
Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease

 ,
,  , and
, and 
Abstract
1. Introduction
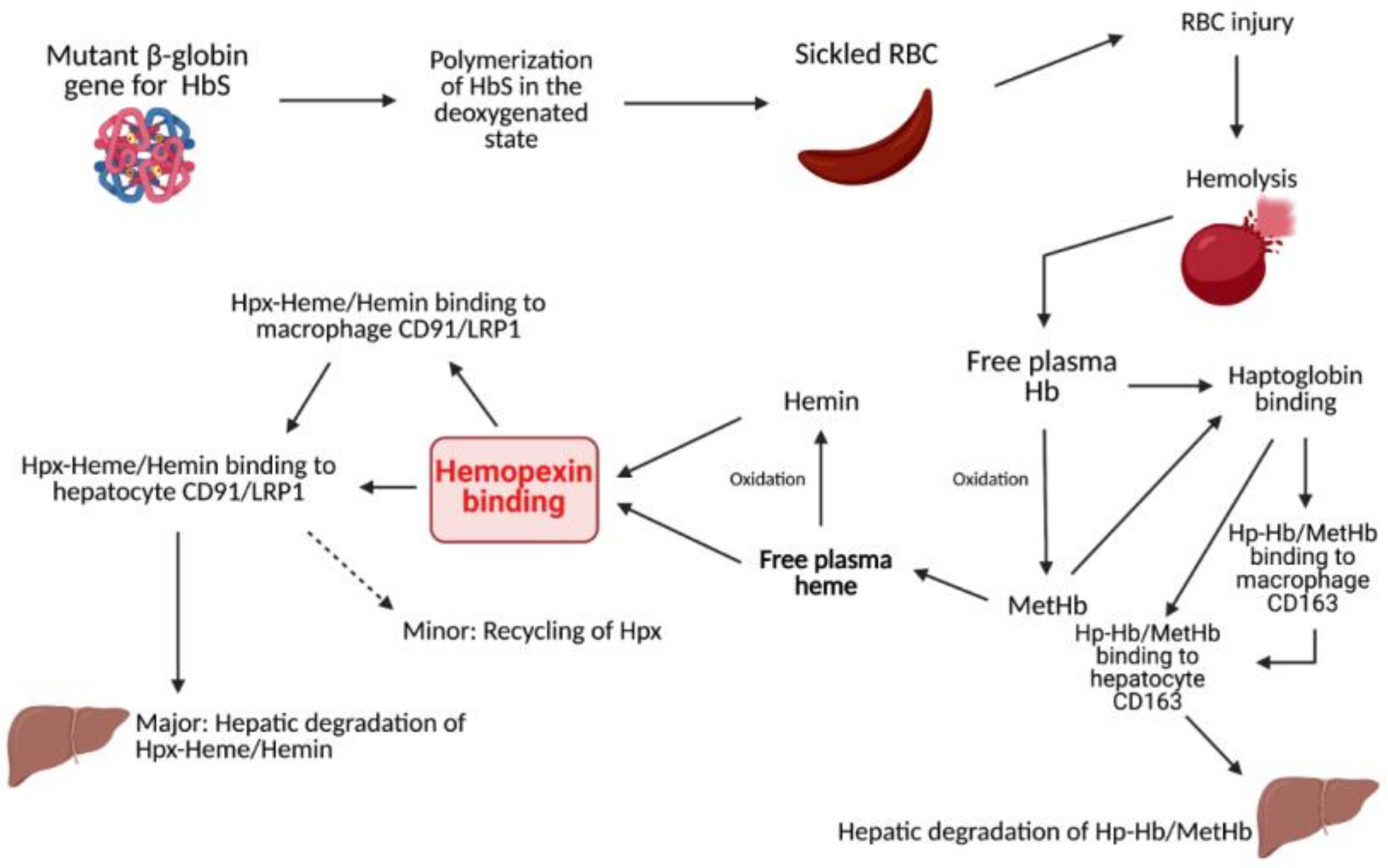
1.1. Heme Clearance
1.2. Sickle Cell Disease
1.3. Sickle Cell Disease and Hemolysis
2. Search Terms and Strategy
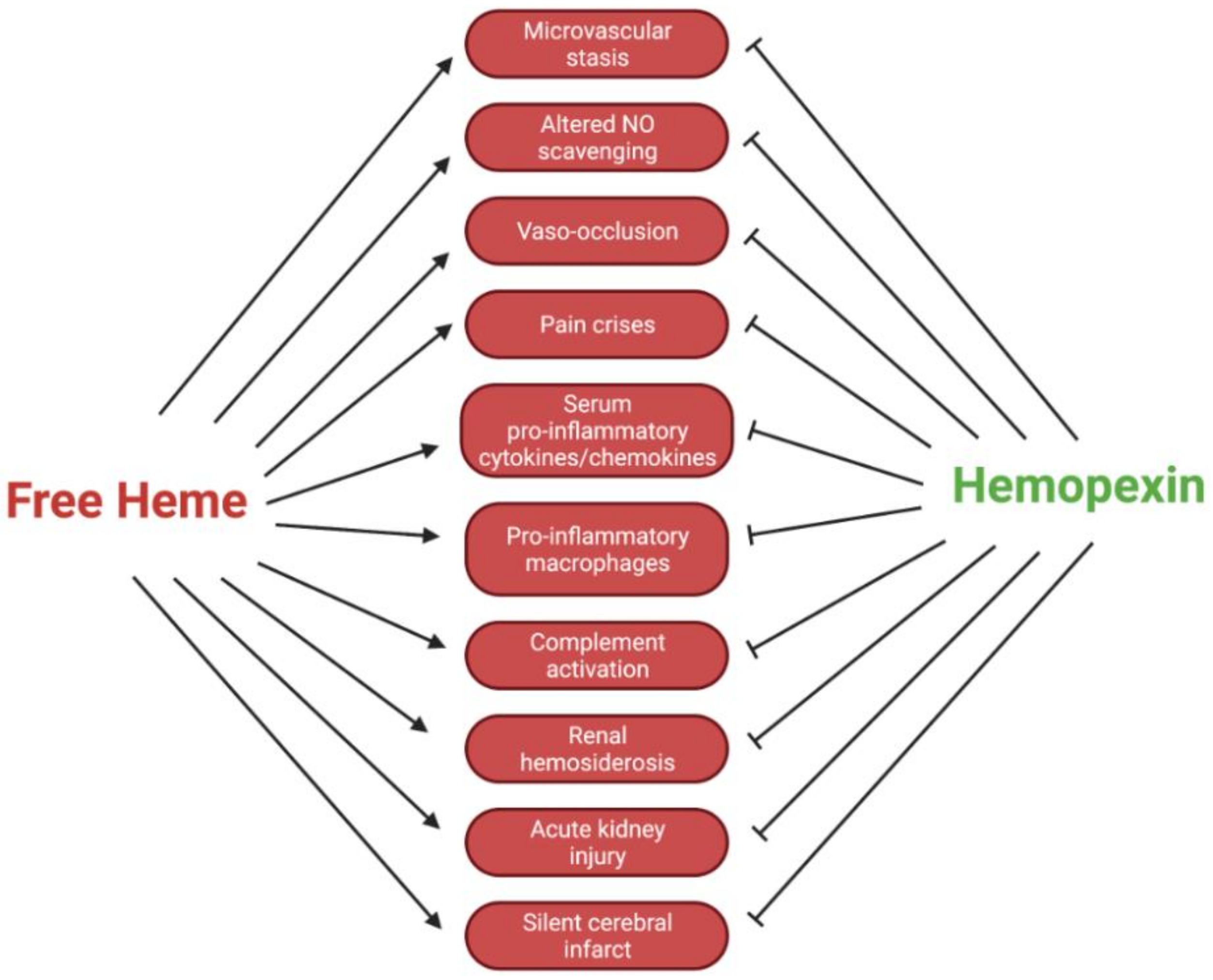
3. Immune Modulation
3.1. Hemopexin Mediation of Pro-Inflammatory Cytokine Expression
3.2. Hemopexin Attenuation of Deleterious Complement Activation
4. Microvascular Stasis and Vaso-Occlusion
5. Heme Oxygenase-1
6. Hemopexin and Lipoproteins
6.1. Hemopexin Effects on Lipoprotein Expression
6.2. Hemopexin Effects on Lipoprotein Oxidation
7. Hemopexin and Acute Kidney Injury
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced pro-inflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef]
- Cox, M.C.; Le Brun, N.; Thomson, A.J.; Smith, A.; Morgan, W.T.; Moore, G.R. MCD, EPR and NMR Spectroscopic Studies of Rabbit Hemopexin and Its Heme Binding Domain. Biochim. Biophys. Acta 1995, 1253, 215–223. [Google Scholar] [CrossRef]
- Shipulina, N.; Smith, A.; Morgan, W.T. Heme Binding by Hemopexin: Evidence for Multiple Modes of Binding and Functional Implications. J. Protein Chem. 2000, 19, 239–248. [Google Scholar] [CrossRef]
- Detzel, M.S.; Schmalohr, B.F.; Steinbock, F.; Hopp, M.-T.; Ramoji, A.; Paul George, A.A.; Neugebauer, U.; Imhof, D. Revisiting the Interaction of Heme with Hemopexin. Biol. Chem. 2021, 402, 675–691. [Google Scholar] [CrossRef]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE 2018, 13, e0196455. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Paul, J.; Wang, Y.; Gupta, M.; Vang, D.; Thompson, S.; Jha, R.; Nguyen, J.; Valverde, Y.; Lamarre, Y.; et al. Heme Causes Pain in Sickle Mice via Toll-Like Receptor 4-Mediated Reactive Oxygen Species- and Endoplasmic Reticulum Stress-Induced Glial Activation. Antioxid. Redox Signal. 2021, 34, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Sansing, L.H.; Harris, T.H.; Welsh, F.A.; Kasner, S.E.; Hunter, C.A.; Kariko, K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 2011, 70, 646–656. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Adisa, O.A.; Hu, Y.; Ghosh, S.; Aryee, D.; Osunkwo, I.; Ofori-Acquah, S.F. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br. J. Haematol. 2013, 162, 702–705. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718. [Google Scholar] [CrossRef]
- Li, R.; Saleem, S.; Zhen, G.; Cao, W.; Zhuang, H.; Lee, J.; Smith, A.; Altruda, F.; Tolosano, E.; Doré, S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J. Cereb. Blood Flow Metab. 2009, 29, 953–964. [Google Scholar] [CrossRef]
- Dong, B.; Yang, Y.; Zhang, Z.; Xie, K.; Su, L.; Yu, Y. Hemopexin alleviates cognitive dysfunction after focal cerebral ischemia-reperfusion injury in rats. BMC Anesthesiol. 2019, 19, 13. [Google Scholar] [CrossRef]
