
Annexin A2 in Fibrinolysis, Inflammation and Fibrosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
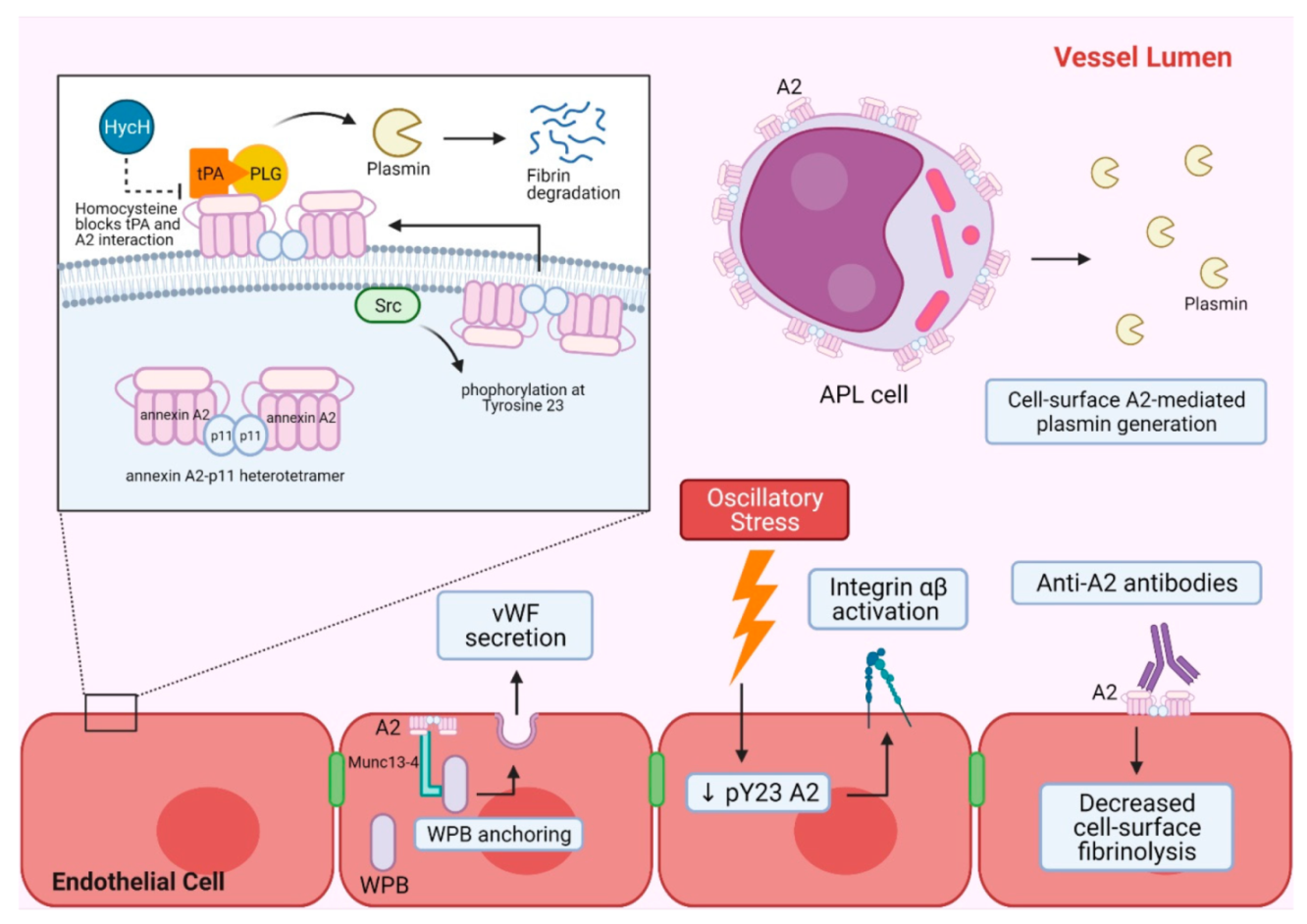
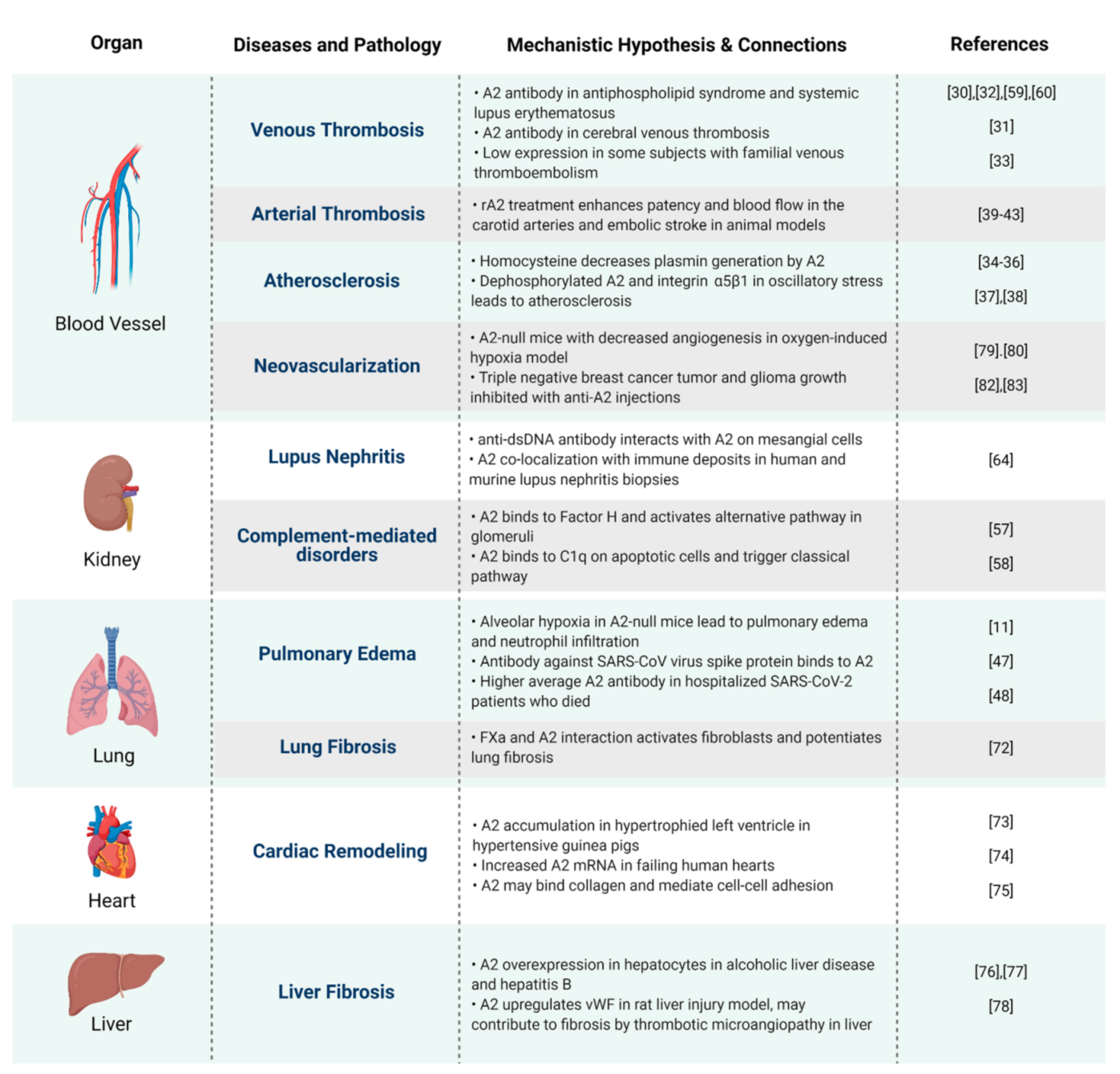
2. A2 in Hemostasis and Vascular Homeostasis
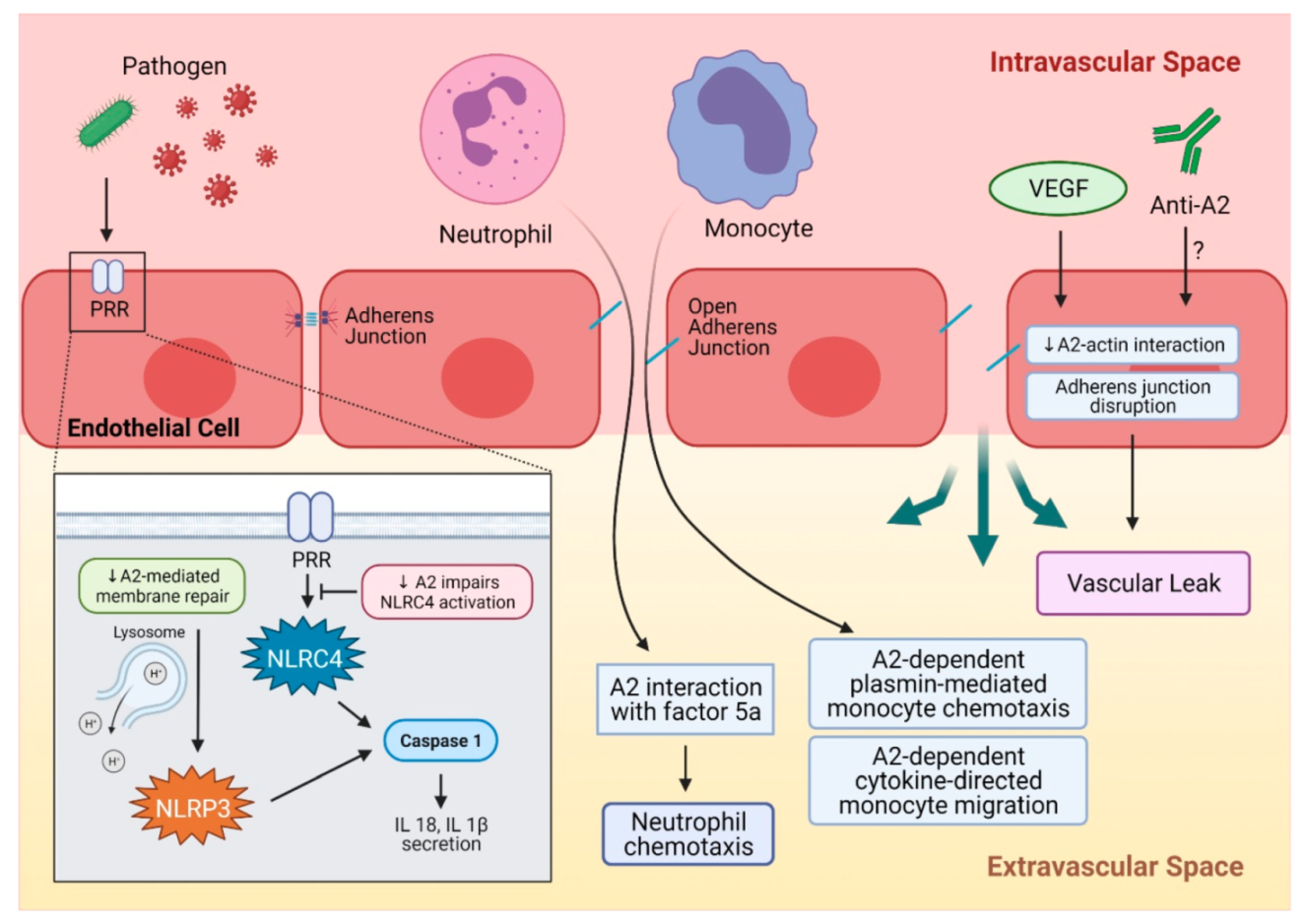
3. A2 in Inflammation and Autoimmune Disorders
4. A2 in Tissue Repair and Fibrosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A2 | annexin A2 |
| tPA | tissue plasminogen activator |
| uPA | urokinase plasminogen activator |
| APL | acute promyelocytic leukemia |
| IgG | immunoglobulin G |
| IL | Interleukin |
| APS | antiphospholipid syndrome |
| VEC | vascular endothelial cadherin |
| VEGF | vascular endothelial growth factor |
| COVID-19 | corona virus disease-19 |
| SARS | severe acute respiratory syndrome |
| PRR | pattern recognition receptor |
| ROS | reactive oxygen species |
| AP | alternative pathway of complement |
| PML | promyelocytic leukemia |
| CD | cluster of differentiation |
| vWF | von Willebrand factor |
| OIR | oxygen-induced retinopathy |
| HIF | hypoxia-inducible factor |
References
- Hajjar, K.A.; Ruan, J. Fibrinolysis and thrombolysis. In Williams Hematology; Kaushansky, K., Prchal, J.T., Burns, L.J., Lichtman, M.A., Levi, M., Linch, D.C., Eds.; McGraw Hill: New York, NY, USA, 2021. [Google Scholar]
- Pepper, M.; Sappino, A.P.; Stöcklin, R.; Montesano, R.; Orci, L.; Vassalli, J.D. Upregulation of urokinase receptor expression on migrating endothelial cells. J. Cell Biol. 1993, 122, 673–684. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The multifaceted role of plasminogen in inflammation. Cell. Signal. 2020, 75, 109761. [Google Scholar] [CrossRef] [PubMed]
- Schuliga, M. The inflammatory actions of coagulant and fibrinolytic proteases in disease. Mediat. Inflamm. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Hajjar, K.A.; Hamel, N.M. Identification and characterization of human endothelial cell membrane binding sites for tissue plasminogen activator and urokinase. J. Biol. Chem. 1990, 265, 2908–2916. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Acharya, S.S. Annexin II and regulation of cell surface fibrinolysis. Ann. N. Y. Acad. Sci. 2006, 902, 265–271. [Google Scholar] [CrossRef]
