
Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts


Abstract
1. Introduction
2. Results
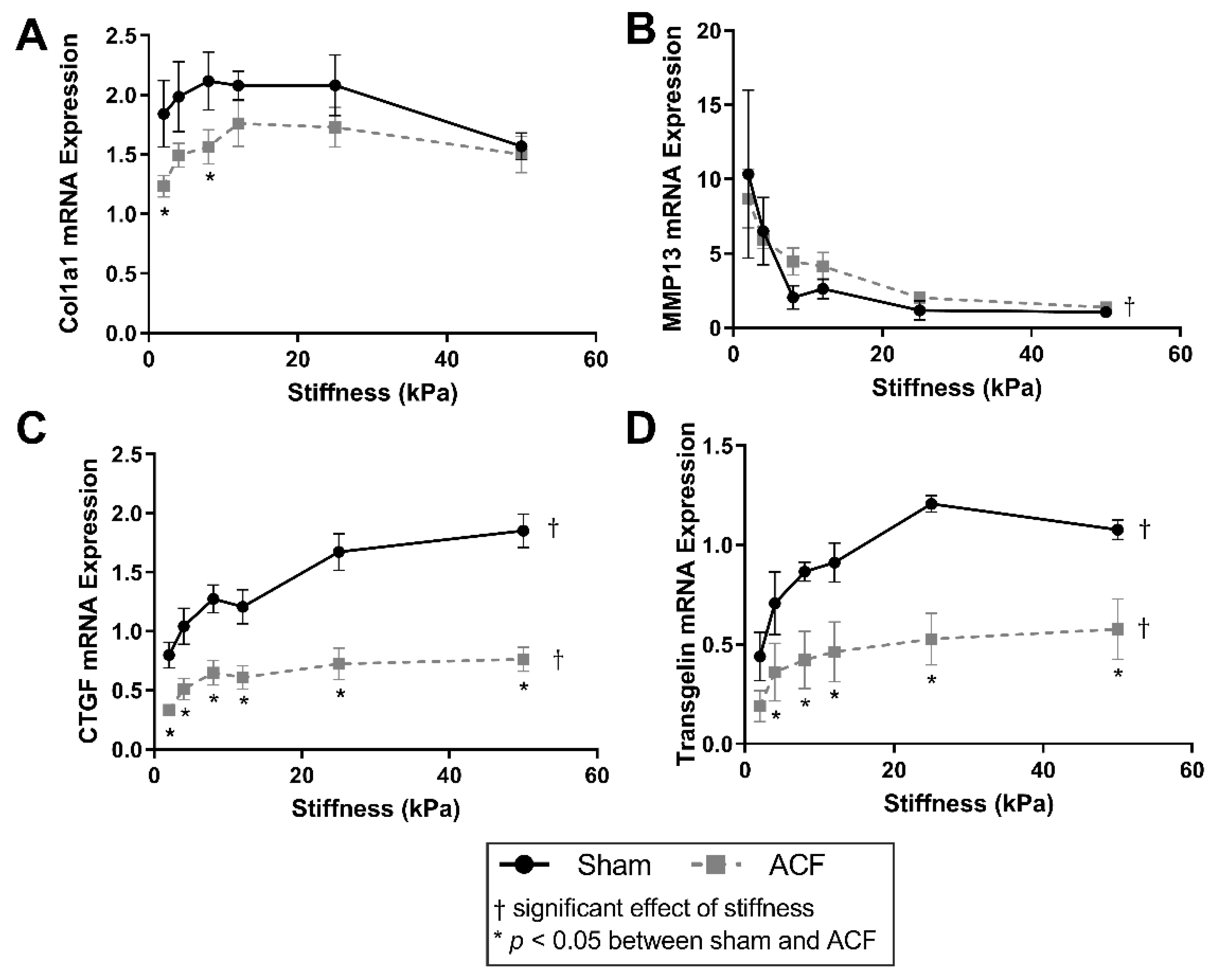
2.1. Soft Stiffness Promotes Hypofibrotic Phenotype
2.2. Increasing Stiffness Causes an Increase in the F/G-Actin Ratios
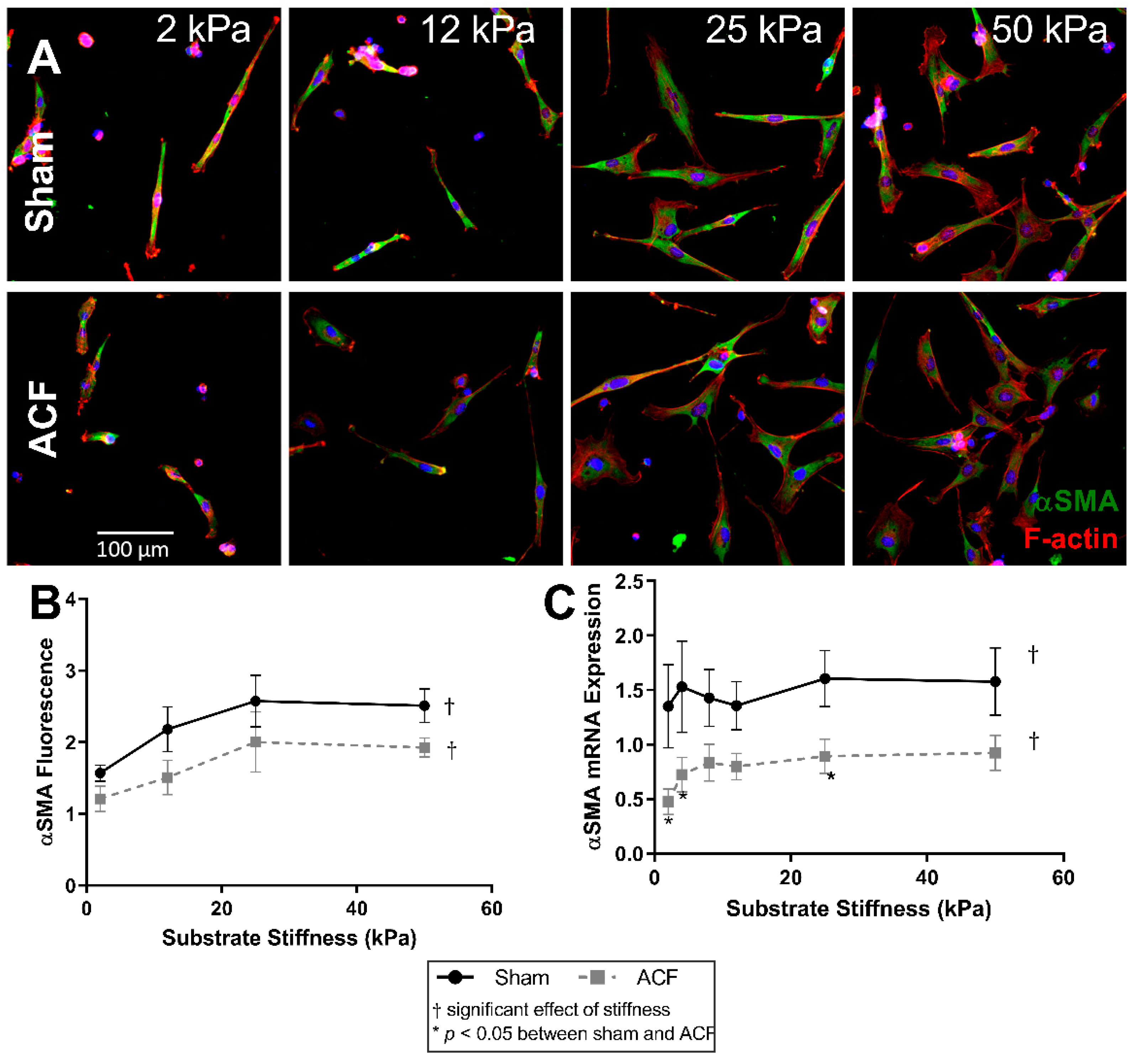
2.3. αSMA Is Decreased in ACF CFs and at Lower Stiffness
2.4. MRTF-A Translocates to the Nucleus on Higher Stiffness
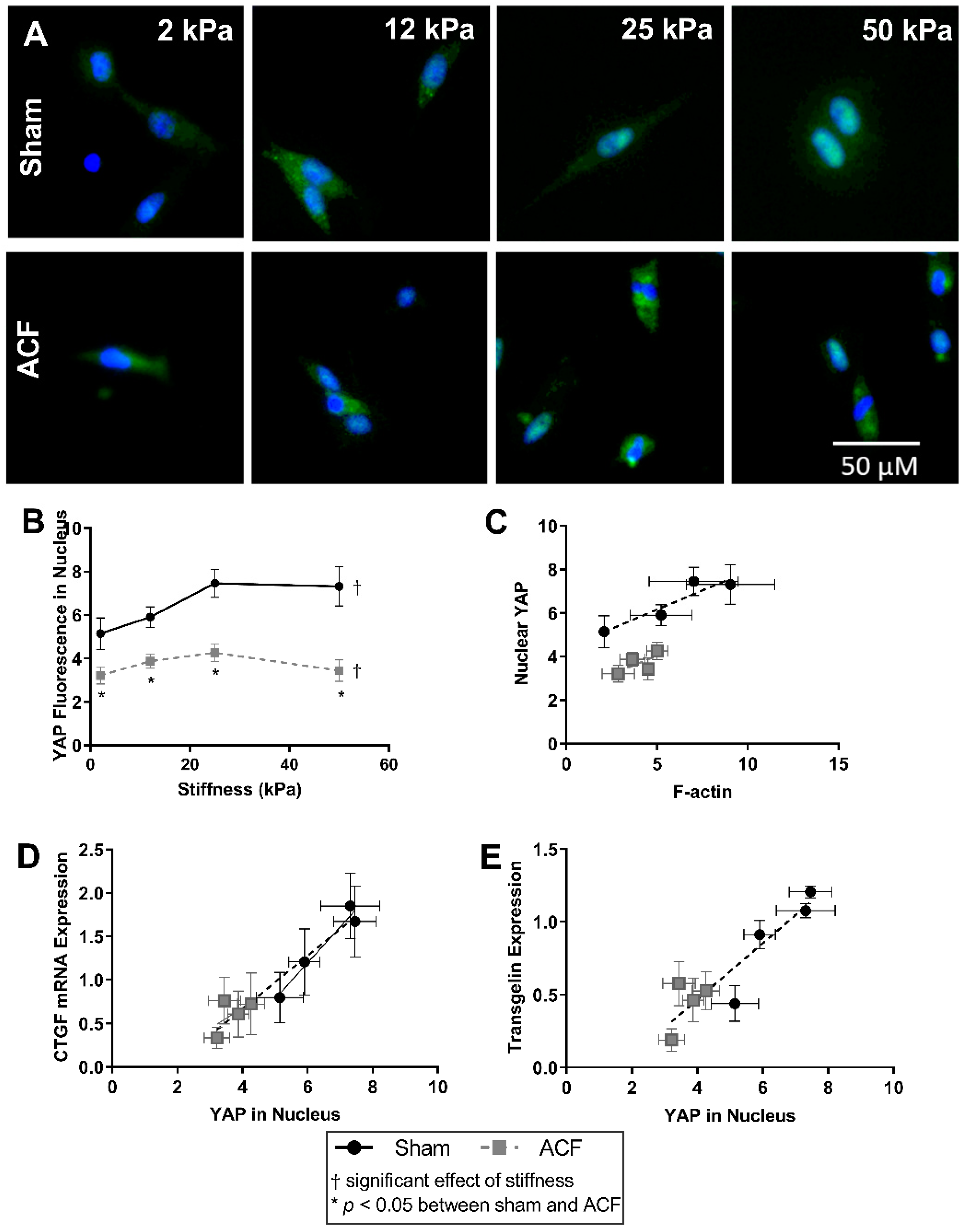
2.5. YAP Localizes to the Nucleus More So in Sham CFs Than ACF CFs on Stiff Substrates
2.6. PPAR-γ Expression Decreases with Increased Stiffness
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cardiac Fibroblasts
4.3. Fluorescent Staining
4.4. Real Time Quantitative PCR
4.5. G-Actin and F-Actin Immunoblot
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| αSMA | Alpha Smooth Muscle Actin |
| ACF | Aortocaval Fistula |
| CF | Cardiac Fibroblast |
| CTGF | Connective Tissue Growth Factor |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| ERK | Extracellular signal-related protein kinase |
| FBS | Fetal bovine serum |
| HF | Heart Failure |
| HRP | Horseradish peroxidase |
| LVEDD | Left ventricular end-diastolic diameter |
| MMP | Matrix Metalloproteinase |
| MRTF-A | Myocardin-Related Transcription Factor A |
| PAGE | Polyacrylamide gel electrophoresis |
| PDGF | Platelet-derived growth factor |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PVDF | Polyvinylidene difluoride |
| SDS | Sodium Dodecyl Sulphate |
| SRF | Serum response factor |
| TEAD | Transcriptional enhancer factor domain |
| TGF-β | Transforming Growth Factor Beta |
| VO | Volume Overload |
| YAP | Yes-Associated Protein |
References
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef]