- Dong, B.; Cai, M.; Fang, Z.; Wei, H.; Zhu, F.; Li, G.; Dong, H.; Xiong, L. Hemopexin induces neuroprotection in the rat subjected to focal cerebral ischemia. BMC Neurosci. 2013, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dong, B.; Lu, J.; Wang, G.; Yu, Y. Hemopexin reduces blood-brain barrier injury and protects synaptic plasticity in cerebral ischemic rats by promoting EPCs through the HO-1 pathway. Brain Res. 2018, 1699, 177–185. [Google Scholar] [CrossRef]
- Redinus, K.; Baek, J.H.; Yalamanoglu, A.; Shin, H.K.H.; Moldova, R.; Harral, J.W.; Swindle, D.; Pak, D.; Ferguson, S.K.; Nuss, R.; et al. An Hb-mediated circulating macrophage contributing to pulmonary vascular remodeling in sickle cell disease. JCI Insight 2019, 4, e127860. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Kapetanaki, M.G.; Ghosh, S.; Villanueva, F.S.; Ofori-Acquah, S.F.; Kato, G.J. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front. Immunol. 2020, 11, 1910. [Google Scholar] [CrossRef]
- Poillerat, V.; Gentinetta, T.; Leon, J.; Wassmer, A.; Edler, M.; Torset, C.; Luo, D.; Tuffin, G.; Roumenina, L.T. Hemopexin as an Inhibitor of Hemolysis-Induced Complement Activation. Front. Immunol. 2020, 11, 1684. [Google Scholar] [CrossRef] [PubMed]
- Vercellotti, G.M.; Zhang, P.; Nguyen, J.; Abdulla, F.; Chen, C.; Nguyen, P.; Nowotny, C.; Steer, C.J.; Smith, A.; Belcher, J.D. Hepatic overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease. Mol. Med. 2016, 22, 437–451. [Google Scholar] [CrossRef]
- Yalamanoglu, A.; Deuel, J.W.; Hunt, R.C.; Baek, J.H.; Hassell, K.; Redinius, K.; Irwin, D.C.; Schaer, D.J.; Buehler, P.W. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L765–L774. [Google Scholar] [CrossRef]
- Ofori-Acquah, S.F.; Hazra, R.; Orikogbo, O.O.; Crosby, D.; Flage, B.; Ackah, E.B.; Lenhart, D.; Tan, R.J.; Vitturi, D.A.; Paintsil, V.; et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood 2020, 135, 1044–1048. [Google Scholar] [CrossRef]
- Chintagari, N.R.; Nguyen, J.; Belcher, J.D.; Vercellotti, G.M.; Alayash, A.I. Haptoglobin attenuates hemoglobin-induced heme oxygenase-1 in renal proximal tubule cells and kidneys of a mouse model of sickle cell disease. Blood Cells Mol. Dis. 2015, 54, 302–306. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.-C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Chadebech, P.; Bodivit, G.; Vieira-Martins, P.; Grunenwald, A.; Boudhabhay, I.; Poillerat, V.; Pakdaman, S.; Kiger, L.; Jouard, A.; et al. Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am. J. Hematol. 2020, 95, 456–464. [Google Scholar] [CrossRef]
- Santiago, R.P.; Guarda, C.C.; Figueiredo, C.V.B.; Fiuza, L.M.; Aleluia, M.M.; Adanho, C.S.A.; Carvalho, M.O.S.; Pitanga, T.N.; Zanette, D.L.; Lyra, I.M.; et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol. Dis. 2018, 72, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Whyte-Stewart, D. The Association between Hemopexin and Silent Cerebral Infarcts in Pediatric Sickle Cell Disease. Ph.D. Thesis, Johns Hopkins, Baltimore, MD, USA, 19 December 2017. [Google Scholar]