- Rao, J.S.; Gujrati, M.; Chetty, C. Tumor-associated soluble uPAR-directed endothelial cell motility and tumor angiogenesis. Oncogenesis 2013, 2, e53. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Hajjar, K.A. Annexin A2 system in human biology: Cell surface and beyond. Semin. Thromb. Hemost. 2013, 39, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Flood, E.C.; Almeida, D.; Yan, L.; Berlin, D.A.; Heerdt, P.M.; Hajjar, K.A. Annexin A2 supports pulmonary microvascular integrity by linking vascular endothelial cadherin and protein tyrosine phosphatases. J. Exp. Med. 2017, 214, 2535–2545. [Google Scholar] [CrossRef]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Grill, D.; Matos, A.L.L.; De Vries, W.C.; Kudruk, S.; Heflik, M.; Dörner, W.; Mootz, H.D.; Ravoo, B.J.; Galla, H.-J.; Gerke, V. Bridging of membrane surfaces by annexin A2. Sci. Rep. 2018, 8, 14662. [Google Scholar] [CrossRef] [Green Version]
- Thiel, C.; Osborn, M.; Gerke, V. The tight association of the tyrosine kinase substrate annexin II with the submembranous cytoskeleton depends on intact p11- and Ca(2+)-binding sites. J. Cell Sci. 1992, 103, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Grindheim, A.K.; Saraste, J.; Vedeler, A. Protein phosphorylation and its role in the regulation of annexin A2 function. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 2515–2529. [Google Scholar] [CrossRef]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem. 2004, 279, 43411–43418. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, K. The endothelial cell tissue plasminogen activator receptor. Specific interaction with plasminogen. J. Biol. Chem. 1991, 266, 21962–21970. [Google Scholar] [CrossRef]
- Hajjar, K.; Jacovina, A.; Chacko, J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with Annexin II. J. Biol. Chem. 1994, 269, 21191–21197. [Google Scholar] [CrossRef]
- Cesarman, G.; Guevara, C.; Hajjar, K. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-Mediated enhancement of t-PA-dependent plasminogen activation. J. Biol. Chem. 1994, 269, 21198–21203. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Mauri, L.; Jacovina, A.T.; Zhong, F.; Mirza, U.A.; Padovan, J.C.; Chait, B.T. Tissue plasminogen activator binding to the annexin II tail domain. J. Biol. Chem. 1998, 273, 9987–9993. [Google Scholar] [CrossRef] [Green Version]
- Dassah, M.; Deora, A.B.; He, K.; Hajjar, K.A. The endothelial cell annexin A2 system and vascular fibrinolysis. Gen. Physiol. Biophys. 2009, 28, F20–F28. [Google Scholar] [PubMed]
