Abstract
Intrinsically disordered proteins and regions typically lack a well-defined structure and thus fall outside the scope of the classic sequence–structure–function relationship. Hence, classic sequence- or structure-based bioinformatic approaches are often not well suited to identify homology or predict the function of unknown intrinsically disordered proteins. Here, we give selected examples of intrinsic disorder in plant proteins and present how protein function is shared, altered or distinct in evolutionary distant organisms. Furthermore, we explore how examining the specific role of disorder across different phyla can provide a better understanding of the common features that protein disorder contributes to the respective biological mechanism.
1. Introduction
Despite the progress made in recent decades, a large proportion of plant protein sequences still lacks useful functional annotation. These proteins represent a promising source for basic research pursuing functional novelty or for translational research seeking new perspectives on biological mechanisms and their disruption in disease. Intriguingly, despite 1.6 billion years of divergent evolution, the majority of genes in Arabidopsis thaliana have conserved orthologs in humans, highlighting the similarity of fundamental biological processes between the two organisms [1]. As a result, research on Arabidopsis often enhanced our understanding of many molecular mechanisms associated with human diseases like cancer, Parkinson’s disease and Alzheimer’s disease [2,3]. Conversely, homology detection allows for information transfer from the heavily studied mammalian organisms to protein sequences that lack annotation in plants.
Resulting from the recent advances in high-throughput sequencing techniques, the genomes of more than 200 plant species were sequenced, outpacing the more laborious process of experimental protein classification. Hence, only approximately 1% of the protein sequences in the UniProt database have experimentally verified functions [4]. To address this discrepancy, putative or hypothetical proteins are typically classified into protein families that may share evolutionary relationships or molecular function by sequence-based computational analysis. However, this approach works insufficiently for proteins lacking sequence conservation or experimentally verified, functional annotation in orthologous proteins.
2. Functional Annotation of Intrinsically Disordered Proteins
Intrinsically disordered proteins (IDPs) and regions (IDRs) lack a well-defined and folded three-dimensional structure in the absence and/or presence of a binding partner. Disorder is a fundamental property of the proteome and can be robustly predicted from primary sequence relying on characteristic patterns of amino acid distribution and overall amino acid content [5,6]. This class of proteins operates largely outside the classic structure–function relationship, with their functionality being defined by a separate set of parameters compared to their structured and globular counterparts [7]. Hence, identification of distantly related IDPs cannot rely on structure, which can help to detect relationships of folded proteins that would remain undetected by conventional sequence-based methods [8]. Methods that use antibodies to metazoan proteins to identify putative plant homologues may result in a higher incidence of artefacts when compared to folded protein targets, due to the smaller size of disordered epitopes and their comparably higher sensitivity to epitope variation [9].
In contrast to folded proteins, IDPs and IDRs show an overall increased rate of evolution, while these rates can strongly vary between different parts of the IDP [10,11]. Moreover, disordered regions appear to be more permissive to mutation, further complicating sequence–function analysis. Overall, genome duplication events seem to influence the distribution of IDRs within genomes, as the amount of identical paralogous IDRs positively correlates with the number of chromosomes [12]. Interestingly, proteins can display different modes of how sequence conservation can relate to disorder as a functional feature [13]. While in some cases only the disorder itself is conserved, but not the sequence facilitating it (flexible disorder), other IDPs retain disorder together with a highly conserved sequence (constrained disorder). Short-linear motifs (SLiMs) represent conserved functional modules that can mediate low-affinity interactions and are often interspersed by regions of flexible disorder [14]. As many alignment algorithms require long stretches of sequence similarity to satisfy statistical significance, the high modality and low complexity of SLiMs can obscure the search for homologous sequences considerably. Due to their limited size (7–12 residues), these motifs can easily develop in unrelated proteins in convergent evolution. Furthermore, post-translational modifications (PTMs) that often regulate IDP function complicate the analysis, as they can drastically alter the biophysical features of a protein and phosphorylation sites display short and weakly conserved motifs that are often difficult to detect via computational sequence analysis [15].
Within protein interaction networks, IDPs often represent hubs carrying multispecific binding sites that adapt to different binding partners [16]. These complexes regularly retain wide conformational flexibility (e.g., fuzzy complexes) even if the binding induces folding in parts of the IDP [17]. Depending on the state of the cell, this flexibility can be rapidly modulated, providing the organism a sensitive framework to respond to changing environments under stress conditions [18]. Indeed, protein disorder is significantly increased in signalling and stress-related processes, providing flexible and rapid adaptation networks for sessile organisms like plants in response to environmental cues [18]. However, despite the numerous IDPs characterized in the animal kingdom and particularly in human diseases, the study of protein intrinsic disorder in plants is still in its infancy.
3. Different Frameworks of Protein Similarity
Determining the similarity or difference between entities pre-requires a conceptual system that inevitably creates a hierarchy within sets of features deemed ‘relevant’. The different frameworks of protein similarity, like function, biological context, biophysical properties or evolutionary relationship, will operate on a set of categories that may or may not be compatible with other frameworks. Beyond this, the features within a defined system may not be fixed but draw meaning from reciprocal determination of features (e.g., a protein with a folded domain will likely be classified by that domain rather than by a non-folded, multivalent IDR ‘tail’). Given the ambiguous modes of sequence conservation of disordered regions, it is tempting to assume that the determination of similarity or difference along the lines of function and biophysics will provide a better understanding of the common features that protein disorder contributes to biological mechanisms. However, disregarding evolutionary relationships artificially divorces molecular function from its developmental constraints. In this review, we exemplify the functional kinship of disordered proteins across evolutionary distant organisms and give insights into the essential features that underpin it.
4. LEA Proteins as Disordered Spatial Organizers under Cell-Stress Conditions
Although first identified in cotton seeds, Late Embryogenesis Abundant (LEA) proteins are no longer viewed as being restricted to the plant kingdom [19,20]. The members of this family represent one of the most prominent examples of IDP-mediated stress responses in plants [19]. Most notably, LEA proteins accumulate in response to water-deficit conditions and are particularly abundant in anhydrobiotic plants [21]. LEA proteins have been shown to play important roles for the plant responses to salinity, drought, freezing, heat and desiccation [22]. Overexpression studies of selected LEA proteins resulted in increased resistance against diverse abiotic stresses, which underscores their importance as safeguarding proteins [23,24,25,26,27]. Within plants, LEA proteins localize ubiquitously in the cytoplasm and subcellular organelles like the nucleus, mitochondria, endoplasmic reticulum, plasma membrane, chloroplast, and peroxisome [28].
LEA proteins do not display any clear sequence similarity with other proteins of known function and can be classified into several broadly defined families or classes based on conserved protein domains [29], the occurrence of defined sequence motifs [28] or their physico-chemical features [30]. Overall, their amino acid composition is dominated by hydrophilic residues, charged amino acids, small amino acids like glycine or serine and a high proportion of disorder-promoting amino acids, although there is some significant variation within the LEA protein family regarding overall hydropathy and charge (Figure 1 A). Indeed, experimental and bioinformatic evidence suggests that a key feature of the LEA protein family is their total or partial lack of stable structure, resulting in a flexible conformation [31]. Some bacterial and plant LEA proteins also carry folded domains like the Water stress and Hypersensitive response (WHy) domain, which is involved in the desiccation response and may originate from an ancestral domain in Archaea [32,33]. As seen in other disorder-containing protein families, LEA proteins display a diverse multifunctionality, for example as protein and membrane chaperones, as DNA-binding proteins, or sequesters of metal ions and radicals [20]. As many interactions and functions have only been characterized using in vitro approaches, the full scope of this family’s functional repertoire remains obscure.
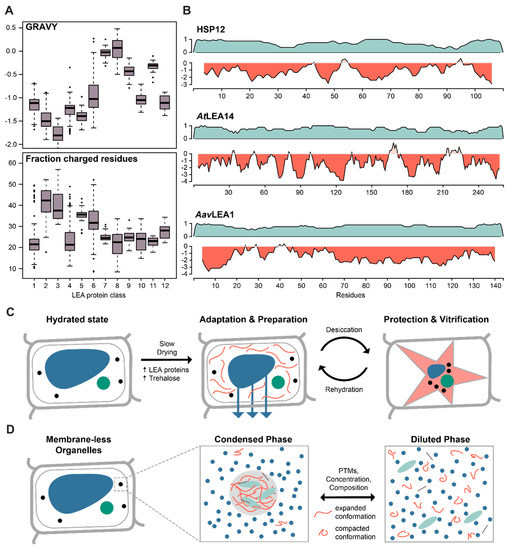
Figure 1.
Late Embryogenesis Abundant (LEA) proteins and the role of intrinsically disordered proteins (IDPs) in desiccation and liquid–liquid phase separation. (A) Boxplot of the Grand Average of Hydropathy (GRAVY) and the fraction of charged residues (DEKR) calculated for the 12 LEA protein classes, based on statistical analysis of physico-chemical properties and amino acid usage [30]. Adapted and modified with permission from http://forge.info.univ-angers.fr/~gh/Leadb/index.php. (B) Disorder prediction (teal) and hydrophobicity plot (red) for hydrophilin representatives from yeast (HSP12), plant (AtLEA14) and nematode (AavLEA1), all involved in desiccation tolerance. (C) Cartoon representation of the molecular processes involved in desiccation. Upon slow drying, trehalose and LEA proteins accumulate in the cell and vitrify, forming an amorphous matrix to stabilize and protect other proteins and membranes during desiccation. (D) Cartoon representation of the molecular processes involved in phase separation. Upon reaching a critical concentration, certain IDPs undergo liquid–liquid phase separation to form membrane-less organelles. They undergo a transition from a compacted state in solution to an expanded state in the phase separated droplet. The process can furthermore be modified by other features such as post-translational modifications (PTMs) or protein composition.
Previous studies showed LEA proteins to be sufficient desiccation protectants both in vivo and in vitro [34,35]. Indeed, LEA proteins exhibit increased expression during the desiccation-tolerant stages of plant seed development and in dividing cells of root tips [36]. Leaves of Arabidopsis are not desiccation tolerant, but experimental evidence suggests that the LEA proteins COR15A and COR15B assert a cryoprotective function by mediating freezing-induced crowding effects [37]. In vitro experiments showed that hydrophilins like the LEA protein family together with trehalose, or other disaccharides, can vitrify, which directly protects cells from protein aggregation and membrane disintegration [38,39,40]. The formation of these intracellular, glass-like matrices is hypothesized to prevent non-specific protein–protein interactions that otherwise lead to denaturation and aggregation, and thus protect seeds and roots during drought. The general mechanism was proposed to be mediated by the protein’s ability to acquire secondary structure during drying, when intra-protein hydrogen bonds become energetically more favourable [41,42,43,44]. However, the structural aspects of the mechanism have yet to be validated in vivo. In recent years, in-cell solid and solution state, Nuclear Magnetic Resonance (NMR) methodologies have advanced to the point that experimental approaches to study structural transitions under physiological conditions are now conceivable [45,46].
Besides plant organisms, LEA proteins have also been found in other desiccation-tolerant organisms, such as bacteria, fungi, and some invertebrates, suggesting a common mechanism across distinct life forms [47]. However, the full scope of LEA involvement in desiccation tolerance across species is hard to estimate since relatedness identified by sequence-based methods can be compromised given the low sequence complexity of IDPs. Moreover, many organisms carry multiple types of LEA proteins that display diverse subcellular localisation and divergent functions within different species and cellular contexts. Typically, a desiccation-tolerant state is triggered by pre-exposure to slow drying that induces changes in the expression profile of the cell (Figure 1C). Thus, the functional annotation of many LEA and LEA-like proteins was often based on gene expression analyses under stress conditions. These include the two divergent LEA-like proteins (ArLEA1A, ArLEA1B) in the freshwater rotifer Adineta ricciae [48], or the desiccation-induced expression of AavLEA1 in the nematode Aphelenchus avenae [49]. Another example are the desiccation-tolerant larval stages of the brine shrimp Artemia franciscana, in which mRNA levels of AfrLEA1 and AfrLEA2 transcripts are increased 7- and 14-fold when compared to the stages that do not share the capacity for anhydrobiosis [50]. Although LEA proteins are not represented in mammalian genomes, AfrLEA2 transfected into human HepG2 cells enhanced desiccation tolerance in the presence of intracellular trehalose, and resulted in increased membrane integrity after rehydration [51]. Similarly, eight out of fifteen Arabidopsis LEA proteins increased tolerance to desiccation when heterologously expressed in Saccharomyces cerevisiae [34]. Furthermore, the disordered yeast hydrophilin HSP12 alleviated the damage caused by severe water loss, indicating a synergistic and independent activity together with trehalose [52]. Interestingly, although HSP12 shares the general biophysical features (size, charge, disorder) of the eleven other hydrophilins in yeast, it appears to be unique in mediating desiccation tolerance amongst the hydrophilin family.