- Vendrame, F.; Olops, L.; Saad, S.T.O.; Costa, F.F.; Fertrin, K.Y. Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease. J. Clin. Lipidol. 2018, 12, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Detterich, J.A.; Liu, H.; Suriany, S.; Kato, R.M.; Chalacheva, P.; Tedla, B.; Shah, P.M.; Khoo, M.C.; Wood, J.C.; Coates, T.D.; et al. Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents. Free Radic. Biol. Med. 2019, 141, 408–415. [Google Scholar] [CrossRef]
- Santaterra, V.A.G.; Fiusa, M.M.L.; Hounkpe, B.W.; Chenou, F.; Tonasse, W.V.; da Costa, L.N.G.; Garcia-Weber, D.; Domingos I de, F.; de Lima, F.; Borba-Junior, I.T.; et al. Endothelial barrier integrity is disrupted in vitro by heme and by serum from sickle cell disease patients. Front. Immunol. 2020, 11, 535147. [Google Scholar] [CrossRef]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.-L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3, e96910. [Google Scholar] [CrossRef] [PubMed]
- Effenberger-Neidnicht, K.; Bornmann, S.; Jägers, J.; Patyk, V.; Kirsch, M. Microvascular stasis and hemolysis: Coincidence or causality? J. Inflamm. Res. 2019, 12, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Talahma, M.; Strbian, D.; Sundararajan, S. Sickle cell disease and stroke. Stroke 2014, 45, e98–e100. [Google Scholar] [CrossRef][Green Version]
- Verduzco, L.A.; Nathan, D.G. Sickle cell disease and stroke. Blood 2009, 114, 5117–5125. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J. Stroke prevention and treatment in sickle cell disease. Arch. Neurol. 2001, 58, 565–568. [Google Scholar] [CrossRef]
- Hasson, C.; Veling, L.; Rico, J.; Mhaskar, R. The role of hydroxyurea to prevent silent stroke in sickle cell disease: Systematic review and meta-analysis. Medicine 2019, 98, e18225. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, F.; Verlhac, S.; Arnaud, C.; Kamdem, A.; Chevret, S.; Hau, I.; Coïc, L.; Leveillé, E.; Lemarchand, E.; Lesprit, E.; et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 2011, 117, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Houwing, M.E.; Grohssteiner, R.L.; Dremmen, M.H.G.; Atiq, F.; Bramer, W.M.; de Pagter, A.P.J.; Zwaan, C.M.; White, T.J.H.; Vernooij, M.W.; Cnossen, M.H. Silent cerebral infarcts in patients with sickle cell disease: A systematic review and meta-analysis. BMC Med. 2020, 18, 393. [Google Scholar] [CrossRef]
- Casella, J.F.; King, A.A.; Barton, B.; White, D.A.; Noetzel, M.J.; Ichord, R.N.; Terrill, C.; Hirtz, D.; McKinstry, R.C.; Strouse, J.J.; et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatr. Hematol. Oncol. 2010, 27, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.J.; Midwinter, R.G.; Cassano, C.; Beck, K.; Wang, Y.; Changsiri, D.; Gamble, J.R.; Stocker, R. Heme oxygenase-1 increases endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1537–1542. [Google Scholar] [CrossRef]