- Flood, E.C.; Hajjar, K.A. The annexin A2 system and vascular homeostasis. Vasc. Pharmacol. 2011, 54, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Ling, Q.; Jacovina, A.T.; Deora, A.; Febbraio, M.; Simantov, R.; Silverstein, R.L.; Hempstead, B.; Mark, W.H.; Hajjar, K.A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J. Clin. Investig. 2004, 113, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Brandherm, I.; Disse, J.; Zeuschner, D.; Gerke, V. cAMP-Induced secretion of endothelial von Willebrand factor is regulated by a phosphorylation/dephosphorylation switch in annexin A2. Blood 2013, 122, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehab, T.; Santos, N.C.; Holthenrich, A.; Koerdt, S.; Disse, J.; Schuberth, C.; Nazmi, A.R.; Neeft, M.; Koch, H.; Man, K.N.M.; et al. A novel Munc13-4/S100A10/annexin A2 Complex promotes Weibel–Palade body exocytosis in endothelial cells. Mol. Biol. Cell 2017, 28, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.H. Annexinopathies-A new class of diseases. N. Engl. J. Med. 1999, 340, 1035–1036. [Google Scholar] [CrossRef]
- Menell, J.S.; Cesarman-Maus, G.C.; Jacovina, A.T.; McLaughlin, M.A.; Lev, E.A.; Hajjar, K.A. Annexin II and bleeding in acute promyelocytic leukemia. N. Engl. J. Med. 1999, 340, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnot, C.; Wilde, J.T. Haemostatic Problems in Acute Promyelocytic Leukaemia. Blood Rev. 2006, 20, 289–297. [Google Scholar] [CrossRef]
- Stein, E.; McMahon, B.; Kwaan, H.; Altman, J.K.; Frankfurt, O.; Tallman, M.S. The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract. Res. Clin. Haematol. 2009, 22, 153–163. [Google Scholar] [CrossRef]
- Cesarman-Maus, G.C.; Ríos-Luna, N.P.; Deora, A.B.; Huang, B.; Villa, R.; Cravioto, M.D.C.; Alarcón-Segovia, D.; Sánchez-Guerrero, J.; Hajjar, K.A. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 2006, 107, 4375–4382. [Google Scholar] [CrossRef]
- Cesarman-Maus, G.; Cantú-Brito, C.; Barinagarrementeria, F.; Villa, R.; Reyes, E.; Sanchez-Guerrero, J.; Hajjar, K.A.; Latorre, E.G. Autoantibodies against the fibrinolytic receptor, annexin A2, in cerebral venous thrombosis. Stroke 2011, 42, 501–503. [Google Scholar] [CrossRef] [Green Version]
- Ao, W.; Zheng, H.; Chen, X.-W.; Shen, Y.; Yang, C.-D. Anti-Annexin II antibody is associated with thrombosis and/or pregnancy morbidity in antiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatol. Int. 2010, 31, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Fassel, H.; Chen, H.; Ruisi, M.; Kumar, N.; DeSancho, M.T.; Hajjar, K.A. Reduced expression of annexin A2 is associated with impaired cell surface fibrinolysis and venous thromboembolism. Blood 2021, 137, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, A.K. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J. Clin. Investig. 1993, 91, 2873–2879. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, K.A.; Jacovina, A.T. Modulation of annexin II by homocysteine: Implications for atherothrombosis. J. Investig. Med. 1998, 46, 364–369. [Google Scholar]
