSCs have yet to be elucidated [76]. BCR-ABL1-independent signaling pathways have also been shown to impact on the biogenesis and/or on the levels of miRNAs implicated in LSC survival and resistance. JAK/STAT-dependent activation of Adenosine Deaminase Acting on RNA1 (ADAR1), implicated in post-transcriptional adenosine-to-inosine RNA editing, has been shown to impair the biogenesis of let-7, ultimately enhancing LSC self-renewal [77]. Additionally, miR-21, that appears to be under the control of the PI3K/AKT pathway, has been implicated in LSC resistance to TKIs [78]. The role of miR-30a in autophagy will be discussed below. Further investigations into the roles of miRNAs as well as of other noncoding RNAs in LSC intrinsic survival mechanisms are warranted.
- -
- Autophagy: it is an evolutionarily conserved catabolic process that physiologically consists in the lysosomal degradation and recycle of unnecessary cellular components to generate adenosine triphosphate (ATP) and essential building blocks during nutrient and/or oxygen deprivation. Autophagy has been implicated in normal HSC maintenance [79], but may also act as a pro-survival pathway that helps tumor cells tolerate metabolic stress and avoid apoptosis induced by anticancer agents [80]. Similarly, in CML, autophagy has been highlighted as a drug resistance pathway employed by LSCs for their survival [81]. BCR-ABL1 represses autophagy, in part via the PI3K/Akt/mTORC1 pathway and in part via induction of miR-30a, that in turn dowregulates two key autophagy genes, Beclin 1 and ATG5. TKI treatment induces autophagy, and inhibition of autophagy results in enhanced TKI-induced killing of stem and progenitor cells [81].
- -
- Epigenetic alterations: Enhancer of Zeste Homolog 2 (EZH2) is a member of the Polycomb Repressive Complex 2 (PRC2) that trimethylates histone H3 at lysine 27 but may also directly regulate gene expression. It has been shown to be highly expressed in CML LSCs and to contribute to their survival [82]. Targeting histone deacetylases was also explored against CML LSCs, in light of the reported efficacy of such a strategy in inducing apoptosis in non-proliferating cells, and gave promising results when tested in combination with TKIs in cellular and mouse models [83]. Sirtuin 1 (SIRT1) deacetylase is a multifunctional protein that has several genes implicated in many cellular pathways, including energy metabolism and stress response, among its targets—including TP53 and FOXOs. SIRT1 has been shown to be overexpressed in CML LSCs and to contribute to LSC resistance to TKIs [84,85]. Later on, SIRT1 was found to mediate increased mitochondrial oxidative phosphorylation in CML [86], an important survival mechanism of LSCs [87]—see Section 5. The histone acetyltransferase CBP has also been implicated in LSC self-renewal, as opposed to p300 which rather promotes differentiation and senescence. Both can be recruited by b-catenin upon activation of the Wnt pathway. Inhibition of CBP was found to favor the formation of b-catenin/p300 complexes, switching the balance from cell proliferation to differentiation [88].
- -
- Metabolic changes: one of the most recent and intriguing chapters of LSC survival adaptations is centered on metabolism. Besides the well-known role played by the hypoxic conditions of BMM in metabolic rewiring (that will be discussed more in detail in the dedicated section below), several molecules implicated in cellular metabolism have been shown to be deregulated in CML LSCs. Arachidonate 5-lipoxygenase (ALOX5), involved in fatty acid metabolism (it converts arachidonic acid into leukotrienes), has been shown to be required for CML LSC self-renewal [89]. Later on, ALOX15 was also found to be essential for LSC survival [90].
- -
- Autocrine factors: BCR-ABL1-induced transformation is known to rely, among other mechanisms, on the autocrine production by LSCs of cytokines like Interleukin (IL)-3 and Granulocyte-Colony Stimulating Factor (G-CSF) resulting in growth factor-independent STAT5 activation. Other, kinase-independent autocrine loops have more recently been reported to contribute to LSC persistence. BCR-ABL1 kinase-independent autocrine production of Tumor Necrosis Factor a (TNFa) has been reported to support CML LSC survival via the nuclear factor κB (NFκB)/p65 pathway leading to the expression of the IL-3 and granulocyte/macrophage-colony stimulating factor common β-chain receptor [91]. An autocrine loop enhancing bone morphogenetic protein (BMP) 2 and 4 (normally produced by mesenchymal cells and involved in regulation of proliferation and fate of normal HSCs) concentration in the BMM (see further details in the next section) has also been implicated in LSC resistance to TKIs [92]. Very recently, CML LSC have also been found to upregulate pleiotrophin (PTN), a heparin-binding growth factor normally produced by BM stromal cells and ECs. PTN has been shown to promote CML LSC survival and TKI resistance [93].
