Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
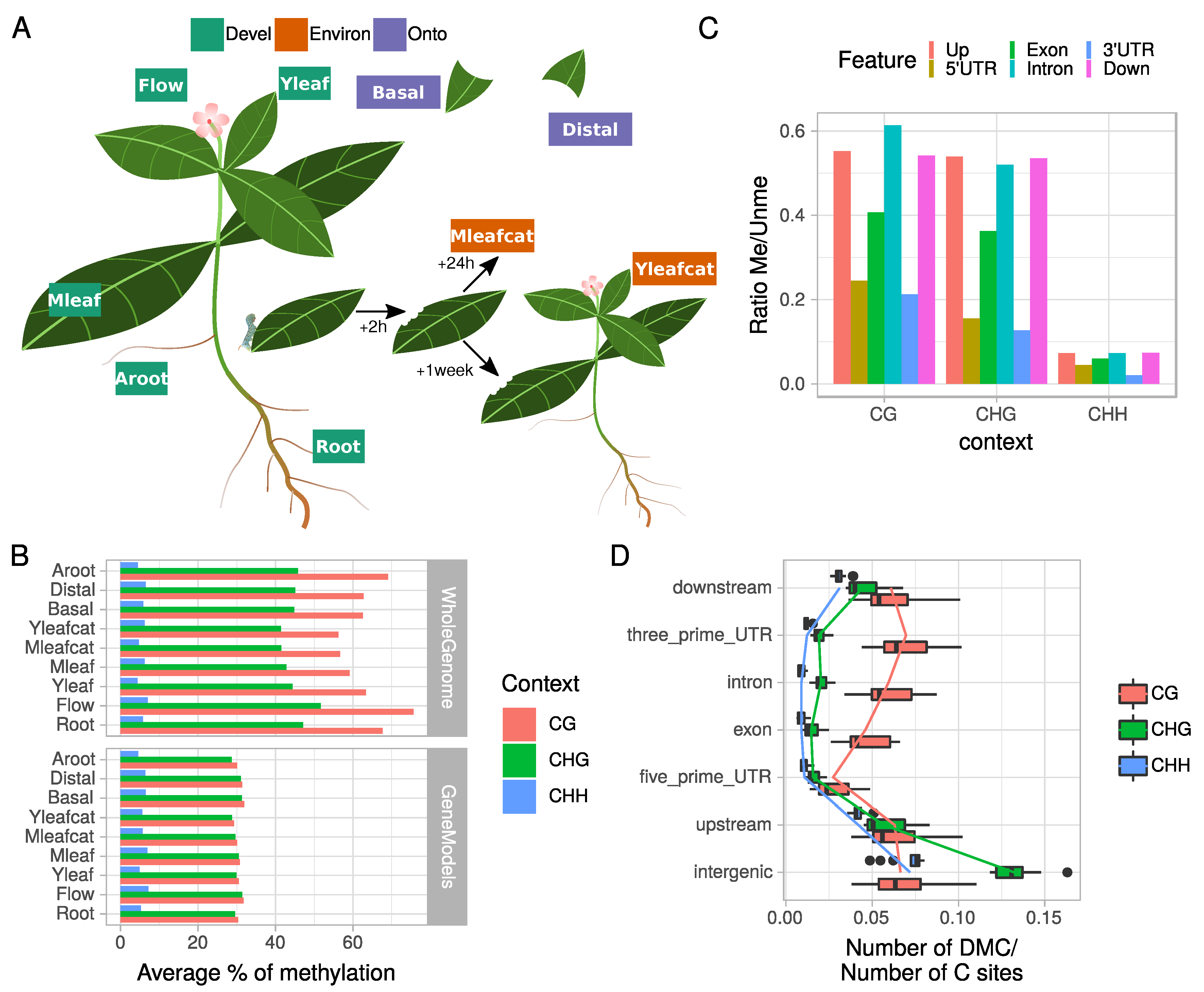
2.1. C. roseus Tissue-Specific Methylome Is Characterized by Context-Dependent Variations within Genes and Their Flanking Regions
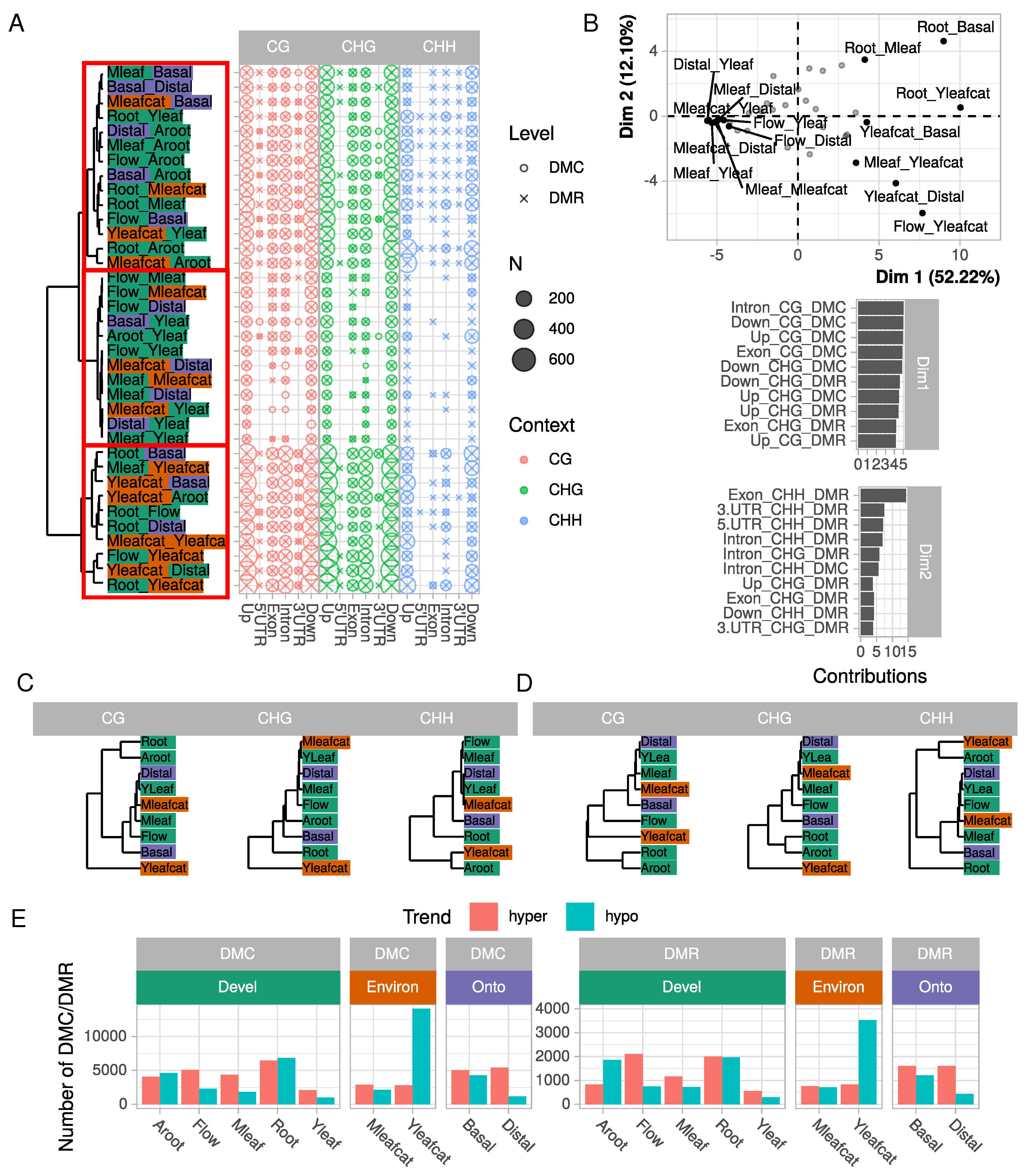
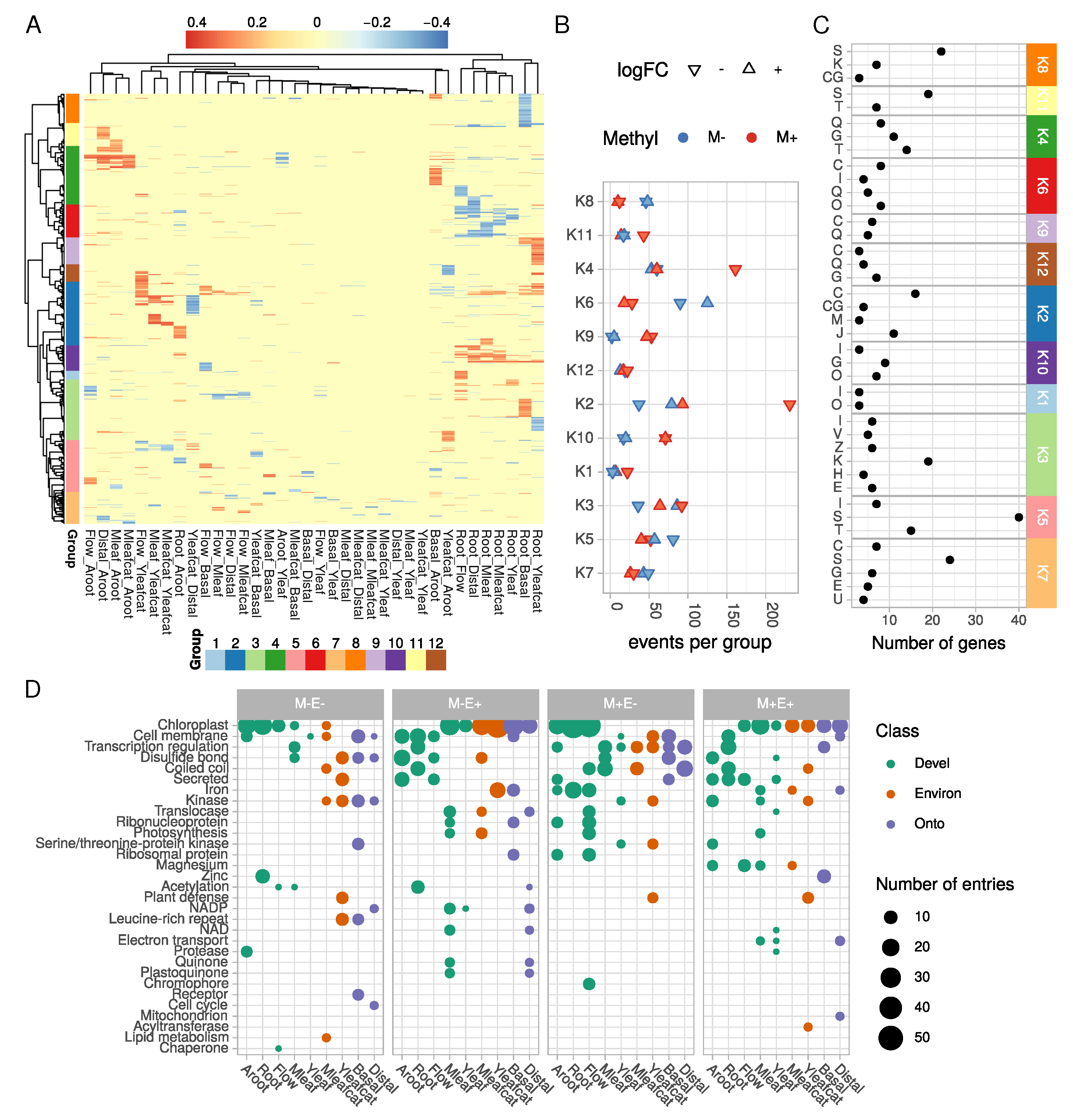
2.2. Tissue-Specific Covariations between Context-Dependent DNA Methylation and Differential Gene Expression May Affect Cellular and Physiological Functions
2.2.1. Photosynthesis
2.2.2. Defense Response
2.2.3. Cell Signaling
2.2.4. Specialized Metabolism
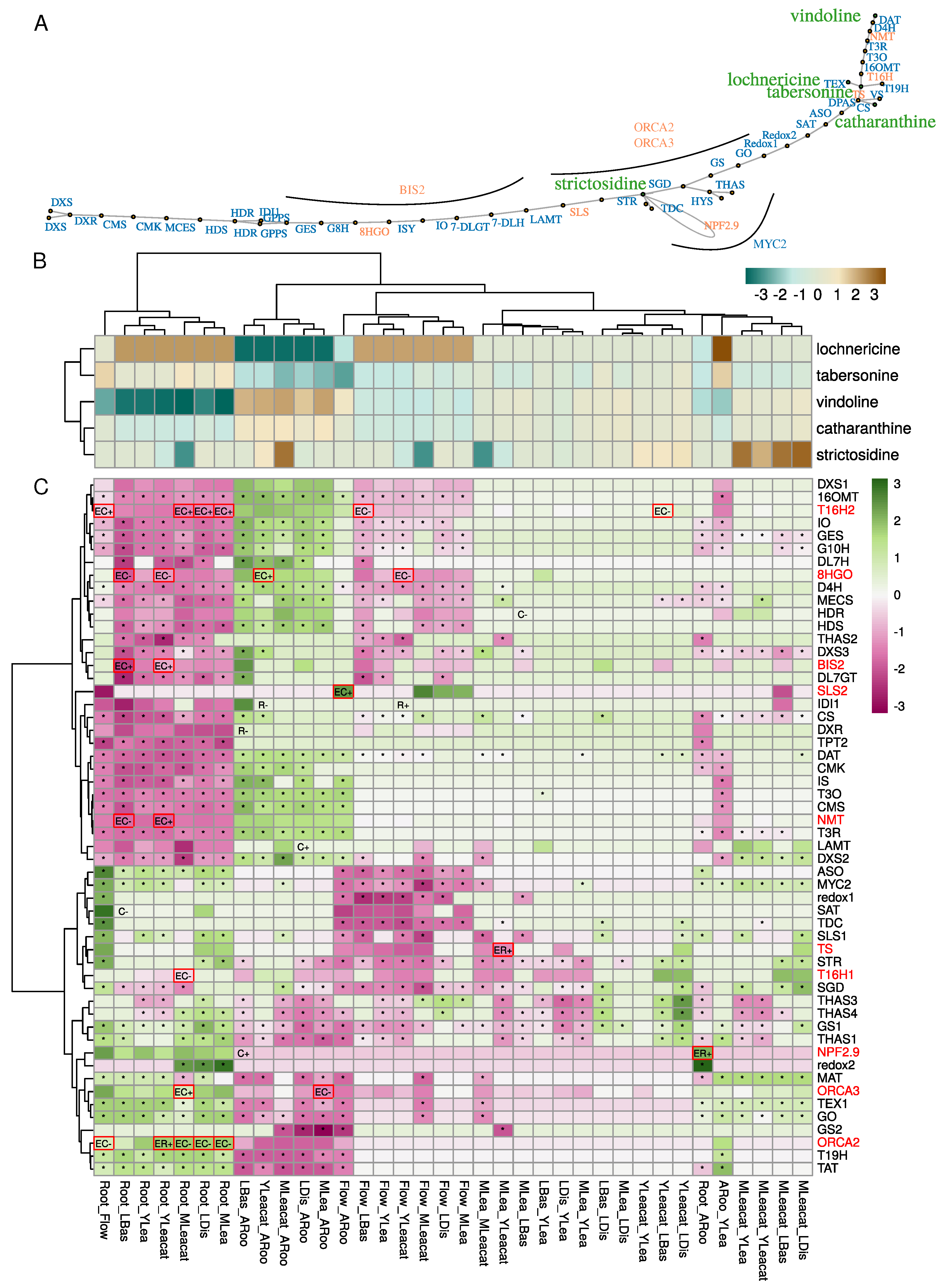
2.3. Control of the Monoterpene Indole Alkaloid (MIA) Pathway Involves Variations in DNA Methylation in Genes Encoding Enzymes and Transcription Factors
3. Materials and Methods
3.1. Plant Material
3.2. Whole Genome Bisulfite Sequencing (WGBS)
3.3. Epigenomics Data Generation and Analysis
3.4. Transcriptome Analysis
3.5. Metabolite Analysis
3.6. Statistical Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| WGBS | Whole-Genome Bisulfite Sequencing |