Similar to rotifers, tardigrades display desiccation tolerance despite naturally lacking trehalose [53]. Only recently, tardigrade-specific IDPs were shown to vitrify in vitro and in vivo, when heterologously expressed in yeast [54]. Moreover, RNA interference experiments reduced survival to desiccation independently of any sugar mediator. The authors proposed that tardigrade-specific IDPs mediate desiccation tolerance by protecting proteins against denaturation, trapping them in an amorphous matrix [54]. Similar to the examples above, these proteins were identified using differential gene expression analyses of hydrated and slowly drying tardigrades. Interestingly, these mechanistic similarities between tardigrade-specific IDPs and LEA proteins seem to have developed independently in convergent evolution, highlighting the importance of IDPs in organizing cellular matter in response to stress across phyla.
5. Protein Disorder as a Driving Force for Liquid–liquid Phase Separation
A classic example of spatio-temporal separation of biochemical processes in eukaryotic cells is the formation of canonical membrane-enclosed organelles. However, eukaryotes also contain numerous membrane-less compartments (MCs), such as the nucleolus or P-bodies [55]. MCs have liquid-like properties and undergo dynamic liquid–liquid phase separation (LLPS). This behaviour enables them to rapidly form on demand, fuse, shear, exchange their content, or disassemble and thus concentrate proteins and biochemical reactions at distinct locations when needed [56,57].
To achieve LLPS and formation of MCs, their contents have to reach a critical concentration to enable a de-mixing effect and form non-miscible phases [58]. Under these conditions, intermolecular interactions between IDPs stabilize the condensed phase overcoming intramolecular and solvent interactions [59,60]. The level of anisotropy remarkably increases in phase separated droplets, indicating an expanded state of IDPs in comparison to a more compacted state in solution [59,60] (Figure 1D). The resulting MCs are highly concentrated in proteinous components and can also rapidly dissociate once the concentration falls below the critical point or protein interactions are altered (e.g., by PTMs) [61,62,63,64,65]. Indeed, the lack of defined structure renders IDPs more accessible to regulation through PTMs that can change the biophysical properties of IDPs and consequently modify protein–protein interaction [64].
Almost all MCs contain a large proportion of IDPs [66,67] and their molecular features were suggested to be essential for LLPS and thus MC formation [67,68]. The lack of defined secondary or tertiary structure and thus high flexibility might provide the needs for the dynamic behaviour of MCs [67,69]. Furthermore, the capacity of IDPs to exhibit promiscuous protein–protein interactions [70] might permit the spontaneous and reversible formation of sufficient, local protein concentrations to initiate LLPS. These multivalent protein interactions are proposed to be mediated by repetitive sequence elements that result in overall IDP sequence simplicity [57,64,71]. A prominent example of an MC-forming IDP is the Essential Pyrenoid Component 1 (EPYC1). It is an indispensable part of the Pyrenoid, a MC in the chloroplast of many algae that concentrates components of the carbon fixation machinery to increase its efficiency [72]. EPYC1 contains four sequentially simplistic regions that have been proposed to form weak multivalent interactions with Rubisco and thus might be central to the liquid-like properties of the pyrenoid [72]. Other examples from the mammalian and fungal kingdoms are DEAD-box helicase 4 (DDx4) proteins that harbour clusters of FG and GF repeats [73] or the polyQ tract of Whi3 [74], respectively.
MCs of plants and other eukaryotes contain overlapping sets of proteins, indicating a common function in eukaryotic metabolism. For example, stress granules, small cytoplasmic MCs that form upon a variety of stresses contain RNA, translation initiation factors, RNA-binding, and other proteins [75,76]. Curiously, also components of the cell cycle regulation machinery (see paragraph below), such as cyclin-dependent kinases localize to stress granules in both plant and human cells [76,77]. However, plants and green algae also contain specific MCs. The chloroplasts of green algae contain the above mentioned pyrenoid and plant photobodies contain light receptors and signalling proteins [78]. Cryptochromes (see paragraph below) are components of photobodies and undergo rapid LLPS upon blue light perception and are thus a valuable optogenetic tool in mammalian cells [79].
Members of the SR protein family were recently shown to adopt similar functions in plant and mammalian MCs. SR proteins are intrinsically disordered, exhibit RNA-binding capacity, are involved in alternative splicing, and contain long repeats of serine and arginine residues [80,81]. The plant-specific SR45 selectively accumulates in nuclear body MCs, in a temperature- and phosphorylation-dependent manner [82]. It was suggested that plant SR proteins might regulate splicing activity in response to stress by undergoing LLPS and thereby concentrating the splicing machinery into MCs [56]. Similarly, the mammalian SR protein SRSF9 was recently shown to regulate nuclear stress body formation upon heat exposure, depending on the phosphorylation state of the protein [83,84]. SR45 and SRSF9 can adopt similar functions in plants and mammals, despite significant differences in protein size (414 vs 221 residues, respectively) and no sequence conservation except in the conserved RNA recognition motifs. Overall, information on plant IDPs that undergo LLPS is still scarce but investigating the composition of plant-derived MCs may represent a promising path towards a better understanding of the fundamental and species-specific features of membrane-less compartmentalization in cell biology. Moreover, features like prion-like domains are important predictors of LLPS in RNA-binding proteins [85]. In Arabidopsis, nearly 500 proteins were predicted to carry such domains [86]. Indeed, the plant-specific Flowering Locus CA and FLL2 proteins have recently been shown to be in vivo regulators of LLPS within the autonomous flowering pathway of Arabidopsis [87].
6. The Role of Disordered Proteins in Microtubule Organisation
Beyond their roles in the structural integrity and division of the cell, plant microtubules also adopt a sensory function for the perception of abiotic stress conditions [88]. Recently, we showed that the intrinsically disordered region of the Cellulose synthase Companion (CC) proteins is critically involved in the salt-stress response of Arabidopsis [89]. Representing one of the primary responses to salt stress, the plant’s cortical microtubule network is re-structured and rendered stress tolerant under saline conditions [90]. Plant cortical microtubules steer the movement of the cellulose synthase complex and thus are essential for the organism’s morphology and growth [91]. The CC protein family is an essential player in the microtubule re-assembly process during salt exposure [92]. Interestingly, the mechanism by which the cytosolic N-terminus of CC1 regulates and interacts with microtubules appear to be remarkably similar to that of the human Tau microtubule-associated protein, which is widely known for its potential role in multiple neurodegenerative diseases [89,93]. Both proteins are intrinsically disordered and contain multiple short hydrophobic microtubule-binding motifs that can bind tubulin and microtubules transiently and independently [94] (Figure 2).
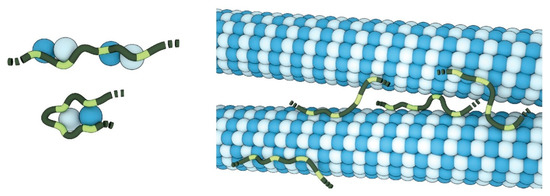
Figure 2.
Disordered microtubule-binding proteins. The dynamic nature of the CC1- and Tau-binding (dark- and light-green) behaviour suggests that both might be able to bind multiple distinct tubulin dimers (light- and dark-blue) via their individual binding motifs (light-green), thereby increasing the local tubulin concentration, connecting and stabilizing protofilaments or bundling microtubules. Image by Barth van Rossum.
Thus, both proteins can promote polymerization and bundling of microtubules, while also being able to diffuse along the microtubule lattice [95,96]. While Tau controls microtubule dynamics and organisation in neurons [97], the CC1-mediated microtubule bundling may underpin microtubule array stabilization of the plant cell during salt stress. Notably, microtubule-associated proteins display an increased content of disorder in eukaryotes [98]. The observation that key biophysical and functional properties are shared across distantly related kingdoms may spark new perspectives on the evolution of microtubule-associated IDPs and their function in stress and disease-related processes.
7. The Multivalent Role of Protein Disorder in Cryptochrome Signalling
Cryptochromes (CRYs) are blue-light receptors that regulate varying functions such as cell growth and circadian rhythm in a range of organisms like plants, insects and bacteria. The conserved and folded N-terminal domain of all CRYs resembles photolyases (Photolyase homology region (PHR); [99]). As such, CRYs are flavoproteins but lost their photolyase activity and hence are not involved in DNA damage repair [99]. Nearly all C-terminal extensions of CRYs are predicted to be disordered and vary greatly in length and sequence between species [100,101]. Nevertheless, the function of cryptochromes is critically dependent on these C-terminal IDRs [102,103,104,105,106]. Arabidopsis harbours two cryptochromes (CRY1 and CRY2), whose C-terminal IDRs differ in sequence, but are functionally equivalent as they can be interchanged [107,108]. Proteolytic analysis of human and Arabidopsis CRYs revealed that the C-terminal IDRs show increased susceptibility to proteolytic digestion after illumination, which suggests a conformational change that exposes the IDR [101]. This observation is in line with the crystal structure of full-length Drosophila cryptochrome, which revealed that the flexible IDR resides in a grove of the PHR domain in the non-excited state [109,110]. Transgenic Arabidopsis plants overexpressing the CRY C-terminal extension phenotypically show a constant light response, indicating that it is sufficient to activate the otherwise light-induced signalling pathway [102]. Indeed, it directly interacts with several downstream regulatory proteins such as Constitutive Photomorphogenic 1 (COP1) or Suppressor of Phytochrome A1 (SPA1) [111,112,113,114] and is heavily phosphorylated upon light perception [115,116,117]. Figure 3A summarizes the general action model of plant CRYs, which includes the following steps: 1. In the dark, PHR and the C-terminal IDR interact and form a closed conformation; 2. Light induces a dimerization of CRYs and phosphorylation of the IDR leads to an exposed IDR conformation; 3. The IDR binds to regulatory proteins that subsequently modulate developmental processes [101,113,114,116,117,118].
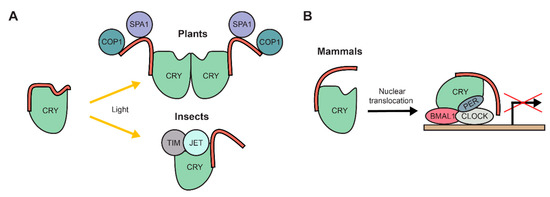
Figure 3.
The intrinsically disordered C-terminal extension of cryptochromes (CRYs) has diverse functions. (A) Upon light perception through a photolyase homology region (PHR), the C-terminal extension (red) is released from the PHR and acquires an exposed conformation. In plants, the C-terminal extension directly binds to partner proteins, e.g. COP1 and SPA1. In insects it is not involved in binding to proteins, instead the PHR binds to partner proteins, e.g. TIM and JET. (B) In mammals, the C-terminal extension is responsible for translocation of CRYs to the nucleus, where they are involved in regulating gene expression as part of the central circadian clock regulation complex together with proteins like BMAL1, CLOCK and PER.
The circadian clock regulation in Drosophila requires the tightly regulated binding of multiple proteins to CRYs, e.g., Timeless (TIM) and Jetlag (JET) [119,120]. In response to light, the C-terminal IDR is released from the PHR, which then allows binding of both TIM and JET to the PHR [109,119,120]. In contrast to plant CRYs, the IDRs of Drosophila CRYs thus do not facilitate binding to regulatory proteins but inhibit it (Figure 3A). Consistent with this conclusion, the Drosophila PHR domain, deficient of its C-terminal IDR, induced a constitutive light response, in contrast to plant CRYs that transmit their response directly via their disordered C-terminus [102,121].