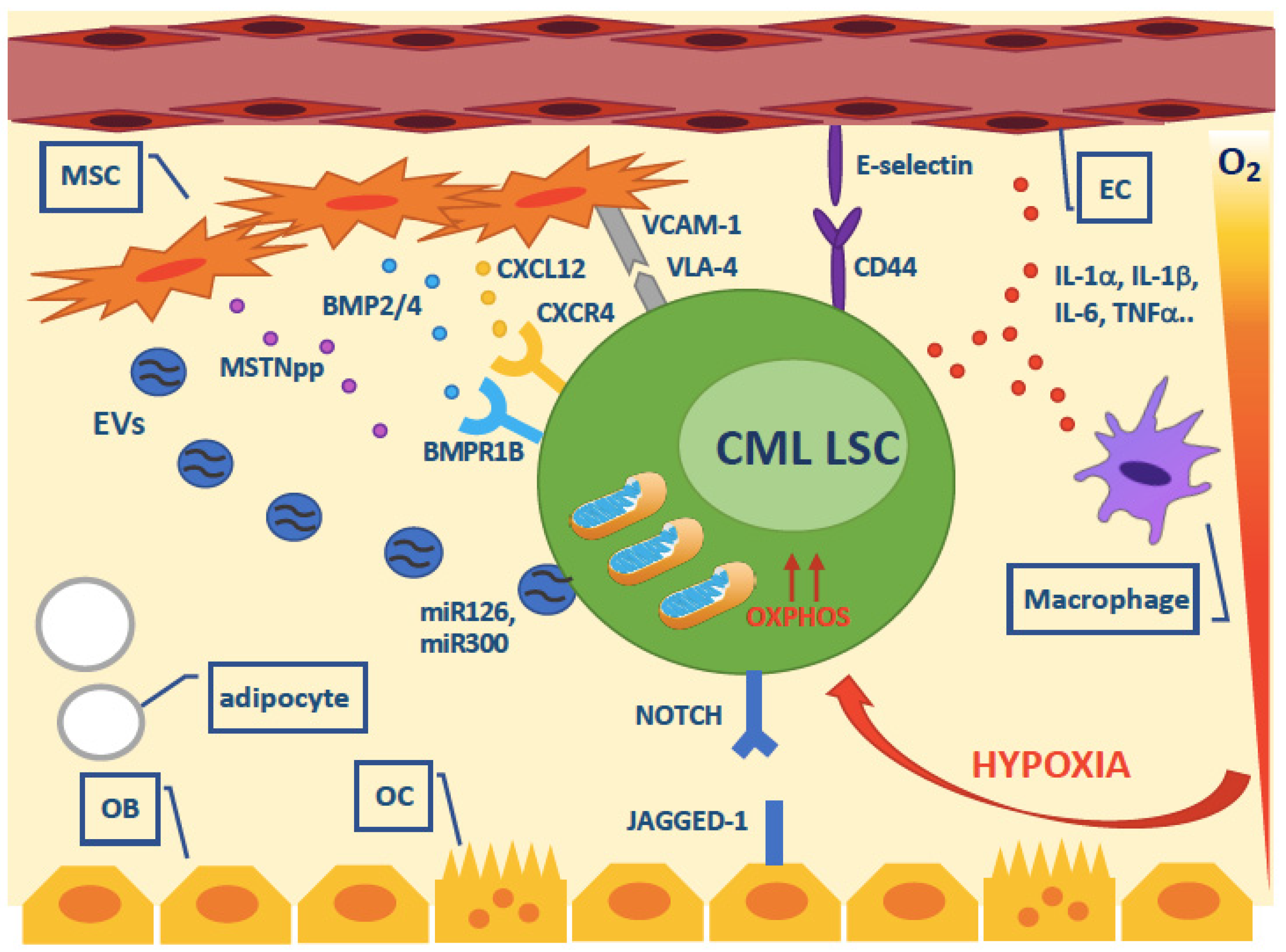
5. Cell-Extrinsic Survival Pathways
6. Will We Ever Be Able to Kill CML LSCs?
7. Do We Really Need to Kill CML LSCs?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Martinelli, G.; Iacobucci, I.; Baccarani, M. Imatinib mesylate for the treatment of chronic myeloid leukemia. Expert Rev. Anticancer Ther. 2008, 8, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; De Benedittis, C.; Mancini, M.; Martinelli, G. Best practices in chronic myeloid leukemia monitoring and management. Oncologist 2016, 21, 626–633. [Google Scholar] [CrossRef]
- Bavaro, L.; Martelli, M.; Cavo, M.; Soverini, S. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: An update. Int. J. Mol. Sci. 2019, 20, 6141. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Bower, H.; Bjorkholm, M.; Dickman, P.W.; Hoglund, M.; Lambert, P.C.; Andersson, T.M. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J. Clin. Oncol. 2016, 34, 2851–2857. [Google Scholar] [CrossRef]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Soverini, S.; Rosti, G.; Baccarani, M.; Martinelli, G. Molecular monitoring. Curr. Hematol. Malig. Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.M.; Hughes, T.P. Treatment-free remission in patients with chronic myeloid leukaemia. Nat. Rev. Clin. Oncol. 2020, 17, 493–503. [Google Scholar] [CrossRef]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 39. [Google Scholar] [CrossRef]
- Eisterer, W.; Jiang, X.; Christ, O.; Glimm, H.; Lee, K.H.; Pang, E.; Lambie, K.; Shaw, G.; Holyoake, T.L.; Petzer, A.L.; et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia 2005, 19, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kinstrie, R.; Karamitros, D.; Goardon, N.; Morrison, H.; Hamblin, M.; Robinson, L.; Clark, R.E.; Copland, M.; Vyas, P. Heterogeneous leukemia stem cells in myeloid blast phase chronic myeloid leukemia. Blood Adv. 2016, 1, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, L.; Ho, Y.; Li, M.; Marcucci, G.; Tong, W.; Bhatia, R. Heterogeneity of leukemia-initiating capacity of chronic myelogenous leukemia stem cells. J. Clin. Investig. 2016, 126, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Deininger, M.; Gora-Tybor, J.; Goldman, J.M.; Melo, J.V. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: Biologic significance and implications for the assessment of minimal residual disease. Blood 1998, 92, 3362–3367. [Google Scholar] [CrossRef]
- Biernaux, C.; Loos, M.; Sels, A.; Huez, G.; Stryckmans, P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995, 86, 3118–3122. [Google Scholar] [CrossRef]
- Huang, W.; Liu, B.; Eklund, E.A. Investigating the role of the innate immune response in relapse or blast crisis in chronic myeloid leukemia. Leukemia 2020, 34, 2364–2374. [Google Scholar] [CrossRef]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999, 94, 2056–2064. [Google Scholar] [CrossRef]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Holtz, M.S.; Slovak, M.L.; Zhang, F.; Sawyers, C.L.; Forman, S.J.; Bhatia, R. Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood 2002, 99, 3792–3800. [Google Scholar] [CrossRef]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Bertrand, A.; Meunier, M.C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.H.; Ame, S.; et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Modi, H.; McDonald, T.; Chu, S.; Yee, J.K.; Forman, S.J.; Bhatia, R. Role of BCR/ABL gene-expression levels in determining the phenotype and imatinib sensitivity of transformed human hematopoietic cells. Blood 2007, 109, 5411–5421. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Brendel, C.; Hochhaus, A.; Neubauer, A.; Burchert, A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood 2012, 119, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Janssen, J.J.; Deenik, W.; Smolders, K.G.; van Kuijk, B.J.; Pouwels, W.; Kelder, A.; Cornelissen, J.J.; Schuurhuis, G.J.; Ossenkoppele, G.J. Residual normal stem cells can be detected in newly diagnosed chronic myeloid leukemia patients by a new flow cytometric approach and predict for optimal response to imatinib. Leukemia 2012, 26, 977–984. [Google Scholar] [CrossRef][Green Version]
- Mustjoki, S.; Rohon, P.; Rapakko, K.; Jalkanen, S.; Koskenvesa, P.; Lundan, T.; Porkka, K. Low or undetectable numbers of Philadelphia chromosome-positive leukemic stem cells (Ph(+)CD34(+)CD38(neg)) in chronic myeloid leukemia patients in complete cytogenetic remission after tyrosine kinase inhibitor therapy. Leukemia 2010, 24, 219–222. [Google Scholar] [CrossRef]
- Mustjoki, S.; Richter, J.; Barbany, G.; Ehrencrona, H.; Fioretos, T.; Gedde-Dahl, T.; Gjertsen, B.T.; Hovland, R.; Hernesniemi, S.; Josefsen, D.; et al. Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia 2013, 27, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Thielen, N.; Richter, J.; Baldauf, M.; Barbany, G.; Fioretos, T.; Giles, F.; Gjertsen, B.T.; Hochhaus, A.; Schuurhuis, G.J.; Sopper, S.; et al. Leukemic stem cell quantification in newly diagnosed patients with chronic myeloid leukemia predicts response to nilotinib therapy. Clin. Cancer Res. 2016, 22, 4030–4038. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Bauer, K.; Herrmann, H.; Gleixner, K.V.; Schulenburg, A.; Rabitsch, W.; Sperr, W.R.; Wolf, D. Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML. Int. J. Mol. Sci. 2019, 20, 4233. [Google Scholar] [CrossRef]
- Jaras, M.; Johnels, P.; Hansen, N.; Agerstam, H.; Tsapogas, P.; Rissler, M.; Lassen, C.; Olofsson, T.; Bjerrum, O.W.; Richter, J.; et al. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. USA 2010, 107, 16280–16285. [Google Scholar] [CrossRef]
- Landberg, N.; Hansen, N.; Askmyr, M.; Agerstam, H.; Lassen, C.; Rissler, M.; Hjorth-Hansen, H.; Mustjoki, S.; Jaras, M.; Richter, J.; et al. IL1RAP expression as a measure of leukemic stem cell burden at diagnosis of chronic myeloid leukemia predicts therapy outcome. Leukemia 2016, 30, 253–257. [Google Scholar] [CrossRef]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML hematopoietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor-engineered T cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef]
- Landberg, N.; von Palffy, S.; Askmyr, M.; Lilljebjorn, H.; Sanden, C.; Rissler, M.; Mustjoki, S.; Hjorth-Hansen, H.; Richter, J.; Agerstam, H.; et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica 2018, 103, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Sadovnik, I.; Racil, Z.; Herrmann, H.; Blatt, K.; Cerny-Reiterer, S.; Eisenwort, G.; Lion, T.; Holyoake, T.; Mayer, J. DPPIV (CD26) as a novel stem cell marker in Ph+ chronic myeloid leukaemia. Eur. J. Clin. Investig. 2014, 44, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rulicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Blatt, K.; Menzl, I.; Eisenwort, G.; Cerny-Reiterer, S.; Herrmann, H.; Herndlhofer, S.; Stefanzl, G.; Sadovnik, I.; Berger, D.; Keller, A.; et al. Phenotyping and target expression profiling of CD34(+)/CD38(-) and CD34(+)/CD38(+) stem- and progenitor cells in acute lymphoblastic leukemia. Neoplasia 2018, 20, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Culen, M.; Borsky, M.; Nemethova, V.; Razga, F.; Smejkal, J.; Jurcek, T.; Dvorakova, D.; Zackova, D.; Weinbergerova, B.; Semerad, L.; et al. Quantitative assessment of the CD26+ leukemic stem cell compartment in chronic myeloid leukemia: Patient-subgroups, prognostic impact, and technical aspects. Oncotarget 2016, 7, 33016–33024. [Google Scholar] [CrossRef]