- Jacovina, A.T.; Deora, A.B.; Ling, Q.; Broekman, M.J.; Almeida, D.; Greenberg, C.B.; Marcus, A.J.; Smith, J.D.; Hajjar, K.A. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2–dependent fibrinolysis. J. Clin. Investig. 2009, 119, 3384–3394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhou, T.; Chen, Z.; Yan, M.; Li, B.; Lv, H.; Wang, C.; Xiang, S.; Shi, L.; Zhu, Y.; et al. Coupling of integrin α5 to annexin A2 by flow drives endothelial activation. Circ. Res. 2020, 127, 1074–1090. [Google Scholar] [CrossRef] [PubMed]
- Demos, C.; Williams, D.; Jo, H. Disturbed flow induces atherosclerosis by annexin A2-mediated integrin activation. Circ. Res. 2020, 127, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Yoshida, M.; Hiraoka, M.; Hajjar, K.A.; Tanaka, A.; Yasukochi, Y.; Numano, F. Recombinant annexin II modulates impaired fibrinolytic activity in vitro and in rat carotid artery. Circ. Res. 2001, 89, 1240–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ishii, H.; Hiraoka, M.; Miyasaka, N.; Kuroiwa, T.; Hajjar, K.A.; Nagaoka, T.; Duong, T.Q.; Ohno, K.; Yoshida, M. Efficacy of recombinant annexin 2 for fibrinolytic therapy in a rat embolic stroke model: A magnetic resonance imaging study. Brain Res. 2007, 1165, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Fan, X.; Yu, Z.; Liu, J.; Murata, Y.; Lu, J.; Zhao, S.; Hajjar, A.K.; Lo, E.H.; Wang, X. Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. Br. J. Pharmacol. 2010, 30, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fan, X.; Yu, Z.; Liao, Z.; Zhao, J.; Mandeville, E.; Guo, S.; Lo, E.H.; Wang, X. Effects of tissue plasminogen activator and annexin A2 combination therapy on long-term neurological outcomes of rat focal embolic stroke. Stroke 2014, 45, 619–622. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Jiang, Y.; Yu, Z.; Liu, Q.; Guo, S.; Sun, X.; Van Leyen, K.; Ning, M.; Gao, X.; Lo, E.H.; et al. Annexin A2 plus low-dose tissue plasminogen activator combination attenuates cerebrovascular dysfunction after focal embolic stroke of rats. Transl. Stroke Res. 2017, 8, 549–559. [Google Scholar] [CrossRef]
- Dallacasagrande, V.; Hajjar, K.A. Annexin A2 in inflammation and host defense. Cells 2020, 9, 1499. [Google Scholar] [CrossRef]