- Vachharajani, T.J.; Work, J.; Issekutz, A.C.; Granger, D.N. Heme oxygenase modulates selectin expression in different regional vascular beds. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1613–H1617. [Google Scholar] [CrossRef]
- Hahl, P.; Davis, T.; Washburn, C.; Rogers, J.T.; Smith, A. Mechanisms of neuroprotection by hemopexin: Modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J. Neurochem. 2013, 125, 89–101. [Google Scholar] [CrossRef]
- Liu, Y.; Jing, F.; Yi, W.; Mendelson, A.; Shi, P.; Walsh, R.; Friedman, D.F.; Minniti, C.; Manwani, D.; Chou, S.T.; et al. HO-1hi patrolling monocytes protect against vaso-occlusion in sickle cell disease. Blood 2018, 131, 1600–1610. [Google Scholar] [CrossRef]
- Kato, G.J.; Nouraie, S.M.; Gladwin, M.T. Lactate dehydrogenase and hemolysis in sickle cell disease. Blood 2013, 122, 1091–1092. [Google Scholar] [CrossRef]
- Zerez, C.R.; Lachant, N.A.; Lee, S.J.; Tanaka, K.R. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood 1988, 71, 512–515. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Age, Species, Sample, Sex, Duration | Model Used | Outcome |
|---|---|---|---|
| Vinchi, et al., 2016 [1] Hemopexin therapy reverts heme-induced pro-inflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease | 2–3 mo B6J & SV129 n = 14 15 day | Raw264.7 cells and bone-marrow derived macrophages (BMDMs) were treated with one of the following treatment groups: hemolytic aged RBCs (2 × 107), heme or Zn-mesoporphyrin (5–15 μM) bound to albumin or Hpx (5–15 μM), iron-nitrilotriacetate (FeNTA) alone (100 μM), or bound to deferoxamine (100 μM) | Hpx−/− + heme mice had higher levels of HO1 (p < 0.05), L-Ferritin (p < 0.01), FPN (p < 0.01), and IL-6 (p < 0.05) than WT + heme group. In vitro treatment of Hpx + heme had lower levels of HO1 (p < 0.001) and Ferritin (p < 0.001), and FPN (p < 0.01) at 10 h incubation and lower levels of ROS (p < 0.001), TNFα (p < 0.01), and IL-6 (p < 0.01) at 15 h. BMDMs exposed to heme or iron sources showed significantly increased M1 (pro-inflammatory) macrophage polarization. This effect diminished with Hpx treatment in vivo. |
| Belcher, et al., 2018 [6] Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction | 12.6 ± 2.5 week, 24.0 ± 5.1 g Townes SS (n = 4/group) & Townes AA (n = 3/group) 50% M 72 h | Via tail vein, Hb was injected by itself or conjointly with saline, albumin (1 μmol/kg), Hp (1 μmol/kg), Hpx (1 μmol/kg), or Hp + Hpx (0.5 μmol Hp/kg + 0.5 μmol Hpx/kg) | Untreated AA-mice showed minimal or no stasis (p < 0.05) and untreated SS-mice displayed 10–11% stasis at all time points, 24, 48 and 72 h (p < 0.01). SS-mice treated with Hp or Hpx at baseline had <1% stasis 24 h after injection, ~2% stasis at 48 h, and 10–11% stasis at 72 h (p < 0.05 for all groups). SS-mice injected Hb and either Hp, Hpx, or both Hp and Hpx showed lower NF-ҡB phospho-65 levels (p < 0.01), displaying anti-inflammatory effects. |
| Redinus, et al., 2019 [24] An Hb-mediated circulating macrophage contributing to pulmonary vascular remodeling in sickle cell disease | 10 week (300–350 g) Sprague-Dawley rats n = 58 100% M 35 day | 35 mg/d Hb infusion and stimulated high altitude hypobaric Hpx (5500 m, 18,000 ft) were continued for 35 day. Hb + Hpx grouped mice were treated with 10 mg/kg gadolinium trichloride (GdCl3) 3 times a week for either 18 or 35 day | Whole lung protein quantification for HO1, IL-6, and ET-1 demonstrated an increased expression of these proteins in the Hb + Hpx cohort compared to control and GdCl3 treated Hb + Hpx groups (p < 0.05 for HO1; p < 0.01 for IL-6 and ET-1). |
| Gbotosho, et al., 2020 [25] HemeiInduces IL-6 and cardiac hypertrophy genes transcripts in sickle cell mice | 12–16 week Townes’ knocked-in (SS), (AA), & C57BL/6J n = 6–10 (SS &AA) 3 h | Mice injected via tail vein with hemin (50 μm/kg/ body weight) for SS and AA mice. A hemin dose of 120 μm/kg/ body weight was used for Nrf2+/+ and Nrf2−/− C57BL/6 mice. Control mice received sterile vehicle (0.25 M NaOH; pH 7.5 using HCl) in preparation of hemin | IL-6 and Hmox1 expression increased in Townes SS mice compared to AA controls (p ≤ 0.05 and p ≤ 0.01, respectively). After heme injection, SS mice showed a 15.4-fold increase in cardiac IL-6 transcripts compared to the vehicle control group (p ≤ 0.05) and roughly a 53% increase when compared to AA control mice (p ≤ 0.001). 3 h after heme treatment, SS mice showed an increase in Nppa (14.8-fold; p ≤ 0.001) and Myh7 (8.1-fold; p ≤ 0.01) transcripts, while AA control mice showed no increase. In heme-treated Nrf2−/− and Nrf2+/+ mice, cardiac IL-6 was 51% higher in the Nrf2−/− group (p < 0.01). |
| Poillerat, et al., 2020 [26] Hemopexin as an inhibitor of hemolysis-induced complement activation | 8 week (0.125 mg/g) C56BL/6 n = 10 (Hpx injected) n = 8 (control) 100% M 72 h | 3 groups of mice were injected IV with 100 or 500 mg/kg of either human plasma derived Hpx (CSL Behring) or PBS. After this, mice were immediately injected IP with phenylhydrazine (PHZ) at a concentration of 0.125 mg/g body weight. Control mice received PBS injections at an equivalent volume of Hpx and PHZ | Clearance of plasma heme by Hpx was 4-fold higher (100 mg/kg group) and 2-fold higher (500 mg/kg group) in mice treated with PHZ than controls. Kidney injury decreased in Hpx pretreated PHZ-infused mice. At 6 h, urea measurements were decreased in 100 mg/kg Hpx mice (p < 0.005) and creatinine measurements were decreased in both 100 and 500 mg/kg Hpx mice (p < 0.05 and p < 0.001, respectively) compared to PBS-infused controls. PHZ-induced deposits of C3b/iC3b were attenuated in both 100 and 500 mg/kg Hpx groups (no p-values reported). |