In contrast to both plant or insect CRYs, their mammalian orthologs lack any known direct photosensory function [122,123]. However, mutant mice deficient in the photoreceptors opsin or melanopsin show impairment in circadian clock regulation, indicating that mammalian CRYs are dependent on light perception of other photoreceptors [124,125]. Indeed, they are part of a transcription/translation feedback loop to establish the circadian rhythm. In a complex with Period (PER), they repress the activity of the circadian transcription activator complex circadian locomotor output cycles kaput (CLOCK)/brain and muscle Arnt-like protein 1 (BMAL1) and thus repress their own transcription [126,127,128]. The C-terminal IDR extension of mammalian CRYs regulates the import of the protein into the nucleus as well as the interaction of the PHR with the CLOCK/BMAL1 complex (Figure 3B) [99,105,106]. Interestingly and reminiscent of plant CRYs, phosphorylation of the C-terminal IDR plays an important role to modulate mammalian CRY proteins. Phosphorylation of the mammalian CRY1 IDR stabilizes the protein [129], while phosphorylation of the CRY2 IDR destabilizes the protein and leads to its degradation [130,131]. Curiously, the C-terminal IDR of the mammalian-like CRY ortholog from the green algae Chlamydomonas reinhardtii was recently suggested to bind to its PHR upon light perception [132]. Thus, a similar mode of action might be possible for mammalian CRYs, potentially through a light dependent signalling pathway in conjunction with other photoreceptors.
In summary, despite having a conserved PHR domain, the members of the CRY protein family have adopted varying, species-dependent functions, which are mediated by their highly variable C-terminal IDR extensions and may have developed independently. The CRY proteins are thus an excellent example of how protein disorder, despite the presence of other, folded regions with a conserved mechanism, can represent the primary determining factor of molecular function between kingdoms of life.
8. Disordered Proteins Represent Key Regulators in Cell Cycle Progression
The cell cycle is one of the most intensively studied processes in biology, especially due to its misregulation in many human diseases. Unlike animal development, plants largely develop post-embryonically and, thus, organ formation, like flowers, leaves, stems, or roots, continuously develops throughout the plant life cycle. Plant cell division is located in meristems, containing pluripotent stem cells whose progeny is subsequently developing into specialized cells. Despite these striking differences in developmental organisation, all eukaryotic cells essentially undergo the same cell cycle that is defined by characteristic phases. Cyclin-dependent kinases (CDKs) play an essential role in the progression of the cell cycle and are conserved in all eukaryotes. A multitude of CDK–cyclin complexes control the transition from the post-mitotic gap phase (G1) to the synthetic (S) phase and second gap phase (G2) to mitosis (M) phases by phosphorylating downstream target proteins [133]. Cyclins hereby act as mediators between the CDKs and multiple substrates. Because of the essential role in the continuation of the cell cycle, CDK–cyclin complexes are heavily regulated by several mechanisms, like phosphorylation and proteolysis initiated by ubiquitination, all of which were reviewed elsewhere [133,134,135].
Cyclin-dependent kinase inhibitors (CKIs) bind CDKs and inhibit their activity to regulate the progression of the cell cycle. Consequently, CKI misregulation is associated with a multitude of diseases [136,137,138,139]. CKIs are IDPs that only share a conserved inhibitory domain (CID), which acquires a folded state when involved in cooperative binding to both CDKs and cyclins [140,141,142,143]. The budding yeast CKI SIC1 and its mammalian counterparts of the p27kip1 family share very low sequence homology [141]. However, prediction tools indicated structural similarity of the CIDs. Intriguingly, the positioning of the CID within the overall topology of the CKIs varies between different species. While the domain is located at the C-terminus of SIC1, its position is N-terminal in p27kip1 (Figure 4A) [140,144]. Despite these positional differences, heterologously expressed SIC1 can bind and inhibit the activity of the Cdk2–cyclin A complex, the mammalian binding partner of p27kip1, in vitro and overexpression of mammalian p27kip1 in a SIC1 deficient yeast strains can rescue its cell cycle related phenotype [141]. The first plant CKI was identified by a yeast two-hybrid assay employing A. thaliana CDKA;1 as a bait protein and subsequent sequence analysis, in which the CID was found to be distantly homologous to that of mammalian p27kip1 [145]. This Kip-related protein (KRP) family subsequently expanded to seven members in Arabidopsis [146,147] and fulfils an analogous function in cell cycle control as their yeast and mammalian counterparts [147,148,149,150]. Curiously, like the yeast protein, they carry the conserved inhibitory domain at their C-terminus (Figure 4A) [145,147,149] and heterologously expressed Zea mays KRPs furthermore decrease overall cell size when expressed in fission yeast [151], hinting at a delay in cell cycle progression. In contrast to the functional conservation between yeast and plant CKIs, classic sequence alignment of the CIDs show no homology better than expected by chance, while KRPs and mammalian p27kip1 are at least distantly related [152]. However, the CIDs of all three CKIs acquire or are predicted to acquire a common fold of two α-helices interspaced by a flexible linker (Figure 4B).
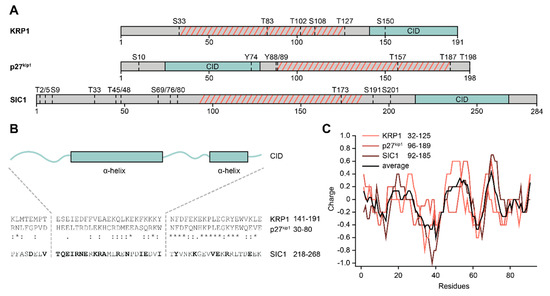
Figure 4.
Features of cyclin-dependent kinase inhibitors (CKIs). (A) Overview of general CKI architecture across organisms. Arabidopsis KRP1, human p27kip1, and yeast SIC1 all share a large intrinsically disordered region (grey) that shows no sequence conservation. A sequentially conserved inhibitory domain (CID; green) is located at different positions in the proteins. Dotted lines denote known (p27kip1, SIC1) or predicted (KRP1; NetPhos 3.1 score > 0.9) phosphorylation sites in the IDR. (B) Sequential comparison of the conserved CID. KRP1 and p27kip1 are sequentially conserved, while SIC1 only shows similar amino acids (bold letters) in several positions to either protein. Two alpha helices (boxes) comprise the binding domain of the CIDs, as shown by the crystal structure of p27kip1, modelling of SIC1, or prediction with JPRED for KRP1. (C) All three CKIs show conserved, charge-based features in the IDR region between important phosphorylation sites (highlighted by hatched, red lines in (A).
The IDR domains of the CKIs presented here, especially p27kip1, have been associated with many functions, e.g., transcriptional regulation, regulation of the cytoskeleton or tumour development, but these were reviewed elsewhere [153,154,155]. One of the most investigated functions is the regulation of their own CID domain. To release CKIs from their respective CDK–cyclin complexes and thus activate them, phosphorylation of the IDR domains at multiple positions is required. The process is best described in budding yeast. SIC1 is tightly bound to Clb (B-type cyclins)-CDK complexes at the beginning of the cell cycle and therefore inhibits them [156,157]. To initiate deactivation of SIC1 through ubiquitination and subsequent degradation through the 26S proteasome [158,159,160], at least six out of multiple phosphorylation sites in its IDR domain (Figure 4A) have to be phosphorylated in a specific cascade [161,162,163,164]. Yeast cells enter the (S) phase of their cell cycle afterwards. Furthermore, phosphorylation of other, differential positions in SIC1 seems to play a role in cell cycle progression [165,166].
The mammalian analogue p27kip1 is also heavily regulated by phosphorylation. An overview of important p27kip1 phosphorylation sites can be found in Figure 4A. While phosphorylation of residues S10 and T198 is mainly involved in protein stability in interphase cells [167,168,169], together with T157 they also contribute to the subcellular localization of the protein [170]. When progressing through the cell cycle Y88 gets phosphorylated, which partially releases p27kip1 from the CDK2/cyclin A complex [171]. Further phosphorylation of Y74 leads to CDK2 activation and phosphorylation of T187 in p27kip1 in an intra-complex manner [172,173]. This leads to deactivation of p27kip1 through ubiquitination and subsequent degradation through the 26S proteasome [174] and ultimately entry into (S) phase [171].
The regulation of plant KRPs is not well understood to date. However, KRPs are phosphorylated before deactivation, although the key sites remain unknown [175]. Phosphorylation in all three protein classes is thus an important regulatory factor. Indeed, SIC1 acquires different transient folding states when being phosphorylated or dephosphorylated, influencing the electrostatic field experienced by the SIC1 binding partner [176]. Furthermore, a region in p27kip1, in between the important phosphorylation sites Y88 and T187, shows a charge pattern that is important for the selective phosphorylation of T187 in the cascade of events leading to deactivation of p27kip1 and (S) phase entry [177]. Remarkably, SIC1 exhibits a comparable charge pattern in between annotated, important phosphorylation sites. Furthermore, predicted phosphorylation sites in KRP1, identified with the NetPhos 3.1 server [178], frame a region with such a charge pattern. The region thus seems to be functionally conserved through biophysical properties, despite no classic sequence conservation (Figure 4C).
9. The Role of Protein Disorder in Transcriptional Regulation
While the general principles of protein–DNA recognition are well conserved among eukaryotes, transcriptional control that relies on protein–protein interactions is more species-specific [179]. This situation is further complicated by the fact that these regulatory domains often employ disordered SLiMs that are difficult to robustly predict de novo from sequence and can develop functionality through both rapid convergent and divergent evolution [180]. The overall disorder content of TFs may be linked to the capacity of to establish more complex gene regulatory networks in multicellular organisms [181]. In Arabidopsis thaliana, a large proportion of transcription factors (TFs) contains extended regions of disorder (82%–94%, [182], disorder in eukaryotic TFs is reviewed in [183]). In particular, their transactivation domains (TADs), scaffolding domains responsible for recruiting transcriptional co-regulators that are critical for transcription initiation, display a high degree of disorder (73%–95%, [182]). The “Nine amino acid Transactivation Domain” (9aaTAD) family is a prominent example of an important generic TAD in eukaryotes. Its motif is defined by a tandem of hydrophobic clusters, hydrophilic residues with proportional positive/negative charge and a 3 aa hydrophobic region towards its N-terminus [184]. Despite showing wide variability across species, the 9aaTAD represents a universal module that mediates binding to the transcriptional machinery. For instance, plant (e.g., MYB63), yeast (e.g., Oaf1p) and animal (e.g., SREBP) TFs were found to harbour 9aaTADs that facilitate interaction with the Med15 KIX domain of the Med Mediator Tail module [185,186,187].
Understanding the mechanistic details of TAD recruitment promises the opportunity of protein design for crop optimization in food and feed production due to their prominent involvement in plant stress responses. TAD engineering by design principles taken from other organisms was applied early on in plant research. Many studies used the viral protein 16 (VP16) acidic activation domain from the herpes simplex virus to change transcript levels of specific target genes [188,189]. For instance, a zinc finger DNA-binding protein-VP16 fusion construct targeting the b-KETOACYL- ACP-SYNTHASE II allowed for the modification of the oil content in rapeseed leaves and seeds [190]. Much like the TAD of human tumour suppressor p53, the VP16 TAD contains two disordered activation subdomains, each with transcription activation potential that may form amphipathic α-helices upon complex formation [191,192]. Recently, Krois and co-workers gave remarkable structural insights into the binding specificity of p53 by showing how the p53 TADs directly compete with non-specific DNA sequences for binding to the DNA-binding core domain [193]. As neither VP16 nor p53 are present in the genome of vascular plants, the VP16 design principles were used to screen for TADs of plant regulatory regions [194]. Interestingly, some of the identified domains significantly improved transcriptional activation and exhibited higher efficacy in planta when compared to VP16. Approaching the design challenge in a high-throughput manner, Ravarani and co-workers developed an IDR-Screen framework for TADs [195]. Using a yeast transcription factor assay with Heat shock factor protein 1 as a bait protein, the authors screened a random sequence library and variants of known TADs to derive sequence patterns that underlie TAD function. The surprisingly large amount of functional TAD sequences was enriched in negatively charged amino acids and aromatic hydrophobic residues. However, also highly degenerate and redundant sequences were sufficiently functional within the assay, which may indicate broad compatibility with co-factor interaction and/or non-specific binding to the components of the transcriptional machinery. Moreover, the high sequence degeneracy of the TAD sequences hints for a binding mechanism that is primarily mediated by multiple and fuzzy interactions rather than strong specific binding.