- Raspadori, D.; Pacelli, P.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Cattaneo, D.; Gozzini, A.; Galimberti, S.; Barate, C.; Pregno, P.; et al. Flow cytometry assessment of CD26(+) leukemic stem cells in peripheral blood: A simple and rapid new diagnostic tool for chronic myeloid leukemia. Cytom. B Clin. Cytom. 2019, 96, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Bocchia, M.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Sirianni, S.; Gozzini, A.; Galimberti, S.; Aprile, L.; Martino, B.; Pregno, P.; et al. Residual peripheral blood CD26(+) leukemic stem cells in chronic myeloid leukemia patients during TKI therapy and during treatment-free remission. Front. Oncol. 2018, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Bocchia, M.; Sicuranza, A.; Pacelli, P.; Iurlo, A.; Abruzzese, E.; Galimberti, S.; Pregno, P.; Caocci, G.; Capodanno, I.; Crugnola, M.; et al. Peripheral blood CD26+ leukemia stem cells monitoring in chronic myeloid leukemia patients from diagnosis to response to TKIS: Interim results of a multicenter prospective study (PROSPECTIVE FLOWERS). Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Willmann, M.; Sadovnik, I.; Eisenwort, G.; Entner, M.; Bernthaler, T.; Stefanzl, G.; Hadzijusufovic, E.; Berger, D.; Herrmann, H.; Hoermann, G.; et al. Evaluation of cooperative antileukemic effects of nilotinib and vildagliptin in Ph(+) chronic myeloid leukemia. Exp. Hematol. 2018, 57, 50–59. [Google Scholar] [CrossRef]
- Houshmand, M.; Garello, F.; Stefania, R.; Gaidano, V.; Cignetti, A.; Spinelli, M.; Fava, C.; Nikougoftar Zarif, M.; Galimberti, S.; Pungolino, E.; et al. Targeting chronic myeloid leukemia stem/progenitor cells using venetoclax-loaded immunoliposome. Cancers 2021, 13, 1311. [Google Scholar] [CrossRef]
- Goff, D.J.; Court Recart, A.; Sadarangani, A.; Chun, H.J.; Barrett, C.L.; Krajewska, M.; Leu, H.; Low-Marchelli, J.; Ma, W.; Shih, A.Y.; et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013, 12, 316–328. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.I.; Takubo, K.; Kobayashi, H.; Nakamura-Ishizu, A.; Honda, H.; Kataoka, K.; Kumano, K.; Akiyama, H.; Sudo, T.; Kurokawa, M.; et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood 2014, 123, 2540–2549. [Google Scholar] [CrossRef]
- Sadovnik, I.; Hoelbl-Kovacic, A.; Herrmann, H.; Eisenwort, G.; Cerny-Reiterer, S.; Warsch, W.; Hoermann, G.; Greiner, G.; Blatt, K.; Peter, B.; et al. Identification of CD25 as STAT5-dependent growth regulator of leukemic stem cells in Ph+ CML. Clin. Cancer Res. 2016, 22, 2051–2061. [Google Scholar] [CrossRef]
- Sadovnik, I.; Herrmann, H.; Eisenwort, G.; Blatt, K.; Hoermann, G.; Mueller, N.; Sperr, W.R.; Valent, P. Expression of CD25 on leukemic stem cells in BCR-ABL1(+) CML: Potential diagnostic value and functional implications. Exp. Hematol. 2017, 51, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sadovnik, I.; Herrmann, H.; Blatt, K.; Eisenwort, G.; Mueller, N.; Stefanzl, G.; Hoermann, G.; Herndlhofer, S.; Bauer, K.; Peter, B.; et al. Evaluation of cell surface markers and targets in leukemic stem cells (LSC) reveals distinct expression profiles, unique drug effects, and specific checkpoint regulation in AML LSC and CML LSC. Blood 2016, 128, 4234. [Google Scholar] [CrossRef]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Moka, H.A.; Cassels, J.; Dunn, K.; Herzyk, P.; Irvine, D.A.; Copland, M. CD93 is a novel biomarker of leukemia stem cells in chronic myeloid leukemia. Blood 2015, 126, 49. [Google Scholar] [CrossRef]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Irvine, D.; Munje, C.; Castaneda, E.G.; Moka, H.A.; Dunn, K.; Cassels, J.E.; Parry, N.; et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia 2020, 34, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef]