- Murakami, M.; Simons, M. Regulation of vascular integrity. J. Mol. Med. 2009, 87, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyraud, S.; Jaquinod, M.; Durmort, C.; Dambroise, E.; Concord, E.; Schaal, J.P.; Huber, P.; Gulino-Debrac, D. Contribution of annexin 2 to the architecture of mature endothelial adherens junctions. Mol. Cell. Biol. 2008, 28, 1657–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.-T.; Lin, C.-F.; Liao, P.-C.; Kuo, Y.-M.; Wang, S.; Yeh, T.-M.; Shieh, C.-C.K.; Su, I.-J.; Lei, H.-Y.; Lin, Y.-S. Annexin A2 on lung epithelial cell surface is recognized by severe acute respiratory syndrome-associated coronavirus spike domain 2 antibodies. Mol. Immunol. 2010, 47, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, M.; Gomes, C.; Carsons, S.E.; Bender, M.T.; Cotzia, P.; Miao, Q.R.; Lee, D.C.; Rodriguez, A. Autoimmunity to the lung protective phospholipid-binding protein annexin A2 predicts mortality among hospitalized COVID-19 patients. medRxiv 2021. [Google Scholar] [CrossRef]
- Laumonnier, Y.; Syrovets, T.; Burysek, L.; Simmet, T. Identification of the annexin A2 heterotetramer as a receptor for the plasmin-induced signaling in human peripheral monocytes. Blood 2006, 107, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.; Deora, A.B.; Jacovina, A.T.; Weintraub, R.; Gertler, M.; Khan, K.M.F.; Falcone, D.J.; Hajjar, K.A. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: Enhanced expression by macrophages. Blood 2004, 103, 317–324. [Google Scholar] [CrossRef]
- McVoy, L.A.; Kew, R.R. CD44 and Annexin A2 mediate the C5a chemotactic cofactor function of the vitamin D binding protein. J. Immunol. 2005, 175, 4754–4760. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shaw, D.; Sakhon, O.S.; Snyder, G.A.; Sundberg, E.J.; Santambrogio, L.; Sutterwala, F.S.; Dumler, J.S.; Shirey, K.A.; Perkins, D.J.; et al. The tick protein sialostatin L2 binds to annexin A2 and inhibits NLRC4-mediated inflammasome activation. Infect. Immun. 2016, 84, 1796–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharf, B.; Clement, C.; Wu, X.-X.; Morozova, K.; Zanolini, D.; Follenzi, A.; Larocca, J.N.; Levon, K.; Sutterwala, F.S.; Rand, J.; et al. Annexin A2 binds to endosomes following organelle destabilization by particulate wear debris. Nat. Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Li, X.; Li, R.; Fang, L.; Sun, L.; Wang, Y.; Wu, M. Annexin A2 modulates ROS and impacts inflammatory response via IL-17 signaling in polymicrobial sepsis mice. PLoS Pathog. 2016, 12, e1005743. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Renner, B.; Tong, H.H.; Laskowski, J.; Jonscher, K.; Goetz, L.; Woolaver, R.; Hannan, J.; Li, Y.X.; Hourcade, D.; Pickering, M.C.; et al. Annexin A2 enhances complement activation by inhibiting factor H. J. Immunol. 