| DMC | Differentially Methylated Cytosine |
| DMR | Differentially Methylated Region |
| DM | Differentially Methylated |
| DEG | Differentially Expressed Gene |
| DEM | Differentially Expressed and Methylated |
| MIA | Monoterpene Indole Alkaloid |
| GO | Gene Ontology |
References
- Courdavault, V.; Papon, N.; Clastre, M.; Giglioli-Guivarc’h, N.; St-Pierre, B.; Burlat, V. A look inside an alkaloid multisite plant: The Catharanthus logistics. Curr. Opin. Plant Biol. 2014, 19, 43–50. [Google Scholar] [CrossRef]
- Dugé de Bernonville, T.; Clastre, M.; Besseau, S.; Oudin, A.; Burlat, V.; Glévarec, G.; Lanoue, A.; Papon, N.; Giglioli-Guivarc’h, N.; St-Pierre, B.; et al. Phytochemical genomics of the Madagascar periwinkle: Unravelling the last twists of the alkaloid engine. Phytochemistry 2015, 113, 9–23. [Google Scholar] [CrossRef]
- Franke, J.; Kim, J.; Hamilton, J.P.; Zhao, D.; Pham, G.M.; Wiegert-Rininger, K.; Crisovan, E.; Newton, L.; Vaillancourt, B.; Tatsis, E.; et al. Gene discovery in gelsemium highlights conserved gene clusters in monoterpene indole alkaloid biosynthesis. Chembiochem 2019, 20, 83–87. [Google Scholar] [CrossRef]