- Riether, C.; Schurch, C.M.; Flury, C.; Hinterbrandner, M.; Druck, L.; Huguenin, A.L.; Baerlocher, G.M.; Radpour, R.; Ochsenbein, A.F. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci. Transl. Med. 2015, 7, 298ra119. [Google Scholar] [CrossRef]
- Schurch, C.; Riether, C.; Matter, M.S.; Tzankov, A.; Ochsenbein, A.F. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J. Clin. Investig. 2012, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gallipoli, P.; DeGeer, D.; Sloma, I.; Forrest, D.L.; Chan, M.; Lai, D.; Jorgensen, H.; Ringrose, A.; Wang, H.M.; et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J. Natl. Cancer Inst. 2013, 105, 405–423. [Google Scholar] [CrossRef]
- Traer, E.; MacKenzie, R.; Snead, J.; Agarwal, A.; Eiring, A.M.; O’Hare, T.; Druker, B.J.; Deininger, M.W. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia 2012, 26, 1140–1143. [Google Scholar] [CrossRef]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef]
- Kuepper, M.K.; Butow, M.; Herrmann, O.; Ziemons, J.; Chatain, N.; Maurer, A.; Kirschner, M.; Maie, T.; Costa, I.G.; Eschweiler, J.; et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia 2019, 33, 1964–1977. [Google Scholar] [CrossRef] [PubMed]
- Salomoni, P.; Pandolfi, P.P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar] [CrossRef]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Lewis, A.H.; Chen, T.J.; Bridges, C.S.; Shen, Y.; Suppipat, K.; Puppi, M.; Tomolonis, J.A.; Pang, P.D.; Mistretta, T.A.; et al. A KLF4-DYRK2-mediated pathway regulating self-renewal in CML stem cells. Blood 2019, 134, 1960–1972. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef]
- Salati, S.; Salvestrini, V.; Carretta, C.; Genovese, E.; Rontauroli, S.; Zini, R.; Rossi, C.; Ruberti, S.; Bianchi, E.; Barbieri, G.; et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget 2017, 8, 49451–49469. [Google Scholar] [CrossRef]
- Pellicano, F.; Park, L.; Hopcroft, L.E.M.; Shah, M.M.; Jackson, L.; Scott, M.T.; Clarke, C.J.; Sinclair, A.; Abraham, S.A.; Hair, A.; et al. hsa-mir183/EGR1-mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood 2018, 131, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.S.; Sanchez, M.B.; Bonecker, S.; Furtado, C.; Koile, D.; Yankilevich, P.; Cranco, S.; Custidiano, M.D.R.; Freitas, J.; Moiraghi, B.; et al. miRNome profiling of LSC-enriched CD34(+)CD38(-)CD26(+) fraction in Ph(+) CML-CP samples from Argentinean patients: A potential new pharmacogenomic tool. Front. Pharm. 2020, 11, 612573. [Google Scholar] [CrossRef]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Wang, W.Z.; Pu, Q.H.; Lin, X.H.; Liu, M.Y.; Wu, L.R.; Wu, Q.Q.; Chen, Y.H.; Liao, F.F.; Zhu, J.Y.; Jin, X.B. Silencing of miR-21 sensitizes CML CD34+ stem/progenitor cells to imatinib-induced apoptosis by blocking PI3K/AKT pathway. Leuk. Res. 2015, 39, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.; He, J.; Agarwal, P.; Shah, M.; Welner, R.; Darley-Usmar, V.M.; et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investig. 2019, 129, 2685–2701. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Yang, K.; Wang, F.; Zhang, H.; Wang, X.; Chen, L.; Su, X.; Wu, X.; Han, Q.; Chen, Z.; Chen, Z.S.; et al. Target inhibition of CBP induced cell senescence in BCR-ABL- T315I mutant chronic myeloid leukemia. Front. Oncol. 2020, 10, 588641. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Abraham, S.A.; Shan, Y.; Guo, Z.; Desouza, N.; Cheloni, G.; Li, D.; Holyoake, T.L.; Li, S. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J. Clin. Investig. 2014, 124, 3847–3862. [Google Scholar] [CrossRef]