- Van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016. [Google Scholar] [CrossRef]
- Swaney, J.S.; Roth, D.M.; Olson, E.R.; Naugle, J.E.; Meszaros, J.G.; Insel, P.A. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. USA 2004, 102, 437–442. [Google Scholar] [CrossRef]
- Mehta, N.; Chopra, A.; Jamney, P.A.; Kresh, J.Y. Transdifferentiation of Cardiac Fibroblasts To Myofibroblast Phenotype and Its Regulation By Extracellular Matrix Composition and Mechanics. J. Am. Coll. Cardiol. 2014, 63, A851. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Takagi, M.; Kurokawa, Y.; Satomi, S.; Konttinen, Y.T. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab. Investig. 1998, 78, 1077–1087. [Google Scholar]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatol. (Oxf.) 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, K.R.; Stewart, J.A.; Lucchesi, P.A. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J. Mol. Cell. Cardiol. 2010, 48, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, K.R.; Guggilam, A.; Cismowski, M.J.; Galantowicz, M.L.; West, T.A.; Stewart, J.A.; Zhang, X.; Lord, K.C.; Lucchesi, P.A. Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J. Appl. Physiol. 2011, 111, 1778–1788. [Google Scholar] [CrossRef]
- Ryan, T.D.; Rothstein, E.C.; Aban, I.; Tallaj, J.A.; Husain, A.; Lucchesi, P.A.; Dell’Italia, L.J. Left Ventricular Eccentric Remodeling and Matrix Loss Are Mediated by Bradykinin and Precede Cardiomyocyte Elongation in Rats With Volume Overload. J. Am. Coll. Cardiol. 2007, 49, 811–821. [Google Scholar] [CrossRef]