| Vercellotti, et al., 2016 [27] Hepatic overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease | 8–20 week (20–30 g) NY1DD & Townes AA & SS (n = 4 for all groups) 50% M 12 week | High-pressure injection of DNA solution (constructed via plasmid manipulation) into tail vein (10% body weight, up to 2.5 mL). Plasmids were either WT-Hpx, missense-Hpx, or Luc plasmid | 1 h after infusion with heme, Hpx−/− sickle mice developed 33 ± 3% static vessels compared to 21 ± 5% in control sickle mice (p < 0.025). |
| Yalamanoglu, et al., 2018 [28] Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease | 20 week C57BL/6 (n = 5), BERK-SS (n = 9), & Hartley guinea pigs (GP) (n = 8) 50% M 2 week | PE-50 tubing catheters were placed into the left external jugular vein of GPs. High diet (HD) GPs (n = 4) were infused with heme-albumin (5 and 25 mg/kg doses) while another group of HD GPs (n = 4) was dosed with vehicle (0.9% NaCl) via the jugular catheter. Blood samples taken from mice were centrifuged at 2000 rpm for 15 min | BERK-SS mice reveal greater formation of lipid oxidation end products, malondialdehyde (MDA), compared to WT mice. Specifically, HDL and LDL concentrations were 0.3 ± 0.1 μm/80 μg and 0.3 ± 0.4 μm/80 μg (p < 0.05 for both groups). Similar MDA levels were seen in HD GP groups sampled 8 h after infusion for GPs injected with either 5 or 25 mg/kg/body weight heme-albumin (p < 0.05). |
| Ofori-Acquah, et al., 2020 [29] Hemopexin deficiency promotes acute kidney injury in sickle cell disease | 20 μmol/kg Knockin Townes SS & AA n = 8–12 (SS) & 8–12 (AA) 2 day | Hpx−/− and C57BL/6 mice were transplanted with SS bone marrow. Heme infusion (20 μmol/kg) mentioned, although location not specified | After heme infusion, excess heme was preferentially transported to the kidneys in SS mice (p < 0.01) versus the liver in AA mice (p < 0.05). Additionally, in SS mice, plasma creatine (Cr) and urinary albumin-Cr ratio increased 4-fold (p < 0.001) and 2-fold (p < 0.01), respectively, compared to baseline levels. Levels of α-1-microglobulin (A1M) were elevated in SS mice compared to controls (p < 0.001) while Hpx levels were low in both groups. The Hpx−/− SS mice had the lowest baseline glomerular filtration rate (p < 0.05) and increased kidney injury molecule-1 (KIM-1) compared with Hpx−/− and control mice (p < 0.001). |
| Chintagari, et al., 2015 [30] Haptoglobin attenuates hemoglobin-induced heme oxygenase-1 in renal proximal tubule cells and kidneys of a mouse model of sickle cell disease | 8–12 mo (20–30 g) NY1DD mice n = 4 7 day | SCD mice were infused via the tail vein with one bolus of Hb (3.2 μm/kg), Hp (3.2 μm/kg), or Hb-Hp complex (0.012 mL/g) | Hp attenuated Hb-induced HO1 mRNA expression in SCD mice at 4 h after Hb infusion (p < 0.05). Additionally, analyzed via Western blot, Hp attenuated the Hb-induced expression of HO1 protein in the proximal tubule of the kidney of SCD mice. |