2016, 196, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Leffler, J.; Blom, A.M. Annexin A2 and A5 serve as new ligands for c1q on apoptotic cells. J. Biol. Chem. 2012, 287, 33733–33744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; McCrae, K.R. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-Β2 glycoprotein I antibodies. Blood 2005, 105, 1964–1969. [Google Scholar] [CrossRef] [Green Version]
- Romay-Penabad, Z.; Montiel-Manzano, M.G.; Shilagard, T.; Papalardo, E.; Vargas, G.; Deora, A.B.; Wang, M.; Jacovina, A.T.; Garcia-Latorre, E.; Reyes-Maldonado, E.; et al. Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood 2009, 114, 3074–3083. [Google Scholar] [CrossRef] [Green Version]
- Cockrell, E.; Espinola, R.G.; McCrae, K.R. Annexin A2: Biology and relevance to the antiphospholipid syndrome. Lupus 2008, 17, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Salle, V.; Mazière, J.C.; Smail, A.; Cévallos, R.; Mazière, C.; Fuentes, V.; Tramier, B.; Makdassi, R.; Choukroun, G.; Vittecoq, O.; et al. Anti-annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J. Clin. Immunol. 2008, 28, 291–297. [Google Scholar] [CrossRef]
- Cañas, F.; Simonin, L.; Couturaud, F.; Renaudineau, Y. Annexin A2 autoantibodies in thrombosis and autoimmune diseases. Thromb. Res. 2015, 135, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J. Am. Soc. Nephrol. 2010, 21, 1912–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defour, A.; Medikayala, S.; Van Der Meulen, J.H.; Hogarth, M.W.; Holdreith, N.; Malatras, A.; Duddy, W.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 mediates dysferlin accumulation and muscle cell membrane repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Rescher, U.; Ludwig, C.; Konietzko, V.; Kharitonenkov, A.; Gerke, V. Tyrosine phosphorylation of annexin A2 regulates rho-mediated actin rearrangement and cell adhesion. J. Cell Sci. 2008, 121, 2177–2185. [Google Scholar] [CrossRef] [Green Version]
- Rankin, C.R.; Hilgarth, R.S.; Leoni, G.; Kwon, M.; Beste, K.A.D.; Parkos, C.A.; Nusrat, A. Annexin A2 regulates β1 integrin internalization and intestinal epithelial cell migration. J. Biol. Chem. 2013, 288, 15229–15239. [Google Scholar] [CrossRef] [Green Version]
- Madureira, P.A.; Hill, R.; Miller, V.A.; Giacomantonio, C.; Lee, P.W.K.; Waisman, D.M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget 2011, 2, 1075–1093. [Google Scholar] [CrossRef]