- Dell’italia, L.J.; Balcells, E.; Meng, Q.C.; Su, X.; Schultz, D.; Bishop, S.P.; Machida, N.; Straeter-Knowlen, I.M.; Hankes, G.H.; Dillon, R.; et al. Volume-overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am. J. Physiol. 1997, 273, H961–H970. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, Y.; Pat, B.; Dell’Italia, L.A.; Tillson, M.; Dillon, A.R.; Powell, P.C.; Shi, K.; Shah, N.; Denney, T.; et al. Microarray Identifies Extensive Downregulation of Noncollagen Extracellular Matrix and Profibrotic Growth Factor Genes in Chronic Isolated Mitral Regurgitation in the Dog. Circulation 2009, 119, 2086. [Google Scholar] [CrossRef]
- Childers, R.C.; Sunyecz, I.; West, T.A.; Cismowski, M.J.; Lucchesi, P.A.; Gooch, K.J. Role of the cytoskeleton in the development of a hypofibrotic cardiac fibroblast phenotype in volume overload heart failure. Am. J. Physiol. Circ. Physiol. 2019, 316, H596–H608. [Google Scholar] [CrossRef] [PubMed]
- Childers, R.C.; Trask, A.J.; Liu, J.; Lucchesi, P.A.; Gooch, K.J. Paired PV Loop Analysis and Biaxial Mechanical Testing Characterize Differences in Left Ventricular Tissue Stiffness of Volume Overload and Angiotensin-Induced Pressure Overload Hearts. J. Biomech. Eng. 2021. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 884–890. [Google Scholar] [CrossRef]
- Arora, P.D.; Narani, N.; Mcculloch, C.A.G. The Compliance of Collagen Gels Regulates Transforming Growth Factor-β Induction of α-Smooth Muscle Actin in Fibroblasts. Am. J. Pathol. 1999, 154, 871–882. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef] [PubMed]
- Talele, N.P.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of α-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M. The Actin–MRTF–SRF Gene Regulatory Axis and Myofibroblast Differentiation. J. Cardiovasc. Transl. Res. 2012, 5, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Ota, M.; Sasaki, H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Burgess, H.A.; Daugherty, L.E.; Thatcher, T.H.; Lakatos, H.F.; Ray, D.M.; Redonnet, M.; Phipps, R.P.; Sime, P.J. PPARγ Agonists Inhibit TGF-β Induced Pulmonary Myofibroblast Differentiation and Collagen Production: Implications for Therapy of Lung Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1146–L1153. [Google Scholar] [CrossRef]
- Sun, K.; Wang, Q.; Huang, X. PPAR Gamma Inhibits Growth of Rat Hepatic Stellate Cells and TGF Beta-Induced Connective Tissue Growth Factor Expression. Acta Pharmacol. Sin. 2006, 27, 715–723. [Google Scholar] [CrossRef]
- Hogrebe, N.J.; Gooch, K.J. Direct influence of culture dimensionality on human mesenchymal stem cell differentiation at various matrix stiffnesses using a fibrous self-assembling peptide hydrogel. J. Biomed. Mater. Res. Part A 2016, 104, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tibbitt, M.W.; Basta, L.; Anseth, K.S. Mechanical memory and dosing influence stem cell fate. Nat. Mater. 2014, 13, 645. [Google Scholar] [CrossRef]
- Pelham, R.J.; Wang, Y.-L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.; Georges, P.C.; Flanagan, L.A.; Marg, B.; Ortiz, M.; Funaki, M.; Zahir, N.; Ming, W.; Weaver, V.; Janmey, P.A. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton 2005, 60, 24–34. [Google Scholar] [CrossRef]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sévigny, J.; Wells, R.G. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.-J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, Q.; Zhu, T.; Zhang, Y.; Liu, B.; Xu, J.; Zhao, H. Substrate stiffness-regulated matrix metalloproteinase output in myocardial cells and cardiac fibroblasts: Implications for myocardial fibrosis. Acta Biomater. 2014, 10, 2463–2472. [Google Scholar] [CrossRef]