| Camus, et al., 2015 [31] Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease | 10–18 week C57BL/6J mice n = 6 100% M | Microparticles (MP) were purified and then injected via IV administration | MP injection increased circulating erythrocyte MP concentration by 25% and reduced both diastolic and mean blood flow velocity in the renal arteries by 30%, within 5 min (p = 0.009). Heart rate and cardiac output were not affected after injection of MPs. Mice treated with MPs that were pretreated with 1µM Hpx had no change in blow flow velocity (p = 0.72). |
| Reference | Age, Sample, Sex, Duration, Location | Study Design | Outcomes |
|---|---|---|---|
| Yalamanoglu, et al., 2018 [28] Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease | ≥18 year n = 19 (SCD) & n = 11 (controls) 50% M Aurora, CO, USA | Prospective Case-control study | In SCD patients, HDL and LDL plasma levels were lower compared to controls (25 ± 16 and 82 ± 26 mg/dl vs control 53 ± 18 and 128 ± 21 mg/dl). SCD patients had an increase in heme bound to Hb (9.5 ± 4 μM) and total plasma heme levels (60 ± 23 μM) compared to plasma obtained from control patients (4.6 ± 4.2 μM and 19 ± 11 μM). |
| Camus, et al., 2015 [31] Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease | ≥18 year n = 47 (SCD), n = 22 (AA) Paris, France | Retrospective Case-control | In patients with SCD, annexin-a5+ MPs were increased 5-fold and plasma Hb were increased 3-fold compared to controls (p < 0.001 and p = 0.007, respectively). SCD erythrocytes released MPs that contained 2X the concentration of heme compared to control MPs (p = 0.038). |
| Roumenina, et al., 2020 [32] Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy | ≥18 year n = 106 (SCD) & n = 176 (AA) Paris, France | Retrospective | sC5b-9, a marker of complement activation, was increased in 42% of SCD patient plasma (p < 0.001) and was higher in SCD patients treated with hydroxyurea compared to the nontreatment SCD group (p < 0.005). In nontreated-SCD patients, sC5b-9 levels were higher after incubation of SCD RBCs with normal human serum compared hydroxyurea-treated SCD and control RBCs (p = 0.038 and p = 0.006, respectively). Endothelial cells incubated with SCD serum led to the C3 deposits on the surface of cells and were decreased after the addition of Hpx (p = 0.0117). |
| Santiago, et al., 2018 [33] Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients | <18 year n = 179 (SS), n = 93 (SC), n = 28 (AA) University of Bahia, Brazil | Cross-sectional study | SS and SC patient groups showed lower Hp and Hpx levels (SS < SC < AA) and increased reticulocyte counts and serum LDH (SS > SC > AA). SCD patient (SS and SC) serum analysis showed a negative correlation between Hpx and LDH (r = −0.509, p < 0.001) and heme (r = −0.592, p < 0.001). |