With representatives in over 100 land plant species and over 100 genes in Arabidopsis alone, the NAM/ATAF1/CUC2 (NAC) family is one of the largest plant-specific transcription factor families and of vital importance in the stress response and cell wall synthesis of the plant organism [196,197]. These proteins contain a conserved and structured N-terminal DNA-binding domain, while a highly variable C-terminal domain is predicted to be largely disordered [198,199]. NAC proteins interact with a number of different proteins but it is yet unclear which are mediated by the disordered C-terminal domain [200,201]. Upon complexation with the stress regulator and hub-protein Radical-induced Cell Death 1, the C-terminal domains of ANAC046 and ANAC013 do not adopt an induced structure, which is consistent with an inherent conformational flexibility and fuzziness of the interaction [202]. The Suppressor of Gamma Response 1 (SOG1) is a plant-specific NAC transcription factor that regulates the DNA damage response [203]. Similar to p53, SOG1 regulates a variety of genes involved in cell cycle arrest, apoptosis, DNA damage response and repair. Due to these similarities and since the plant genome lacks a p53 ortholog, SOG1 has been put forward as a functional analogue of p53 [204]. Indeed, in an analysis of the Arabidopsis DNA damage response transcriptional network, SOG1 represented a major activator targeting other TFs, repair factors, and cell cycle regulators and thus coordinates the induction of DNA damage repair [205]. Interestingly, many of the identified genes targeted by SOG1 have human orthologs that are p53 targets and thus share a similar role within their respective regulatory networks. One example is the SOG1 target KRP6, a CKI resembling p21 and p27 (see above), which conversely represents a major target of p53 activity and mediates the down-regulation of cell cycle genes [206]. Despite functional similarities, the domain architecture and sequence are not conserved between p53 and SOG1, suggesting that the kinship of the two proteins is not rooted in common ancestry, but may have developed independently in response to unique demands imposed by their respective species’ DNA damage repair networks. Similar to the NAC family members described above, the SOG1 C-terminal domain is predicted to be disordered and appears to be strongly post-transcriptionally regulated [207]. Hence, it is conceivable that a disordered C-terminus may enable SOG1 to display a broad functional and structural repertoire similar to p53, which interacts with a large number of protein partners. The much lower number of confirmed SOG1 interaction partners in comparison to p53 may stem from the less extensive characterization of the plant DNA damage repair mechanism.
10. Methodological Advances and Outlook
From the shape of the cell, the ultrastructure of the cytoskeletal network, down to protein structure at atomic resolution, structural observations have traditionally been the predominant framework of comparisons between the molecular life of plant and metazoan organisms. As the conservation of proteins is more pronounced at the structural than the sequential level, IDPs and IDRs thus represent a challenging target for functional comparison [208]. Computational approaches of high-throughput protein functional annotation are highly desirable to guide more in-depth and laborious in vitro and in vivo analysis. Besides structural considerations, current computational protein function predictions rely either on sequence or information-based methods [209]. In IDPs, the sequence–function relationship is often independent of structural restraints and thus requires novel methods for analysis. Applying an average distance map technique, Shimomura and co-workers were able to identify disordered regions that show a tendency to adopt an ordered structure in their bound state [210]. Zarin and co-workers could show that biophysical features like net charge or hydrophobicity of amino acids rather than sequence seem to be the determining factor in the evolution of IDPs [211]. The authors could demonstrate that intrinsically disordered regions can be readily exchanged based on their physical properties without the need for classic sequence conservation. Interestingly, the pattern of protein disorder itself was put forward as an alternative approach to trace distant relatives in classes of proteins with high levels of intrinsic disorder [212]. These patterns may have developed in response to species-specific requirements and biological context and will thus likely aid in establishing the key determinants of the molecular mechanism. Disregarding the premises of the sequence–structure–function relationship altogether, information-based methods are well suited to identify disordered proteins of analogous function in distantly related phyla. Recent advances made in high-throughput transcriptomics at single-cell resolution and the analysis of elaborate co-expression networks will allow researchers to gain insights into complex regulatory relationships and identify novel players in the targeted biological processes [213,214]. Multiple methods have been developed that exploit protein–protein interaction networks in order to identify protein function based on the topological features of the target proteins interaction network independent of structure or sequence homology [215,216]. Hence, the ever-increasing amount of protein–protein interaction and co-expression data may help to illuminate common biological mechanisms of proteins between distantly related phyla by comparing their position within the respective networks. However, despite recent improvements in the analysis, identifying hub proteins is critically dependent on the quality and origin of the underlying data [217].
The arsenal of biophysical methodologies to characterize structural features of IDPs has significantly expanded in recent years and thus enables a more refined functional comparison between proteins from different phyla. The integration of experimental data from NMR, small-angle X-ray scattering and molecular dynamics simulations can yield a detailed structural description of IDP conformational ensembles [218,219]. Although the capacity to resolve protein disorder with cryogenic electron microscopy (cryo-EM) remains limited, the method can make significant contributions to elucidate IDP binding to higher-order molecular complexes [220]. Magic-angle spinning (MAS) NMR carries the potential to study both rigid and flexible protein regions and can also be applied to study structural properties in living cells although plant cells pose a special challenge for protein delivery due to their cell wall [46,221,222]. Beyond the improvements in high-throughput phosphoproteomics by mass spectrometry, time-resolved solution-state NMR methods have been developed to probe phosphorylation patterns in cell extracts, intact cells, or under defined physiological conditions [223,224]. Furthermore, elaborate single-molecule and microfluidic techniques can describe the collective properties of IDPs in LLPS or during oligomerization [225,226]. Characterization of single molecules and their dynamic behaviour in vivo is still challenging. Live cell confocal imaging based on single-molecule Förster resonance energy transfer (FRET) is an established tool to study biophysical features of IDPs in vitro [227,228], but was recently also proven to be a versatile tool to study biophysical features of IDPs in vivo, e.g., dimensions, submicrosecond chain dynamics or conformational changes upon interaction with binding partners [229,230,231,232]. However, plant proteins are usually imaged at the organismal level and imaging is thus based on expression of fluorescent fusion proteins, rather than on microinjected proteins that are chemically linked with fluorescent dyes. Due to weaker photophysical features of these fusion proteins, their application for extended single molecule FRET is limited [227]. Additionally, fusion proteins are bulky molecules in comparison to fluorescent dyes and might influence the accuracy of measurements. Introduction of non-canonical amino acids and subsequent click chemistry based labelling [233,234] of plant IDPs might circumvent these limitations in the future and enable single-molecule FRET for in vivo biophysical property determination.
Targeted creation of IDP chimeras in vivo with similar functionality and biophysical signatures might reveal regions and distinct features that are crucial for protein function and open novel avenues for protein design. Chimeric protein approaches were successfully used for folded proteins involved in the development of multiple diseases such as breast cancer, neuroinflammation, Alzheimer’s disease, or addiction [235,236,237]. Chimeras of plant and metazoan IDPs may thus reveal potential targets to improve plant growth under stress conditions or to improve our understanding of key players in human diseases.
11. Conclusions
Since protein disorder emerged as a systematically studied field some 20 years ago, the challenge of devising a scheme of classification and functional annotation for the disordered proteome has been widely discussed within the scientific community. Indeed, making meaningful comparisons within the enigmatic realm of disordered proteins that operates outside the classic sequence–structure–function relationship requires navigating between different frameworks of similarity. With this contribution, we want to illustrate the need for curated knowledge transfer across phyla that works in concert with traditional computer-based annotation methods. Describing the functional versatility of IDRs requires a broad and integrative approach that must include evolutionary and structural, as well as, functional and biophysical considerations. Beyond this, deciphering the properties that are common or species specific across evolutionary distant organisms can improve our understanding of how IDP function can evolve in diverse biological contexts and how the interplay between protein structure and disorder creates the diverse functional repertoire found in the proteome.
Author Contributions
A.W. and C.K. contributed equally to the writing, reviewing, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
CK was funded by the Peter und Traudl Engelhorn Stiftung and by an ETH Career Seed Grant SEED-05 19-2 of the ETH Foundation and the Rofonda Stiftung Vaduz.
Acknowledgments
We thank Staffan Persson for critical review of the manuscript prior to submission and we also thank Barth van Rossum for creating Figure 2.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CC | Companion of Cellulose Synthase |
| CDK | Cyclin-Dependent Kinase |
| CID | Conserved Inhibitory Domain |
| CKI | Cyclin-Dependent Kinase Inhibitor |
| CRY | Cryptochrome |
| EPYC | Essential Pyrenoid Component |
| GRAVY | Grand Average of Hydropathy |
| LEA | Late Embryogenesis Abundant |
| LLPS | Liquid–Liquid Phase Separation |
| MC | Membrane-less Compartment |
| IDP | Intrinsically Disordered Protein |
| IDR | Intrinsically Disordered Region |
| PHR | Photolyase Homology Region |
| PTM | Post-Translational Modification |
| Slim | Short-Linear Motif |
| SOG | Suppressor of Gamma Response |
| TAD | Transactivation Domain |
| TF | Transcription Factor |
| TDP | Tardigrade-Specific IDP |
| VP16 | Viral Protein 16 |
References