- Gallipoli, P.; Pellicano, F.; Morrison, H.; Laidlaw, K.; Allan, E.K.; Bhatia, R.; Copland, M.; Jorgensen, H.G.; Holyoake, T.L. Autocrine TNF-alpha production supports CML stem and progenitor cell survival and enhances their proliferation. Blood 2013, 122, 3335–3339. [Google Scholar] [CrossRef]
- Grockowiak, E.; Laperrousaz, B.; Jeanpierre, S.; Voeltzel, T.; Guyot, B.; Gobert, S.; Nicolini, F.E.; Maguer-Satta, V. Immature CML cells implement a BMP autocrine loop to escape TKI treatment. Blood 2017, 130, 2860–2871. [Google Scholar] [CrossRef]
- Himburg, H.A.; Roos, M.; Fang, T.; Zhang, Y.; Termini, C.M.; Schlussel, L.; Kim, M.; Pang, A.; Kan, J.; Zhao, L.; et al. Chronic myeloid leukemia stem cells require cell-autonomous pleiotrophin signaling. J. Clin. Investig. 2020, 130, 315–328. [Google Scholar] [CrossRef]
- Muselli, F.; Peyron, J.F.; Mary, D. Druggable biochemical pathways and potential therapeutic alternatives to target leukemic stem cells and eliminate the residual disease in chronic myeloid leukemia. Int. J. Mol. Sci. 2019, 20, 5616. [Google Scholar] [CrossRef] [PubMed]
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk. Res. 2018, 74, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef]
- Zhao, Y.; Masiello, D.; McMillian, M.; Nguyen, C.; Wu, Y.; Melendez, E.; Smbatyan, G.; Kida, A.; He, Y.; Teo, J.L.; et al. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene 2016, 35, 3705–3717. [Google Scholar] [CrossRef]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef]
- Pagnano, K.B.B.; Lopes, A.B.P.; Miranda, E.C.; Delamain, M.T.; Duarte, G.O.; Rodrigues, B.R.V.; Povoa, V.M.O.; Furlin, G.C.P.; Vianna, J.C.; da Silva, M.A.S.; et al. Efficacy and safety of pioglitazone in a phase 1/2 imatinib discontinuation trial (EDI-PIO) in chronic myeloid leukemia with deep molecular response. Am. J. Hematol. 2020, 95, E321–E323. [Google Scholar] [CrossRef]
- Selleri, C.; Maciejewski, J.P.; Pane, F.; Luciano, L.; Raiola, A.M.; Mostarda, I.; Salvatore, F.; Rotoli, B. Fas-mediated modulation of Bcr/Abl in chronic myelogenous leukemia results in differential effects on apoptosis. Blood 1998, 92, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, E.S.; Ninfali, P. Phytochemicals as innovative therapeutic tools against cancer stem cells. Int. J. Mol. Sci. 2015, 16, 15727–15742. [Google Scholar] [CrossRef]
- Liskova, A.; Kubatka, P.; Samec, M.; Zubor, P.; Mlyncek, M.; Bielik, T.; Samuel, S.M.; Zulli, A.; Kwon, T.K.; Busselberg, D. Dietary phytochemicals targeting cancer stem cells. Molecules 2019, 24, 899. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; O’Brien, S.; Cortes, J. Homoharringtonine/omacetaxine mepesuccinate: The long and winding road to food and drug administration approval. Clin. Lymphoma Myeloma Leuk. 2013, 13, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Mak, D.H.; Schober, W.D.; Chen, W.; Konopleva, M.; Cortes, J.; Kantarjian, H.M.; Andreeff, M.; Carter, B.Z. Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells. Mol. Cancer Ther. 2009, 8, 2509–2516. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, T.; Meng, Z.; Gan, Y.; Xu, X.; Lou, G.; Li, H.; Gan, X.; Zhou, H.; Tang, J.; et al. CaMKII gamma, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood 2012, 120, 4829–4839. [Google Scholar] [CrossRef]