- Kellner, F.; Kim, J.; Clavijo, B.J.; Hamilton, J.P.; Childs, K.L.; Vaillancourt, B.; Cepela, J.; Habermann, M.; Steuernagel, B.; Clissold, L.; et al. Genome-guided investigation of plant natural product biosynthesis. Plant J. Cell Mol. Biol. 2015, 82, 680–692. [Google Scholar] [CrossRef]
- She, J.; Yan, H.; Yang, J.; Xu, W.; Su, Z. croFGD: Catharanthus roseus Functional Genomics Database. Front. Genet. 2019, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Besseau, S.; Kellner, F.; Lanoue, A.; Thamm, A.M.K.; Salim, V.; Schneider, B.; Geu-Flores, F.; Höfer, R.; Guirimand, G.; Guihur, A.; et al. A pair of tabersonine 16-hydroxylases initiates the synthesis of vindoline in an organ-dependent manner in Catharanthus roseus. Plant Physiol. 2013, 163, 1792–1803. [Google Scholar] [CrossRef]
- St-Pierre, B.; Vazquez-Flota, F.; De Luca, V. Multicellular compartmentation of catharanthus roseus alkaloid biosynthesis predicts intercellular translocation of a pathway intermediate. Plant Cell 1999, 11, 887–900. [Google Scholar] [CrossRef]