- Rubin, G.M. Comparative Genomics of the Eukaryotes. Science 2000, 287, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Møller, S.G. The value of Arabidopsis research in understanding human disease states. Curr. Opin. Biotechnol. 2011, 22, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Chory, J.; Dangl, J.L.; Estelle, M.; Jacobsen, S.E.; Meyerowitz, E.M.; Nordborg, M.; Weigel, D. The impact of Arabidopsis on human health: Diversifying our portfolio. Cell 2008, 133, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Orengo, C.A. Protein function annotation using protein domain family resources. Methods 2016, 93, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Pietrosemoli, N.; García-Martín, J.A.; Solano, R.; Pazos, F. Genome-wide analysis of protein disorder in Arabidopsis thaliana: Implications for plant environmental adaptation. PLoS ONE 2013, 8, e55524. [Google Scholar] [CrossRef]
- Atkins, J.D.; Boateng, S.Y.; Sorensen, T.; McGuffin, L.J. Disorder Prediction Methods, Their Applicability to Different Protein Targets and Their Usefulness for Guiding Experimental Studies. Int. J. Mol. Sci. 2015, 16, 19040–19054. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Wallqvist, A.; Fukunishi, Y.; Murphy, L.R.; Fadel, A.; Levy, R.M. Iterative sequence/secondary structure search for protein homologs: Comparison with amino acid sequence alignments and application to fold recognition in genome databases. Bioinformatics 2000, 16, 988–1002. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Richards, J.S.; Anders, R.F.; Norton, R.S. Antibody Recognition of Disordered Antigens. Structure 2016, 24, 148–157. [Google Scholar] [CrossRef]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Daughdrill, G.W. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2010, 27, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Yruela, I.; Contreras-Moreira, B.; Dunker, A.K.; Niklas, K.J. Evolution of Protein Ductility in Duplicated Genes of Plants. Front. Plant Sci. 2018, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Bellay, J.; Han, S.; Michaut, M.; Kim, T.; Costanzo, M.; Andrews, B.J.; Boone, C.; Bader, G.D.; Myers, C.L.; Kim, P.M. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011, 12, R14. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Van Roey, K.; Michael, S.; Davey, N.E.; Weatheritt, R.J.; Born, D.; Speck, T.; Krüger, D.; Grebnev, G.; Kuban, M.; et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014, 42, D259–D266. [Google Scholar] [CrossRef]
- Li, T.; Du, P.; Xu, N. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PLoS ONE 2010, 5, e15411. [Google Scholar] [CrossRef]
- Kim, P.M.; Sboner, A.; Xia, Y.; Gerstein, M. The role of disorder in interaction networks: A structural analysis. Mol. Syst. Biol. 2008, 4, 179. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Dure, L., 3rd; Greenway, S.C.; Galau, G.A. Developmental biochemistry of cottonseed embryogenesis and germination: Changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry 1981, 20, 4162–4168. [Google Scholar] [CrossRef]
- Hand, S.C.; Menze, M.A.; Toner, M.; Boswell, L.; Moore, D. LEA Proteins During Water Stress: Not Just for Plants Anymore. Annu. Rev. Physiol. 2011, 73, 115–134. [Google Scholar] [CrossRef]
- Garay-Arroyo, A.; Colmenero-Flores, J.M.; Garciarrubio, A.; Covarrubias, A.A. Highly hydrophilic proteins in prokaryotes and eukaryotes are common during conditions of water deficit. J. Biol. Chem. 2000, 275, 5668–5674. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Olvera-Carrillo, Y.; Garciarrubio, A.; Campos, F.; Covarrubias, A.A. The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008, 148, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Duan, X.; Wang, B.; Hong, B.; Ho, T.; Wu, R. Expression of a Late Embryogenesis Abundant Protein Gene, HVA1, from Barley Confers Tolerance to Water Deficit and Salt Stress in Transgenic Rice. Plant Physiol. 1996, 110, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-J.; Liu, Z.; Kanno, A.; Kameya, T. Genetic improvement of Chinese cabbage for salt and drought tolerance by constitutive expression of a B. napus LEA gene. Plant Sci. 2005, 169, 553–558. [Google Scholar] [CrossRef]
- Figueras, M.; Pujal, J.; Saleh, A.; Save, R.; Pages, M.; Goday, A. Maize Rabl7 overexpression in Arabidopsis plants promotes osmotic stress tolerance. Ann. Appl. Biol. 2004, 144, 251–257. [Google Scholar] [CrossRef]
- Puhakainen, T.; Hess, M.W.; Mäkelä, P.; Svensson, J.; Heino, P.; Palva, E.T. Overexpression of multiple dehydrin genes enhances tolerance to freezing stress in Arabidopsis. Plant Mol. Biol. 2004, 54, 743–753. [Google Scholar] [CrossRef]
- Artur, M.A.S.; Silva Artur, M.A.; Rienstra, J.; Dennis, T.J.; Farrant, J.M.; Ligterink, W.; Hilhorst, H. Structural Plasticity of Intrinsically Disordered LEA Proteins from Xerophyta schlechteri Provides Protection In Vitro and In Vivo. Front. Plant Sci. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Tunnacliffe, A.; Wise, M.J. The continuing conundrum of the LEA proteins. Naturwissenschaften 2007, 94, 791–812. [Google Scholar] [CrossRef]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.R.; Griffiths-Jones, S.; Howe, K.L.; Marshall, M.; Sonnhammer, E.L.L. The Pfam protein families database. Nucleic Acids Res. 2002, 30, 276–280. [Google Scholar] [CrossRef]
- Jaspard, E.; Macherel, D.; Hunault, G. Computational and statistical analyses of amino acid usage and physico-chemical properties of the twelve late embryogenesis abundant protein classes. PLoS ONE 2012, 7, e36968. [Google Scholar] [CrossRef]
- Tunnacliffe, A.; Hincha, D.K.; Leprince, O.; Macherel, D. LEA Proteins: Versatility of Form and Function. In Dormancy and Resistance in Harsh Environments; Springer: Berlin/Heidelberg, Germany, 2010; pp. 91–108. [Google Scholar]
- Ciccarelli, F.D.; Bork, P. The WHy domain mediates the response to desiccation in plants and bacteria. Bioinformatics 2005, 21, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Aliyu, H.; Cowan, D.A. LEA Proteins and the Evolution of the WHy Domain. Appl. Environ. Microbiol. 2018, 84, e00539-18. [Google Scholar] [CrossRef] [PubMed]
- Dang, N.X.; Popova, A.V.; Hundertmark, M.; Hincha, D.K. Functional characterization of selected LEA proteins from Arabidopsis thaliana in yeast and in vitro. Planta 2014, 240, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Goyal, K.; Walton, L.J.; Tunnacliffe, A. LEA proteins prevent protein aggregation due to water stress. Biochem. J. 2005, 388, 151–157. [Google Scholar] [CrossRef]
- Nylander, M.; Svensson, J.; Palva, E.T.; Welin, B.V. Stress-induced accumulation and tissue-specific localization of dehydrins in Arabidopsis thaliana. Plant Mol. Biol. 2001, 45, 263–279. [Google Scholar] [CrossRef]
- Bremer, A.; Wolff, M.; Thalhammer, A.; Hincha, D.K. Folding of intrinsically disordered plant LEA proteins is driven by glycerol-induced crowding and the presence of membranes. FEBS J. 2017, 284, 919–936. [Google Scholar] [CrossRef]
- Crowe, J.H.; Crowe, L.M.; Chapman, D. Preservation of membranes in anhydrobiotic organisms: The role of trehalose. Science 1984, 223, 701–703. [Google Scholar] [CrossRef]
- Wolkers, W.F.; McCready, S.; Brandt, W.F.; Lindsey, G.G.; Hoekstra, F.A. Isolation and characterization of a D-7 LEA protein from pollen that stabilizes glasses in vitro. Biochim. Biophys. Acta 2001, 1544, 196–206. [Google Scholar] [CrossRef]
- Kaushik, J.K.; Bhat, R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J. Biol. Chem. 2003, 278, 26458–26465. [Google Scholar] [CrossRef]
- Li, D.; He, X. Desiccation Induced Structural Alterations in a 66-Amino Acid Fragment of an Anhydrobiotic Nematode Late Embryogenesis Abundant (LEA) Protein. Biomacromolecules 2009, 10, 1469–1477. [Google Scholar] [CrossRef]
- Nishimoto, T.; Takahashi, Y.; Miyama, S.; Furuta, T.; Sakurai, M. Replica exchange molecular dynamics simulation study on the mechanism of desiccation-induced structuralization of an intrinsically disordered peptide as a model of LEA proteins. Biophys. Phys. 2019, 16, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Koubaa, S.; Bremer, A.; Hincha, D.K.; Brini, F. Structural properties and enzyme stabilization function of the intrinsically disordered LEA_4 protein TdLEA3 from wheat. Sci. Rep. 2019, 9, 3720. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Velazquez, C.L.; Saab-Rincón, G.; Reyes, J.L.; Covarrubias, A.A. The Unstructured N-terminal Region ofArabidopsisGroup 4 Late Embryogenesis Abundant (LEA) Proteins Is Required for Folding and for Chaperone-like Activity under Water Deficit. J. Biol. Chem. 2016, 291, 10893–10903. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Narasimhan, S.; Scherpe, S.; Paioni, A.L.; van der Zwan, J.; Folkers, G.E.; Ovaa, H.; Baldus, M. DNP-Supported Solid-State NMR Spectroscopy of Proteins Inside Mammalian Cells. Angew. Chem. 2019, 131, 13103–13107. [Google Scholar] [CrossRef]
- Koshland, D.; Tapia, H. Desiccation tolerance: An unusual window into stress biology. Mol. Biol. Cell 2019, 30, 737–741. [Google Scholar] [CrossRef]
- Pouchkina-Stantcheva, N.N.; McGee, B.M.; Boschetti, C.; Tolleter, D.; Chakrabortee, S.; Popova, A.V.; Meersman, F.; Macherel, D.; Hincha, D.K.; Tunnacliffe, A. Functional Divergence of Former Alleles in an Ancient Asexual Invertebrate. Science 2007, 318, 268–271. [Google Scholar] [CrossRef]
- Browne, J.A.; Dolan, K.M.; Tyson, T.; Goyal, K.; Tunnacliffe, A.; Burnell, A.M. Dehydration-specific induction of hydrophilic protein genes in the anhydrobiotic nematode Aphelenchus avenae. Eukaryot. Cell 2004, 3, 966–975. [Google Scholar] [CrossRef]
- Hand, S.C.; Jones, D.; Menze, M.A.; Witt, T.L. Life without water: Expression of plant LEA genes by an anhydrobiotic arthropod. J. Exp. Zool. A Ecol. Genet. Physiol. 2007, 307, 62–66. [Google Scholar] [CrossRef]
- Li, S.; Chakraborty, N.; Borcar, A.; Menze, M.A.; Toner, M.; Hand, S.C. Late embryogenesis abundant proteins protect human hepatoma cells during acute desiccation. Proc. Natl. Acad. Sci. USA 2012, 109, 20859–20864. [Google Scholar] [CrossRef]
- Kim, S.X.; Çamdere, G.; Hu, X.; Koshland, D.; Tapia, H. Synergy between the small intrinsically disordered protein Hsp12 and trehalose sustain viability after severe desiccation. Elife 2018, 7, e38337. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, J.; Tunnacliffe, A. Anhydrobiosis without trehalose in bdelloid rotifers. FEBS Lett. 2003, 553, 387–390. [Google Scholar] [CrossRef]
- Boothby, T.C.; Piszkiewicz, S.; Mehta, A.; Brozena, A.; Tapia, H.; Koshland, D.; Holehouse, A.; Pappu, R.; Goldstein, B.; Pielak, G. Tardigrade Disordered Proteins Mediate Desiccation Tolerance. Biophys. J. 2017, 112, 480a. [Google Scholar] [CrossRef][Green Version]
- Aguilera-Gomez, A.; Rabouille, C. Membrane-bound organelles versus membrane-less compartments and their control of anabolic pathways in Drosophila. Dev. Biol. 2017, 428, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Velazquez, C.L.; Dinneny, J.R. Organization out of disorder: Liquid-liquid phase separation in plants. Curr. Opin. Plant Biol. 2018, 45, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef]
- Majumdar, A.; Dogra, P.; Maity, S.; Mukhopadhyay, S. Liquid-Liquid Phase Separation Is Driven by Large-Scale Conformational Unwinding and Fluctuations of Intrinsically Disordered Protein Molecules. J. Phys. Chem. Lett. 2019, 10, 3929–3936. [Google Scholar] [CrossRef]