- Grindheim, A.K.; Hollås, H.; Raddum, A.M.; Saraste, J.; Vedeler, A. Reactive oxygen species exert opposite effects on Tyr23 phosphorylation of the nuclear and cortical pools of annexin A2. J. Cell Sci. 2016, 129, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Vopálenský, V.; Pospíšek, M.; Vedeler, A. Co-Localization of interleukin-1α and Annexin A2 at the plasma membrane in response to oxidative stress. Cytokine 2020, 133, 155141. [Google Scholar] [CrossRef]
- Schuliga, M.; Jaffar, J.; Berhan, A.; Langenbach, S.; Harris, T.; Waters, D.; Lee, P.V.S.; Grainge, C.; Westall, G.; Knight, D.; et al. Annexin A2 contributes to lung injury and fibrosis by augmenting factor Xa Fibrogenic activity. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L772–L782. [Google Scholar] [CrossRef] [Green Version]
- Trouvé, P.; Legot, S.; Bélikova, I.; Marotte, F.; Bénévolensky, D.; Russo-Marie, F.; Samuel, J.-L.; Charlemagne, D. Localization and quantitation of cardiac annexins II, V, and VI in hypertensive guinea pigs. Am. J. Physiol. Content 1999, 276, H1159–H1166. [Google Scholar] [CrossRef]
- Benevolensky, D.; Belikova, Y.; Mohammadzadeh, R.; Trouvé, P.; Marotte, F.; Russo-Marie, F.; Samuel, J.-L.; Charlemagne, D. Expression and localization of the annexins II, V, and VI in myocardium from patients with end-stage heart failure. Lab. Investig. 2000, 80, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Camors, E.; Monceau, V.; Charlemagne, D. Annexins and Ca Handling in the heart. Cardiovasc. Res. 2005, 65, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, D.; Leo, M.A.; McGuinness, P.H.; Lieber, C.S.; Brennan, Y.; Williams, R.; Wang, X.M.; McCaughan, G.W.; Gorrell, M.D.; Haber, P.S. Gene expression profiling of alcoholic liver disease in the baboon (Papio hamadryas) and human liver. Am. J. Pathol. 2003, 163, 2303–2317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Peng, X.; Zhang, Z.; Feng, Y.; Jia, X.; Shi, Y.; Yang, H.; Zhang, Z.; Zhang, X.; Liu, L.; et al. Subcellular proteome analysis unraveled annexin A2 related to immune liver fibrosis. J. Cell. Biochem. 2010, 110, 219–228. [Google Scholar] [CrossRef]
- Yang, M.; Wang, C.; Li, S.; Xv, X.; She, S.; Ran, X.; Li, S.; Hu, H.; Hu, P.; Zhang, D.; et al. Annexin A2 promotes liver fibrosis by mediating von Willebrand factor secretion. Dig. Liver Dis. 2017, 49, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Deora, A.B.; He, K.-L.; Chen, K.; Sui, G.; Jacovina, A.T.; Almeida, D.; Hong, P.; Burgman, P.; Hajjar, K.A. Hypoxia-inducible factor-1 drives annexin A2 system-mediated perivascular fibrin clearance in oxygen-induced retinopathy in mice. Blood 2011, 118, 2918–2929. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Huang, L.; Wu, J.; Zhang, Y.; Pan, D.; Liu, X. Vascular endothelial growth factor upregulates expression of an-nexin A2 in vitro and in a mouse model of ischemic retinopathy. Mol. Vis. 2009, 15, 1231–1242. [Google Scholar]