- Bhana, B.; Iyer, R.K.; Chen, W.L.K.; Zhao, R.; Sider, K.L.; Likhitpanichkul, M.; Simmons, C.A.; Radisic, M. Influence of substrate stiffness on the phenotype of heart cells. Biotechnol. Bioeng. 2010, 105, 1148–1160. [Google Scholar] [CrossRef]
- Wang, H.; Haeger, S.M.; Kloxin, A.M.; Leinwand, L.A.; Anseth, K.S. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS ONE 2012, 7, e39969. [Google Scholar] [CrossRef]
- Kharaziha, M.; Nikkhah, M.; Shin, S.-R.; Annabi, N.; Masoumi, N.; Gaharwar, A.K.; Camci-Unal, G.; Khademhosseini, A. PGS:Gelatin nanofibrous scaffolds with tunable mechanical and structural properties for engineering cardiac tissues. Biomaterials 2013, 34, 6355–6366. [Google Scholar] [CrossRef]
- Gilles, G.; McCulloch, A.D.; Brakebusch, C.H.; Herum, K.M. Maintaining resting cardiac fibroblasts in vitro by disrupting mechanotransduction. PLoS ONE 2020, 15, e0241390. [Google Scholar] [CrossRef]
- Balestrini, J.L.; Chaudhry, S.; Sarrazy, V.; Koehler, A.; Hinz, B. The mechanical memory of lung myofibroblasts. Integr. Biol. 2012, 4, 410. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.-J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M.C. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef]
- Modarressi, A.; Pietramaggiori, G.; Godbout, C.; Vigato, E.; Pittet, B.; Hinz, B. Hypoxia Impairs Skin Myofibroblast Differentiation and Function. J. Investig. Dermatol. 2010, 130, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. Oxygenation state as a driver of myofibroblast differentiation and wound contraction: Hypoxia impairs wound closure. J. Investig. Dermatol. 2010, 130, 2701–2703. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Diebold, S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc. Res. 1990, 24, 430–432. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Gene | Accession Number | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|---|