- Bergeron-Sandoval, L.-P.; Safaee, N.; Michnick, S.W. Mechanisms and Consequences of Macromolecular Phase Separation. Cell 2016, 165, 1067–1079. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.A.; Forman-Kay, J.D. Liquid–liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. [Google Scholar] [CrossRef]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef]
- Aumiller, W.M., Jr.; Keating, C.D. Experimental models for dynamic compartmentalization of biomolecules in liquid organelles: Reversible formation and partitioning in aqueous biphasic systems. Adv. Colloid Interface Sci. 2017, 239, 75–87. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015, 589, 15–22. [Google Scholar] [CrossRef]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Aumiller, W.M., Jr.; Keating, C.D. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat. Chem. 2016, 8, 129–137. [Google Scholar] [CrossRef]
- Freeman Rosenzweig, E.S.; Xu, B.; Kuhn Cuellar, L.; Martinez-Sanchez, A.; Schaffer, M.; Strauss, M.; Cartwright, H.N.; Ronceray, P.; Plitzko, J.M.; Förster, F.; et al. The Eukaryotic CO2-Concentrating Organelle Is Liquid-like and Exhibits Dynamic Reorganization. Cell 2017, 171, 148–162.e19. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Elbaum-Garfinkle, S.; Langdon, E.M.; Taylor, N.; Occhipinti, P.; Bridges, A.A.; Brangwynne, C.P.; Gladfelter, A.S. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Kosmacz, M.; Gorka, M.; Schmidt, S.; Luzarowski, M.; Moreno, J.C.; Szlachetko, J.; Leniak, E.; Sokolowska, E.M.; Sofroni, K.; Schnittger, A.; et al. Protein and metabolite composition of Arabidopsis stress granules. New Phytol. 2019, 222, 1420–1433. [Google Scholar] [CrossRef]
- Moujalled, D.; James, J.L.; Yang, S.; Zhang, K.; Duncan, C.; Moujalled, D.M.; Parker, S.J.; Caragounis, A.; Lidgerwood, G.; Turner, B.J.; et al. Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum. Mol. Genet. 2015, 24, 1655–1669. [Google Scholar] [CrossRef]
- Van Buskirk, E.K.; Decker, P.V.; Chen, M. Photobodies in light signaling. Plant Physiol. 2012, 158, 52–60. [Google Scholar] [CrossRef]
- Bugaj, L.J.; Choksi, A.T.; Mesuda, C.K.; Kane, R.S.; Schaffer, D.V. Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 2013, 10, 249–252. [Google Scholar] [CrossRef]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1. [Google Scholar] [CrossRef]
- Haynes, C.; Iakoucheva, L.M. Serine/arginine-rich splicing factors belong to a class of intrinsically disordered proteins. Nucleic Acids Res. 2006, 34, 305–312. [Google Scholar] [CrossRef]
- Ali, G.S.; Golovkin, M.; Reddy, A.S.N. Nuclear localization and in vivo dynamics of a plant-specific serine/arginine-rich protein. Plant J. 2003, 36, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J. 2020, 39, e102729. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Stoecklin, G. The heat’s on: Nuclear stress bodies signal intron retention. EMBO J. 2020, 39, e104154. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.; Forman-Kay, J.D. First-generation predictors of biological protein phase separation. Curr. Opin. Struct. Biol. 2019, 58, 88–96. [Google Scholar] [CrossRef]
- Chakrabortee, S.; Kayatekin, C.; Newby, G.A.; Mendillo, M.L.; Lancaster, A.; Lindquist, S. Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc. Natl. Acad. Sci. USA 2016, 113, 6065–6070. [Google Scholar] [CrossRef]
- Fang, X.; Wang, L.; Ishikawa, R.; Li, Y.; Fiedler, M.; Liu, F.; Calder, G.; Rowan, B.; Weigel, D.; Li, P.; et al. Arabidopsis FLL2 promotes liquid–liquid phase separation of polyadenylation complexes. Nature 2019, 569, 265–269. [Google Scholar] [CrossRef]
- Nick, P. Microtubules, signalling and abiotic stress. Plant J. 2013, 75, 309–323. [Google Scholar] [CrossRef]
- Kesten, C.; Wallmann, A.; Schneider, R.; McFarlane, H.E.; Diehl, A.; Khan, G.A.; van Rossum, B.-J.; Lampugnani, E.R.; Szymanski, W.G.; Cremer, N.; et al. The companion of cellulose synthase 1 confers salt tolerance through a Tau-like mechanism in plants. Nat. Commun. 2019, 10, 857. [Google Scholar] [CrossRef]
- Wang, C.; Li, J.; Yuan, M. Salt tolerance requires cortical microtubule reorganization in Arabidopsis. Plant Cell Physiol. 2007, 48, 1534–1547. [Google Scholar] [CrossRef]
- Paredez, A.R.; Somerville, C.R.; Ehrhardt, D.W. Visualization of cellulose synthase demonstrates functional association with microtubules. Science 2006, 312, 1491–1495. [Google Scholar] [CrossRef]
- Endler, A.; Kesten, C.; Schneider, R.; Zhang, Y.; Ivakov, A.; Froehlich, A.; Funke, N.; Persson, S. A Mechanism for Sustained Cellulose Synthesis during Salt Stress. Cell 2015, 162, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, M.H.; Jalal, A.; Brenner, B.; Mandelkow, E.; Kumar, S.; Scholz, T. Tau protein diffuses along the microtubule lattice. J. Biol. Chem. 2012, 287, 38559–38568. [Google Scholar] [CrossRef]
- Scott, C.W.; Klika, A.B.; Lo, M.M.S.; Norris, T.E.; Caputo, C.B. Tau protein induces bundling of microtubules in vitro: Comparison of different tau isoforms and a tau protein fragment. J. Neurosci. Res. 1992, 33, 19–29. [Google Scholar] [CrossRef]
- Prezel, E.; Elie, A.; Delaroche, J.; Stoppin-Mellet, V.; Bosc, C.; Serre, L.; Fourest-Lieuvin, A.; Andrieux, A.; Vantard, M.; Arnal, I. Tau can switch microtubule network organizations: From random networks to dynamic and stable bundles. Mol. Biol. Cell 2018, 29, 154–165. [Google Scholar] [CrossRef]
- Guharoy, M.; Szabo, B.; Contreras Martos, S.; Kosol, S.; Tompa, P. Intrinsic structural disorder in cytoskeletal proteins. Cytoskeleton 2013, 70, 550–571. [Google Scholar] [CrossRef]
- Sancar, A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 2003, 103, 2203–2237. [Google Scholar] [CrossRef]
- Cashmore, A.R. Cryptochromes: Enabling plants and animals to determine circadian time. Cell 2003, 114, 537–543. [Google Scholar] [CrossRef]
- Partch, C.L.; Clarkson, M.W.; Ozgür, S.; Lee, A.L.; Sancar, A. Role of structural plasticity in signal transduction by the cryptochrome blue-light photoreceptor. Biochemistry 2005, 44, 3795–3805. [Google Scholar] [CrossRef]
- Yang, H.Q.; Wu, Y.J.; Tang, R.H.; Liu, D.; Liu, Y.; Cashmore, A.R. The C termini of Arabidopsis cryptochromes mediate a constitutive light response. Cell 2000, 103, 815–827. [Google Scholar] [CrossRef]
- Green, C.B. Cryptochromes: Tailored for distinct functions. Curr. Biol. 2004, 14, R847–R849. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, X.; Liu, H.; Klejnot, J.; Lin, C. The Cryptochrome Blue Light Receptors. Arab. Book 2010, 8, e0135. [Google Scholar] [CrossRef] [PubMed]
- Chaves, I.; Yagita, K.; Barnhoorn, S.; Okamura, H.; van der Horst, G.T.J.; Tamanini, F. Functional evolution of the photolyase/cryptochrome protein family: Importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Mol. Cell. Biol. 2006, 26, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Chaves, I.; Pokorny, R.; Byrdin, M.; Hoang, N.; Ritz, T.; Brettel, K.; Essen, L.-O.; van der Horst, G.T.J.; Batschauer, A.; Ahmad, M. The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 2011, 62, 335–364. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Jarillo, J.A.; Cashmore, A.R. Chimeric proteins between cry1 and cry2 Arabidopsis blue light photoreceptors indicate overlapping functions and varying protein stability. Plant Cell 1998, 10, 197–207. [Google Scholar] [CrossRef]
- Lin, C.; Shalitin, D. Cryptochrome structure and signal transduction. Annu. Rev. Plant Biol. 2003, 54, 469–496. [Google Scholar] [CrossRef]
- Zoltowski, B.D.; Vaidya, A.T.; Top, D.; Widom, J.; Young, M.W.; Crane, B.R. Structure of full-length Drosophila cryptochrome. Nature 2011, 480, 396–399. [Google Scholar] [CrossRef]
- Czarna, A.; Berndt, A.; Singh, H.R.; Grudziecki, A.; Ladurner, A.G.; Timinszky, G.; Kramer, A.; Wolf, E. Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 2013, 153, 1394–1405. [Google Scholar] [CrossRef]
- Wang, H.; Ma, L.G.; Li, J.M.; Zhao, H.Y.; Deng, X.W. Direct interaction of Arabidopsis cryptochromes with COP1 in light control development. Science 2001, 294, 154–158. [Google Scholar] [CrossRef]
- Yang, H.Q.; Tang, R.H.; Cashmore, A.R. The signaling mechanism of Arabidopsis CRY1 involves direct interaction with COP1. Plant Cell 2001, 13, 2573–2587. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Liu, H.; Liu, B.; Liu, X.; Lin, C. Blue light-dependent interaction of CRY2 with SPA1 regulates COP1 activity and floral initiation in Arabidopsis. Curr. Biol. 2011, 21, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zuo, Z.; Liu, H.; Liu, X.; Lin, C. Arabidopsis cryptochrome 1 interacts with SPA1 to suppress COP1 activity in response to blue light. Genes Dev. 2011, 25, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, B.; Zhao, C.; Pepper, M.; Lin, C. The action mechanisms of plant cryptochromes. Trends Plant Sci. 2011, 16, 684–691. [Google Scholar] [CrossRef]
- Wang, Q.; Barshop, W.D.; Bian, M.; Vashisht, A.A.; He, R.; Yu, X.; Liu, B.; Nguyen, P.; Liu, X.; Zhao, X.; et al. The blue light-dependent phosphorylation of the CCE domain determines the photosensitivity of Arabidopsis CRY2. Mol. Plant 2015, 8, 631–643. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, Q.; Deng, W.; Wang, X.; Piao, M.; Cai, D.; Li, Y.; Barshop, W.D.; Yu, X.; Zhou, T.; et al. Molecular basis for blue light-dependent phosphorylation of Arabidopsis cryptochrome 2. Nat. Commun. 2017, 8, 15234. [Google Scholar] [CrossRef]
- Yu, X.; Shalitin, D.; Liu, X.; Maymon, M.; Klejnot, J.; Yang, H.; Lopez, J.; Zhao, X.; Bendehakkalu, K.T.; Lin, C. Derepression of the NC80 motif is critical for the photoactivation of Arabidopsis CRY2. Proc. Natl. Acad. Sci. USA 2007, 104, 7289–7294. [Google Scholar] [CrossRef]
- Busza, A.; Emery-Le, M.; Rosbash, M.; Emery, P. Roles of the two Drosophila CRYPTOCHROME structural domains in circadian photoreception. Science 2004, 304, 1503–1506. [Google Scholar] [CrossRef]
- Peschel, N.; Chen, K.F.; Szabo, G.; Stanewsky, R. Light-dependent interactions between the Drosophila circadian clock factors cryptochrome, jetlag, and timeless. Curr. Biol. 2009, 19, 241–247. [Google Scholar] [CrossRef]
- Dissel, S.; Codd, V.; Fedic, R.; Garner, K.J.; Costa, R.; Kyriacou, C.P.; Rosato, E. A constitutively active cryptochrome in Drosophila melanogaster. Nat. Neurosci. 2004, 7, 834–840. [Google Scholar] [CrossRef]
- Ozturk, N. Phylogenetic and Functional Classification of the Photolyase/Cryptochrome Family. Photochem. Photobiol. 2017, 93, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Nießner, C.; Denzau, S.; Malkemper, E.P.; Gross, J.C.; Burda, H.; Winklhofer, M.; Peichl, L. Cryptochrome 1 in Retinal Cone Photoreceptors Suggests a Novel Functional Role in Mammals. Sci. Rep. 2016, 6, 21848. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Provencio, I.; Tu, D.C.; Pires, S.S.; Rollag, M.D.; Castrucci, A.M.; Pletcher, M.T.; Sato, T.K.; Wiltshire, T.; Andahazy, M.; et al. Melanopsin is required for non-image-forming photic responses in blind mice. Science 2003, 301, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Hattar, S.; Lucas, R.J.; Mrosovsky, N.; Thompson, S.; Douglas, R.H.; Hankins, M.W.; Lem, J.; Biel, M.; Hofmann, F.; Foster, R.G.; et al. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature 2003, 424, 76–81. [Google Scholar] [CrossRef]
- Brown, S.A.; Ripperger, J.; Kadener, S.; Fleury-Olela, F.; Vilbois, F.; Rosbash, M.; Schibler, U. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 2005, 308, 693–696. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kwak, P.B.; Weitz, C.J. Specificity in circadian clock feedback from targeted reconstitution of the NuRD corepressor. Mol. Cell 2014, 56, 738–748. [Google Scholar] [CrossRef]