- Genetos, D.C.; Wong, A.; Watari, S.; Yellowley, C.E. Hypoxia increases annexin A2 expression in osteoblastic cells via VEGF and ERK. Bone 2010, 47, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Blackman, M.R.; Sharma, M. Antibody-directed neutralization of annexin II (ANX II) inhibits neoangiogenesis and human breast tumor growth in a xenograft model. Exp. Mol. Pathol. 2012, 92, 175–184. [Google Scholar] [CrossRef]
- Sharma, M.C.; Jain, D. Important role of annexin A2 (ANXA2) in new blood vessel development in vivo and human triple negative breast cancer (TNBC) growth. Exp. Mol. Pathol. 2020, 116, 104523. [Google Scholar] [CrossRef]
- Zhai, H.; Acharya, S.; Gravanis, I.; Mehmood, S.; Seidman, R.J.; Shroyer, K.R.; Hajjar, K.A.; Tsirka, S.E. Annexin A2 promotes glioma cell invasion and tumor progression. J. Neurosci. 2011, 31, 14346–14360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Hajjar, K.A. The annexin A2 system and angiogenesis. Biol. Chem. 2016, 397, 1005–1016. [Google Scholar] [CrossRef]
- Barker, T.H.; Engler, A.J. The provisional matrix: Setting the stage for tissue repair outcomes. Matrix Biol. 2017, 60-61, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Loskutoff, D.J.; Quigley, J.P. PAI-1, Fibrosis, and the elusive provisional fibrin matrix. J. Clin. Investig. 2000, 106, 1441–1443. [Google Scholar] [CrossRef] [PubMed]
- Wynn, A.T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2007, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nat. Cell Biol. 2020, 587, 555–566. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.I.; Hajjar, K.A. Annexin A2 in Fibrinolysis, Inflammation and Fibrosis. Int. J. Mol. Sci. 2021, 22, 6836. https://doi.org/10.3390/ijms22136836
Lim HI, Hajjar KA. Annexin A2 in Fibrinolysis, Inflammation and Fibrosis. International Journal of Molecular Sciences. 2021; 22(13):6836. https://doi.org/10.3390/ijms22136836
Chicago/Turabian StyleLim, Hana I., and Katherine A. Hajjar. 2021. "Annexin A2 in Fibrinolysis, Inflammation and Fibrosis" International Journal of Molecular Sciences 22, no. 13: 6836. https://doi.org/10.3390/ijms22136836
APA StyleLim, H. I., & Hajjar, K. A. (2021). Annexin A2 in Fibrinolysis, Inflammation and Fibrosis. International Journal of Molecular Sciences, 22(13), 6836. https://doi.org/10.3390/ijms22136836