- Wu, E.J.; Goussetis, D.J.; Beauchamp, E.; Kosciuczuk, E.M.; Altman, J.K.; Eklund, E.A.; Platanias, L.C. Resveratrol enhances the suppressive effects of arsenic trioxide on primitive leukemic progenitors. Cancer Biol. Ther. 2014, 15, 473–478. [Google Scholar] [CrossRef]
- Du, Y.; Xia, Y.; Pan, X.; Chen, Z.; Wang, A.; Wang, K.; Li, J.; Zhang, J. Fenretinide targets chronic myeloid leukemia stem/progenitor cells by regulation of redox signaling. Antioxid. Redox Signal. 2014, 20, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef]
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jorgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 2016, 128, 2671–2682. [Google Scholar] [CrossRef]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell 2019, 24, 769–784 e766. [Google Scholar] [CrossRef]
- Laperrousaz, B.; Jeanpierre, S.; Sagorny, K.; Voeltzel, T.; Ramas, S.; Kaniewski, B.; Ffrench, M.; Salesse, S.; Nicolini, F.E.; Maguer-Satta, V. Primitive CML cell expansion relies on abnormal levels of BMPs provided by the niche and on BMPRIb overexpression. Blood 2013, 122, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Von Palffy, S.; Landberg, N.; Sanden, C.; Zacharaki, D.; Shah, M.; Nakamichi, N.; Hansen, N.; Askmyr, M.; Lilljebjorn, H.; Rissler, M.; et al. A high-content cytokine screen identifies myostatin propeptide as a positive regulator of primitive chronic myeloid leukemia cells. Haematologica 2020, 105, 2095–2104. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef]
- Silvestri, G.; Trotta, R.; Stramucci, L.; Ellis, J.J.; Harb, J.G.; Neviani, P.; Wang, S.; Eisfeld, A.K.; Walker, C.J.; Zhang, B.; et al. Persistence of drug-resistant leukemic stem cells and impaired NK cell immunity in CML patients depend on MIR300 antiproliferative and PP2A-activating functions. Blood Cancer Discov. 2020, 1, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef]
- Godavarthy, P.S.; Kumar, R.; Herkt, S.C.; Pereira, R.S.; Hayduk, N.; Weissenberger, E.S.; Aggoune, D.; Manavski, Y.; Lucas, T.; Pan, K.T.; et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin—SCL/TAL1—CD44 axis. Haematologica 2020, 105, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.C.; Bhatia, R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Stromberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Yung, Y.; Lee, E.; Chu, H.T.; Yip, P.K.; Gill, H. Targeting abnormal hematopoietic stem cells in chronic myeloid leukemia and philadelphia chromosome-negative classical myeloproliferative neoplasms. Int. J. Mol. Sci. 2021, 22, 659. [Google Scholar] [CrossRef]
- Hehlmann, R.; Muller, M.C.; Lauseker, M.; Hanfstein, B.; Fabarius, A.; Schreiber, A.; Proetel, U.; Pletsch, N.; Pfirrmann, M.; Haferlach, C.; et al. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: Results from the randomized CML-study IV. J. Clin. Oncol. 2014, 32, 415–423. [Google Scholar] [CrossRef]
- Falchi, L.; Kantarjian, H.M.; Wang, X.; Verma, D.; Quintas-Cardama, A.; O’Brien, S.; Jabbour, E.J.; Ravandi-Kashani, F.; Borthakur, G.; Garcia-Manero, G.; et al. Significance of deeper molecular responses in patients with chronic myeloid leukemia in early chronic phase treated with tyrosine kinase inhibitors. Am. J. Hematol. 2013, 88, 1024–1029. [Google Scholar] [CrossRef]