- Koike, N.; Yoo, S.-H.; Huang, H.-C.; Kumar, V.; Lee, C.; Kim, T.-K.; Takahashi, J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 2012, 338, 349–354. [Google Scholar] [CrossRef]
- Gao, P.; Yoo, S.-H.; Lee, K.-J.; Rosensweig, C.; Takahashi, J.S.; Chen, B.P.; Green, C.B. Phosphorylation of the cryptochrome 1 C-terminal tail regulates circadian period length. J. Biol. Chem. 2013, 288, 35277–35286. [Google Scholar] [CrossRef]
- Hirano, A.; Kurabayashi, N.; Nakagawa, T.; Shioi, G.; Todo, T.; Hirota, T.; Fukada, Y. In vivo role of phosphorylation of cryptochrome 2 in the mouse circadian clock. Mol. Cell. Biol. 2014, 34, 4464–4473. [Google Scholar] [CrossRef]
- Harada, Y.; Sakai, M.; Kurabayashi, N.; Hirota, T.; Fukada, Y. Ser-557-phosphorylated mCRY2 is degraded upon synergistic phosphorylation by glycogen synthase kinase-3 beta. J. Biol. Chem. 2005, 280, 31714–31721. [Google Scholar] [CrossRef]
- Franz-Badur, S.; Penner, A.; Straß, S.; von Horsten, S.; Linne, U.; Essen, L.-O. Structural changes within the bifunctional cryptochrome/photolyase CraCRY upon blue light excitation. Sci. Rep. 2019, 9, 9896. [Google Scholar] [CrossRef] [PubMed]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761.e16. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef]
- Casimiro, M.C.; Velasco-Velázquez, M.; Aguirre-Alvarado, C.; Pestell, R.G. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: Past and present. Expert Opin. Investig. Drugs 2014, 23, 295–304. [Google Scholar] [CrossRef]
- Van De Pette, M.; Tunster, S.J.; McNamara, G.I.; Shelkovnikova, T.; Millership, S.; Benson, L.; Peirson, S.; Christian, M.; Vidal-Puig, A.; John, R.M. Cdkn1c Boosts the Development of Brown Adipose Tissue in a Murine Model of Silver Russell Syndrome. PLoS Genet. 2016, 12, e1005916. [Google Scholar] [CrossRef]
- Di Sante, G.; Pagé, J.; Jiao, X.; Nawab, O.; Cristofanilli, M.; Skordalakes, E.; Pestell, R.G. Recent advances with cyclin-dependent kinase inhibitors: Therapeutic agents for breast cancer and their role in immuno-oncology. Expert Rev. Anticancer Ther. 2019, 19, 569–587. [Google Scholar] [CrossRef]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massagué, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A–Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef]
- Barberis, M.; De Gioia, L.; Ruzzene, M.; Sarno, S.; Coccetti, P.; Fantucci, P.; Vanoni, M.; Alberghina, L. The yeast cyclin-dependent kinase inhibitor Sic1 and mammalian p27Kip1 are functional homologues with a structurally conserved inhibitory domain. Biochem. J. 2005, 387, 639–647. [Google Scholar] [CrossRef][Green Version]
- McGrath, D.A.; Fifield, B.-A.; Marceau, A.H.; Tripathi, S.; Porter, L.A.; Rubin, S.M. Structural basis of divergent cyclin-dependent kinase activation by Spy1/RINGO proteins. EMBO J. 2017, 36, 2251–2262. [Google Scholar] [CrossRef]
- Brocca, S.; Samalíková, M.; Uversky, V.N.; Lotti, M.; Vanoni, M.; Alberghina, L.; Grandori, R. Order propensity of an intrinsically disordered protein, the cyclin-dependent-kinase inhibitor Sic1. Proteins Struct. Funct. Bioinform. 2009, 76, 731–746. [Google Scholar] [CrossRef]
- Hodge, A.; Mendenhall, M. The cyclin-dependent kinase inhibitory domain of the yeast Sic1 protein is contained within the C-terminal 70 amino acids. Mol. Gen. Genet. 1999, 262, 55–64. [Google Scholar] [CrossRef]
- Wang, H.; Fowke, L.C.; Crosby, W.L. A plant cyclin-dependent kinase inhibitor gene. Nature 1997, 386, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Wang, H.; Delong, C.; Fowke, L.C.; Crosby, W.L.; Fobert, P.R. The Arabidopsis Cdc2a-interacting protein ICK2 is structurally related to ICK1 and is a potent inhibitor of cyclin-dependent kinase activity in vitro. Plant J. 2000, 21, 379–385. [Google Scholar] [CrossRef] [PubMed]
- De Veylder, L.; Beeckman, T.; Beemster, G.T.; Krols, L.; Terras, F.; Landrieu, I.; van der Schueren, E.; Maes, S.; Naudts, M.; Inzé, D. Functional analysis of cyclin-dependent kinase inhibitors of Arabidopsis. Plant Cell 2001, 13, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Verkest, A.; Weinl, C.; Inzé, D.; De Veylder, L.; Schnittger, A. Switching the cell cycle. Kip-related proteins in plant cell cycle control. Plant Physiol. 2005, 139, 1099–1106. [Google Scholar] [CrossRef]
- Zhou, Y.; Fowke, L.; Wang, H. Plant CDK inhibitors: Studies of interactions with cell cycle regulators in the yeast two-hybrid system and functional comparisons in transgenic Arabidopsis plants. Plant Cell Rep. 2002, 20, 967–975. [Google Scholar] [CrossRef]
- Nakai, T.; Kato, K.; Shinmyo, A.; Sekine, M. Arabidopsis KRPs have distinct inhibitory activity toward cyclin D2-associated kinases, including plant-specific B-type cyclin-dependent kinase. FEBS Lett. 2006, 580, 336–340. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, C.; Li, H.; Wei, B.; Wang, Y.; Huang, H.; Li, Y.; Yu, G.; Liu, H.; Zhang, J.; et al. Identification and functional analysis of the ICK gene family in maize. Sci. Rep. 2017, 7, 43818. [Google Scholar] [CrossRef]
- Cross, F.R.; Buchler, N.E.; Skotheim, J.M. Evolution of networks and sequences in eukaryotic cell cycle control. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.S. The function of p27 KIP1 during tumor development. Exp. Mol. Med. 2009, 41, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Bachs, O.; Gallastegui, E.; Orlando, S.; Bigas, A.; Morante-Redolat, J.M.; Serratosa, J.; Fariñas, I.; Aligué, R.; Pujol, M.J. Role of p27Kip1 as a transcriptional regulator. Oncotarget 2018, 9, 26259–26278. [Google Scholar] [CrossRef] [PubMed]
- Rampioni Vinciguerra, G.L.; Citron, F.; Segatto, I.; Belletti, B.; Vecchione, A.; Baldassarre, G. p27kip1 at the crossroad between actin and microtubule dynamics. Cell Div. 2019, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Schwob, E.; Böhm, T.; Mendenhall, M.D.; Nasmyth, K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 1994, 79, 233–244. [Google Scholar] [CrossRef]
- Schneider, B.L.; Yang, Q.H.; Futcher, A.B. Linkage of replication to start by the Cdk inhibitor Sic1. Science 1996, 272, 560–562. [Google Scholar] [CrossRef]
- Feldman, R.M.; Correll, C.C.; Kaplan, K.B.; Deshaies, R.J. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 1997, 91, 221–230. [Google Scholar] [CrossRef]
- Skowyra, D.; Craig, K.L.; Tyers, M.; Elledge, S.J.; Harper, J.W. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 1997, 91, 209–219. [Google Scholar] [CrossRef]
- Verma, R.; Annan, R.S.; Huddleston, M.J.; Carr, S.A.; Reynard, G.; Deshaies, R.J. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 1997, 278, 455–460. [Google Scholar] [CrossRef]
- Nash, P.; Tang, X.; Orlicky, S.; Chen, Q.; Gertler, F.B.; Mendenhall, M.D.; Sicheri, F.; Pawson, T.; Tyers, M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 2001, 414, 514–521. [Google Scholar] [CrossRef]
- Tripodi, F.; Zinzalla, V.; Vanoni, M.; Alberghina, L.; Coccetti, P. In CK2 inactivated cells the cyclin dependent kinase inhibitor Sic1 is involved in cell-cycle arrest before the onset of S phase. Biochem. Biophys. Res. Commun. 2007, 359, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Kõivomägi, M.; Valk, E.; Venta, R.; Iofik, A.; Lepiku, M.; Balog, E.R.M.; Rubin, S.M.; Morgan, D.O.; Loog, M. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 2011, 480, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Kõivomägi, M.; Ord, M.; Iofik, A.; Valk, E.; Venta, R.; Faustova, I.; Kivi, R.; Balog, E.R.M.; Rubin, S.M.; Loog, M. Multisite phosphorylation networks as signal processors for Cdk1. Nat. Struct. Mol. Biol. 2013, 20, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Coccetti, P.; Zinzalla, V.; Tedeschi, G.; Russo, G.L.; Fantinato, S.; Marin, O.; Pinna, L.A.; Vanoni, M.; Alberghina, L. Sic1 is phosphorylated by CK2 on Ser201 in budding yeast cells. Biochem. Biophys. Res. Commun. 2006, 346, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Coccetti, P.; Rossi, R.L.; Sternieri, F.; Porro, D.; Russo, G.L.; Di Fonzo, A.; Magni, F.; Vanoni, M.; Alberghina, L. Mutations of the CK2 phosphorylation site of Sic1 affect cell size and S-Cdk kinase activity in Saccharomyces cerevisiae. Mol. Microbiol. 2004, 51, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Besson, A. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006, 20, 47–64. [Google Scholar] [CrossRef]
- Deng, X.; Mercer, S.E.; Shah, S.; Ewton, D.Z.; Friedman, E. The Cyclin-dependent Kinase Inhibitor p27Kip1 Is Stabilized in G0 by Mirk/dyrk1B Kinase. J. Biol. Chem. 2004, 279, 22498–22504. [Google Scholar] [CrossRef]
- Kossatz, U.; Vervoorts, J.; Nickeleit, I.; Sundberg, H.A.; Arthur, J.S.C.; Manns, M.P.; Malek, N.P. C-terminal phosphorylation controls the stability and function of p27kip1. EMBO J. 2006, 25, 5159–5170. [Google Scholar] [CrossRef]
- Vervoorts, J.; Lüscher, B. Post-translational regulation of the tumor suppressor p27KIP1. Cell. Mol. Life Sci. 2008, 65, 3255. [Google Scholar] [CrossRef]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilensek, Z.; Keidel, E.-M.; Waddell, M.B.; Jäkel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef]
- Orlicky, S.; Tang, X.; Willems, A.; Tyers, M.; Sicheri, F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 2003, 112, 243–256. [Google Scholar] [CrossRef]
- Tsytlonok, M.; Sanabria, H.; Wang, Y.; Felekyan, S.; Hemmen, K.; Phillips, A.H.; Yun, M.-K.; Waddell, M.B.; Park, C.-G.; Vaithiyalingam, S.; et al. Dynamic anticipation by Cdk2/Cyclin A-bound p27 mediates signal integration in cell cycle regulation. Nat. Commun. 2019, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Shirane, M.; Harumiya, Y.; Ishida, N.; Hirai, A.; Miyamoto, C.; Hatakeyama, S.; Nakayama, K.-I.; Kitagawa, M. Down-regulation of p27 Kip1 by Two Mechanisms, Ubiquitin-mediated Degradation and Proteolytic Processing. J. Biol. Chem. 1999, 274, 13886–13893. [Google Scholar] [CrossRef] [PubMed]
- Godínez-Palma, S.K.; Rosas-Bringas, F.R.; Rosas-Bringas, O.G.; García-Ramírez, E.; Zamora-Zaragoza, J.; Vázquez-Ramos, J.M. Two maize Kip-related proteins differentially interact with, inhibit and are phosphorylated by cyclin D–cyclin-dependent kinase complexes. J. Exp. Bot. 2017, 68, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Marsh, J.; Grishaev, A.; Orlicky, S.; Lin, H.; Sicheri, F.; Tyers, M.; Forman-Kay, J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure 2010, 18, 494–506. [Google Scholar] [CrossRef]
- Das, R.K.; Huang, Y.; Phillips, A.H.; Kriwacki, R.W.; Pappu, R.V. Cryptic sequence features within the disordered protein p27Kip1 regulate cell cycle signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 5616–5621. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Walhout, A.J.M. Unraveling transcription regulatory networks by protein-DNA and protein-protein interaction mapping. Genome Res. 2006, 16, 1445–1454. [Google Scholar] [CrossRef]
- Gould, C.M.; Diella, F.; Via, A.; Puntervoll, P.; Gemünd, C.; Chabanis-Davidson, S.; Michael, S.; Sayadi, A.; Bryne, J.C.; Chica, C.; et al. ELM: The status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010, 38, D167–D180. [Google Scholar] [CrossRef]
- Yruela, I.; Oldfield, C.J.; Niklas, K.J.; Keith Dunker, A. Evidence for a Strong Correlation Between Transcription Factor Protein Disorder and Organismic Complexity. Genome Biol. Evol. 2017, 9, 1248–1265. [Google Scholar] [CrossRef]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Keith Dunker, A. Intrinsic Disorder in Transcription Factors†. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [PubMed]