- Qu, Y.; Easson, M.L.A.E.; Froese, J.; Simionescu, R.; Hudlicky, T.; De Luca, V. Completion of the seven-step pathway from tabersonine to the anticancer drug precursor vindoline and its assembly in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 6224–6229. [Google Scholar] [CrossRef]
- Carqueijeiro, I.; Brown, S.; Chung, K.; Dang, T.T.; Walia, M.; Besseau, S.; Dugé de Bernonville, T.; Oudin, A.; Lanoue, A.; Billet, K.; et al. Two Tabersonine 6,7-Epoxidases Initiate Lochnericine-Derived Alkaloid Biosynthesis in Catharanthus roseus. Plant Physiol. 2018, 177, 1473–1486. [Google Scholar] [CrossRef]
- Carqueijeiro, I.; Dugé de Bernonville, T.; Lanoue, A.; Dang, T.T.; Teijaro, C.N.; Paetz, C.; Billet, K.; Mosquera, A.; Oudin, A.; Besseau, S.; et al. A BAHD acyltransferase catalyzing 19-O-acetylation of tabersonine derivatives in roots of Catharanthus roseus enables combinatorial synthesis of monoterpene indole alkaloids. Plant J. Cell Mol. Biol. 2018, 94, 469–484. [Google Scholar] [CrossRef]