- Kantarjian, H.; Cortes, J.E. Complete cytogenetic response, not deep molecular response, is associated with survival in chronic myeloid leukemia. J. Clin. Oncol. 2014, 32, 3077. [Google Scholar] [CrossRef]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Bartley, P.A.; Slader, C.; Field, C.; Dang, P.; Filshie, R.J.; et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia 2010, 24, 1719–1724. [Google Scholar] [CrossRef]
- Bernardi, S.; Malagola, M.; Zanaglio, C.; Polverelli, N.; Dereli Eke, E.; D’Adda, M.; Farina, M.; Bucelli, C.; Scaffidi, L.; Toffoletti, E.; et al. Digital PCR improves the quantitation of DMR and the selection of CML candidates to TKIs discontinuation. Cancer Med. 2019, 8, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Pagani, I.S.; Dang, P.; Saunders, V.A.; Grose, R.; Shanmuganathan, N.; Kok, C.H.; Carne, L.; Rwodzi, Z.; Watts, S.; McLean, J.; et al. Lineage of measurable residual disease in patients with chronic myeloid leukemia in treatment-free remission. Leukemia 2020, 34, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Kirschner, K.; Copland, M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia 2021, 35, 1229–1242. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | Abbreviation | Definition |
|---|---|---|
| Complete Hematological Response | CHR | Normalization of blood cell count |
| Complete Cytogenetic Response | CCyR | No metaphases positive for the Philadelphia chromosome out of 20 total metaphases examined by chromosome banding analysis |
| Major Molecular Response | MMR | 3-log reduction in BCR-ABL1 transcript levels (from a standardized baseline) as assessed by real time quantitative PCR |
| Deep Molecular Response | MR4 | 4-log reduction in BCR-ABL1 transcript levels |
| MR4.5 | 4.5-log reduction in BCR-ABL1 transcript levels | |
| MR5 | 5-log reduction in BCR-ABL1 transcript levels |
| Drug | Target | Trial ID | Status |
|---|---|---|---|
| LDE225 (sonidegib) | SMO | NCT01456676 | Completed |
| PF-04449913 (glasdegib) | SMO | NCT00953758 | Completed |
| BMS-833923 | SMO | NCT01218477 | Completed |
| RAD001 | mTOR | NCT01188889 | Withdrawn |
| LBH589 (panobinostat) | Histone deacetylases | NCT00686218 NCT00451035 | Completed Completed |
| As2O3 | PML | NCT01397734 | Terminated |
| Chloroquine | Autophagy | NCT01227135 | Completed |
| Zileuton | ALOX5 | NCT01130688 | Terminated |
| Ruxolitinib | JAK2 | NCT01751425 NCT01702064 NCT02253277 NCT02973711 NCT03610971 NCT03654768 | Terminated Completed Completed Withdrawn Recruiting Recruiting |
| PRI-274 | CBP/β-catenin | NCT01606579 | Completed |
| Pioglitazone | PPARγ/STAT5 | NCT02730195 NCT04883125 NCT02852486 NCT02852486 NCT02889003 NCT02767063 | Terminated Completed Active, not recruting Active, not recruiting Recruiting Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soverini, S.; De Santis, S.; Monaldi, C.; Bruno, S.; Mancini, M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? Int. J. Mol. Sci. 2021, 22, 7093. https://doi.org/10.3390/ijms22137093
Soverini S, De Santis S, Monaldi C, Bruno S, Mancini M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? International Journal of Molecular Sciences. 2021; 22(13):7093. https://doi.org/10.3390/ijms22137093
Chicago/Turabian StyleSoverini, Simona, Sara De Santis, Cecilia Monaldi, Samantha Bruno, and Manuela Mancini. 2021. "Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort?" International Journal of Molecular Sciences 22, no. 13: 7093. https://doi.org/10.3390/ijms22137093
APA StyleSoverini, S., De Santis, S., Monaldi, C., Bruno, S., & Mancini, M. (2021). Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? International Journal of Molecular Sciences, 22(13), 7093. https://doi.org/10.3390/ijms22137093