- Staby, L.; O’Shea, C.; Willemoës, M.; Theisen, F.; Kragelund, B.B.; Skriver, K. Eukaryotic transcription factors: Paradigms of protein intrinsic disorder. Biochem. J. 2017, 474, 2509–2532. [Google Scholar] [CrossRef] [PubMed]
- Piskacek, M.; Havelka, M.; Rezacova, M.; Knight, A. The 9aaTAD Transactivation Domains: From Gal4 to p53. PLoS ONE 2016, 11, e0162842. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Waseem, M.; Dwivedi, N.; Maji, S.; Kumar, A.; Thakur, J.K. KIX domain of AtMed15a, a Mediator subunit of Arabidopsis, is required for its interaction with different proteins. Plant Signal. Behav. 2018, 13, e1428514. [Google Scholar] [CrossRef] [PubMed]
- Thakur, J.K.; Arthanari, H.; Yang, F.; Chau, K.H.; Wagner, G.; Näär, A.M. Mediator subunit Gal11p/MED15 is required for fatty acid-dependent gene activation by yeast transcription factor Oaf1p. J. Biol. Chem. 2009, 284, 4422–4428. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Vought, B.W.; Satterlee, J.S.; Walker, A.K.; Jim Sun, Z.-Y.; Watts, J.L.; DeBeaumont, R.; Saito, R.M.; Hyberts, S.G.; Yang, S.; et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 2006, 442, 700–704. [Google Scholar] [CrossRef]
- Guan, X.; Stege, J.; Kim, M.; Dahmani, Z.; Fan, N.; Heifetz, P.; Barbas, C.F., 3rd; Briggs, S.P. Heritable endogenous gene regulation in plants with designed polydactyl zinc finger transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 13296–13301. [Google Scholar] [CrossRef]
- Sánchez, J.P.; Ullman, C.; Moore, M.; Choo, Y.; Chua, N.-H. Regulation of Arabidopsis thaliana 4-coumarate:coenzyme-A ligase-1 expression by artificial zinc finger chimeras. Plant Biotechnol. J. 2006, 4, 103–114. [Google Scholar] [CrossRef]
- Gupta, M.; DeKelver, R.C.; Palta, A.; Clifford, C.; Gopalan, S.; Miller, J.C.; Novak, S.; Desloover, D.; Gachotte, D.; Connell, J.; et al. Transcriptional activation of Brassica napus β-ketoacyl-ACP synthase II with an engineered zinc finger protein transcription factor. Plant Biotechnol. J. 2012, 10, 783–791. [Google Scholar] [CrossRef]
- Di Lello, P.; Jenkins, L.M.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar] [CrossRef]
- Langlois, C.; Mas, C.; Di Lello, P.; Jenkins, L.M.M.; Legault, P.; Omichinski, J.G. NMR structure of the complex between the Tfb1 subunit of TFIIH and the activation domain of VP16: Structural similarities between VP16 and p53. J. Am. Chem. Soc. 2008, 130, 10596–10604. [Google Scholar] [CrossRef] [PubMed]
- Krois, A.S.; Dyson, H.J.; Wright, P.E. Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA. 2018, 115, E11302–E11310. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Blue, R.; Zeitler, B.; Strange, T.L.; Pearl, J.R.; Huizinga, D.H.; Evans, S.; Gregory, P.D.; Urnov, F.D.; Petolino, J.F. Activation domains for controlling plant gene expression using designed transcription factors. Plant Biotechnol. J. 2013, 11, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Ravarani, C.N.; Erkina, T.Y.; De Baets, G.; Dudman, D.C.; Erkine, A.M.; Babu, M.M. High-throughput discovery of functional disordered regions: Investigation of transactivation domains. Mol. Syst. Biol. 2018, 14, e8190. [Google Scholar] [CrossRef]
- Ooka, H.; Satoh, K.; Doi, K.; Nagata, T.; Otomo, Y.; Murakami, K.; Matsubara, K.; Osato, N.; Kawai, J.; Carninci, P.; et al. Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 2003, 10, 239–247. [Google Scholar] [CrossRef]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC proteins: Regulation and role in stress tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef]
- Jensen, M.K.; Kjaersgaard, T.; Nielsen, M.M.; Galberg, P.; Petersen, K.; O’Shea, C.; Skriver, K. The Arabidopsis thaliana NAC transcription factor family: Structure-function relationships and determinants of ANAC019 stress signalling. Biochem. J. 2010, 426, 183–196. [Google Scholar] [CrossRef]
- Ernst, H.A.; Olsen, A.N.; Larsen, S.; Lo Leggio, L. Structure of the conserved domain of ANAC, a member of the NAC family of transcription factors. EMBO Rep. 2004, 5, 297–303. [Google Scholar] [CrossRef]
- Kim, H.S.; Park, B.O.; Yoo, J.H.; Jung, M.S.; Lee, S.M.; Han, H.J.; Kim, K.E.; Kim, S.H.; Lim, C.O.; Yun, D.-J.; et al. Identification of a calmodulin-binding NAC protein as a transcriptional repressor in Arabidopsis. J. Biol. Chem. 2007, 282, 36292–36302. [Google Scholar] [CrossRef]
- Kjaersgaard, T.; Jensen, M.K.; Christiansen, M.W.; Gregersen, P.; Kragelund, B.B.; Skriver, K. Senescence-associated Barley NAC (NAM, ATAF1,2, CUC) Transcription Factor Interacts with Radical-induced Cell Death 1 through a Disordered Regulatory Domain. J. Biol. Chem. 2011, 286, 35418–35429. [Google Scholar] [CrossRef]
- O’Shea, C.; Staby, L.; Bendsen, S.K.; Tidemand, F.G.; Redsted, A.; Willemoës, M.; Kragelund, B.B.; Skriver, K. Structures and Short Linear Motif of Disordered Transcription Factor Regions Provide Clues to the Interactome of the Cellular Hub Protein Radical-induced Cell Death1. J. Biol. Chem. 2017, 292, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.; Conklin, P.A.; Huefner, N.D.; Britt, A.B. Suppressor of gamma response 1 (SOG1) encodes a putative transcription factor governing multiple responses to DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, K.O.; Kimura, S.; Maki, H.; Britt, A.B.; Umeda, M. The role of SOG1, a plant-specific transcriptional regulator, in the DNA damage response. Plant Signal. Behav. 2014, 9, e28889. [Google Scholar] [CrossRef] [PubMed]
- Bourbousse, C.; Vegesna, N.; Law, J.A. SOG1 activator and MYB3R repressors regulate a complex DNA damage network in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E12453–E12462. [Google Scholar] [CrossRef]
- Fischer, M. p21 governs p53′s repressive side. Cell Cycle 2016, 15, 2852–2853. [Google Scholar] [CrossRef]
- Yoshiyama, K.O.; Kobayashi, J.; Ogita, N.; Ueda, M.; Kimura, S.; Maki, H.; Umeda, M. ATM-mediated phosphorylation of SOG1 is essential for the DNA damage response in Arabidopsis. EMBO Rep. 2013, 14, 817–822. [Google Scholar] [CrossRef]
- Mills, C.L.; Beuning, P.J.; Ondrechen, M.J. Biochemical functional predictions for protein structures of unknown or uncertain function. Comput. Struct. Biotechnol. J. 2015, 13, 182–191. [Google Scholar] [CrossRef]
- Cruz, L.M.; Trefflich, S.; Weiss, V.A.; Castro, M.A.A. Protein Function Prediction. Methods Mol. Biol. 2017, 1654, 55–75. [Google Scholar]
- Shimomura, T.; Nishijima, K.; Kikuchi, T. A new technique for predicting intrinsically disordered regions based on average distance map constructed with inter-residue average distance statistics. BMC Struct. Biol. 2019, 19, 3. [Google Scholar] [CrossRef]
- Zarin, T.; Strome, B.; Nguyen Ba, A.N.; Alberti, S.; Forman-Kay, J.D.; Moses, A.M. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. Elife 2019, 8, e46883. [Google Scholar] [CrossRef]
- Mahani, A.; Henriksson, J.; Wright, A.P.H. Origins of Myc proteins--using intrinsic protein disorder to trace distant relatives. PLoS ONE 2013, 8, e75057. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.A.R.; Nijveen, H.; Hilhorst, H.W.M.; Ligterink, W. Learning from Co-expression Networks: Possibilities and Challenges. Front. Plant Sci. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Flammini, A.; Maritan, A.; Vespignani, A. Global protein function prediction from protein-protein interaction networks. Nat. Biotechnol. 2003, 21, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Kourmpetis, Y.A.I.; van Dijk, A.D.J.; Bink, M.C.A.M.; van Ham, R.C.H.J.; ter Braak, C.J.F. Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS ONE 2010, 5, e9293. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Van Leene, J.; De Coninck, B.; Cammue, B.P.A. Hub Protein Controversy: Taking a Closer Look at Plant Stress Response Hubs. Front. Plant Sci. 2018, 9, 694. [Google Scholar] [CrossRef]
- Schneider, R.; Huang, J.-R.; Yao, M.; Communie, G.; Ozenne, V.; Mollica, L.; Salmon, L.; Jensen, M.R.; Blackledge, M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol. Biosyst. 2012, 8, 58–68. [Google Scholar] [CrossRef]
- Chan-Yao-Chong, M.; Durand, D.; Ha-Duong, T. Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles. J. Chem. Inf. Model. 2019, 59, 1743–1758. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Cedeño, C.; Pauwels, K.; Tompa, P. Protein Delivery into Plant Cells: Toward In vivo Structural Biology. Front. Plant Sci. 2017, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Rose, H.M.; Liokatis, S.; Binolfi, A.; Thongwichian, R.; Stuiver, M.; Selenko, P. Site-specific NMR mapping and time-resolved monitoring of serine and threonine phosphorylation in reconstituted kinase reactions and mammalian cell extracts. Nat. Protoc. 2013, 8, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.R.; Carroll, N.J.; Rubinstein, M.; Chilkoti, A.; López, G.P. Programming molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat. Chem. 2017, 9, 509–515. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Nasir, I.; Onuchic, P.L.; Labra, S.R.; Deniz, A.A. Single-molecule fluorescence studies of intrinsically disordered proteins and liquid phase separation. Biochim. Biophys. Acta: Proteins Proteom. 2019, 1867, 980–987. [Google Scholar] [CrossRef]
- Brucale, M.; Schuler, B.; Samorì, B. Single-molecule studies of intrinsically disordered proteins. Chem. Rev. 2014, 114, 3281–3317. [Google Scholar] [CrossRef]
- König, I.; Zarrine-Afsar, A.; Aznauryan, M.; Soranno, A.; Wunderlich, B.; Dingfelder, F.; Stüber, J.C.; Plückthun, A.; Nettels, D.; Schuler, B. Single-molecule spectroscopy of protein conformational dynamics in live eukaryotic cells. Nat. Methods 2015, 12, 773–779. [Google Scholar] [CrossRef]
- Soranno, A.; Holla, A.; Dingfelder, F.; Nettels, D.; Makarov, D.E.; Schuler, B. Integrated view of internal friction in unfolded proteins from single-molecule FRET, contact quenching, theory, and simulations. Proc. Natl. Acad. Sci. USA 2017, 114, E1833–E1839. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Förster resonance energy transfer. Science 2018, 359, eaan11333. [Google Scholar] [CrossRef]
- Fakhree, M.A.A.; Nolten, I.S.; Blum, C.; Claessens, M.M.A.E. Different Conformational Subensembles of the Intrinsically Disordered Protein α-Synuclein in Cells. J. Phys. Chem. Lett. 2018, 9, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Kang, J.H.; Girona, G.E.; Aramburu, I.V.; Lemke, E.A. Labeling proteins on live mammalian cells using click chemistry. Nat. Protoc. 2015, 10, 780–791. [Google Scholar] [CrossRef]
- Das, D.K.; Govindan, R.; Nikić-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018, 174, 926–937.e12. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Chang, M.-Y.; Jeon, D.-J.; Oh, D.-Y.; Son, H.; Lee, C.-H.; Lee, Y.-S.; Lee, Y.-S. The functional domains of dopamine transporter for cocaine analog, CFT binding. Exp. Mol. Med. 2002, 34, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Graeber, E.; Korkhov, V.M. Characterisation of the ligand binding sites in the translocator protein TSPO using the chimeric bacterial-mammalian constructs. Protein Expr. Purif. 2019, 164, 105456. [Google Scholar] [CrossRef]
- Guilfoyle, A.P.; Deshpande, C.N.; Font Sadurni, J.; Ash, M.-R.; Tourle, S.; Schenk, G.; Maher, M.J.; Jormakka, M. A GTPase chimera illustrates an uncoupled nucleotide affinity and release rate, providing insight into the activation mechanism. Biophys. J. 2014, 107, L45–L48. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).