| α-smooth muscle actin | ACTA2 | NM_031004.2 | TGCCATGTATGTGGCTATTCA | ACCAGTTGTACGTCCAGAAGC |
| collagen type-1 α-1 | COL1A1 | NM_053304 | TCTGGTCTCCAGGGTCCTC | GTCCATCTTTGCCAGGAGAA |
| connective tissue growth factor | CTGF | NM_022266 | GCTGACCTAGAGGAAAACATTAAGA | CCGGTAGGTCTTCACACTGG |
| lactate dehydrogenase A | LDHA | NM_017025.1 | GATGATGGATCTTCAGCATGG | GCTTGGAGTTTGCAGTCACA |
| MMP-13 | MMP13 | NM_133530 | GGACAAGCAGCTCCAAAGG | GGTCCAGACCGAGGGAGT |
| PPAR-γ | Pparg | NM_013124.3 | GGTGAAACTCTGGGAGATCCT | AATGGCATCTCTGTGTCAACC |
| ribosomal protein L13A | Rpl13a | NM_173340.2 | CCCTCCACCCTATGACAAGA | GGTACTTCCACCCGACCTC |
| transgelin (SM22) | TAGLN | NM_031549.2 | AGTGTGGCCCTGATGTGG | TCACCAACTTGCTCAGAATCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Childers, R.C.; Lucchesi, P.A.; Gooch, K.J. Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts. Int. J. Mol. Sci. 2021, 22, 6231. https://doi.org/10.3390/ijms22126231
Childers RC, Lucchesi PA, Gooch KJ. Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts. International Journal of Molecular Sciences. 2021; 22(12):6231. https://doi.org/10.3390/ijms22126231
Chicago/Turabian StyleChilders, Rachel C., Pamela A. Lucchesi, and Keith J. Gooch. 2021. "Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts" International Journal of Molecular Sciences 22, no. 12: 6231. https://doi.org/10.3390/ijms22126231
APA StyleChilders, R. C., Lucchesi, P. A., & Gooch, K. J. (2021). Decreased Substrate Stiffness Promotes a Hypofibrotic Phenotype in Cardiac Fibroblasts. International Journal of Molecular Sciences, 22(12), 6231. https://doi.org/10.3390/ijms22126231