| Whyte-Stewart, et al., 2017 [34] The association between hemopexin and silent cerebral infarcts in pediatric sickle cell disease | 5–15 year n = 340 Baltimore, Maryland | Prospective Cross-sectional study | In patients who experienced silent cerebral infarcts, Hpx levels were lower than controls (p = 0.0082). Hpx was shown to be associated with an increased risk of silent cerebral infarct (p = 0.03). |
| Vendrame, et al., 2018 [35] Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease | 35 year (AA; n = 37; 32% F), 45 year (SC; n = 39; 62% F), 37 year (SS; n = 40; 48% F) Campinas, Brazil & Seattle, WA, USA | Prospective Case-control study | In SCD groups (SC and SS) compared to controls (AA), erythrocytes and Hb concentrations were lower while reticulocyte, bilirubin, and serum LDH levels were increased (p = 0.0001). In SS group alone, heme was reported at higher levels compared to AA or SC groups (p = 0.0001). In SC and SS patient groups, soluble VCAM-1 levels were increased compared to controls (p = 0.0001) and were shown to be higher in SS patients over SC patients (p = 0.02). Additionally, in SS and SC groups, total cholesterol and LDL levels were decreased compared to controls (p = 0.001). |
| Detterich, et al., 2019 [36] Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents | 23 ± 8.8 year (SCD; n = 55) 27 ± 8.9 year (AA; n = 44) 28 day Los Angeles, California | Retrospective Case-control | Measured at the time of phlebotomy, SCD patients demonstrated higher % metHb (p = 0.004). In the plasma, free Hb and heme, were elevated in SCD patients compared to controls and increased rapidly at Hpx levels less than 200 μg/mL SCD patients. |
| Santaterra, et al., 2020 [37] Endothelial barrier integrity is disrupted in vitro by heme and by serum from sickle cell disease patients | >18 year n = 20 (SCD), n = 10 (AA) Campinas, Brazil | Retrospective Case-control In vitro study | In SCD patients, Hpx levels were lower than comparative healthy volunteers (0.33 ± 0.32 vs 1.29 ± 0.23; p < 0.001). Heme-induced endothelial barrier disruption was correlated with Hpx levels, most significantly at 12 min (Rs = 0.68; p < 0.0001). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Doré, S. Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease. Int. J. Mol. Sci. 2021, 22, 6408. https://doi.org/10.3390/ijms22126408
Ashouri R, Fangman M, Burris A, Ezenwa MO, Wilkie DJ, Doré S. Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease. International Journal of Molecular Sciences. 2021; 22(12):6408. https://doi.org/10.3390/ijms22126408
Chicago/Turabian StyleAshouri, Rani, Madison Fangman, Alicia Burris, Miriam O. Ezenwa, Diana J. Wilkie, and Sylvain Doré. 2021. "Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease" International Journal of Molecular Sciences 22, no. 12: 6408. https://doi.org/10.3390/ijms22126408
APA StyleAshouri, R., Fangman, M., Burris, A., Ezenwa, M. O., Wilkie, D. J., & Doré, S. (2021). Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease. International Journal of Molecular Sciences, 22(12), 6408. https://doi.org/10.3390/ijms22126408