- Caputi, L.; Franke, J.; Farrow, S.C.; Chung, K.; Payne, R.M.E.; Nguyen, T.D.; Dang, T.T.T.; Soares Teto Carqueijeiro, I.; Koudounas, K.; Dugé de Bernonville, T.; et al. Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 2018, 360, 1235–1239. [Google Scholar] [CrossRef]
- Qu, Y.; Easson, M.E.A.M.; Simionescu, R.; Hajicek, J.; Thamm, A.M.K.; Salim, V.; De Luca, V. Solution of the multistep pathway for assembly of corynanthean, strychnos, iboga, and aspidosperma monoterpenoid indole alkaloids from 19E-geissoschizine. Proc. Natl. Acad. Sci. USA 2018, 115, 3180–3185. [Google Scholar] [CrossRef]
- Srivastava, S.; Pandey, R.; Kumar, S.; Nautiyal, C.S. Correspondence between flowers and leaves in terpenoid indole alkaloid metabolism of the phytoplasma-infected Catharanthus roseus plants. Protoplasma 2014, 251, 1307–1320. [Google Scholar] [CrossRef]
- Dugé de Bernonville, T.; Carqueijeiro, I.; Lanoue, A.; Lafontaine, F.; Sánchez Bel, P.; Liesecke, F.; Musset, K.; Oudin, A.; Glévarec, G.; Pichon, O.; et al. Folivory elicits a strong defense reaction in Catharanthus roseus: Metabolomic and transcriptomic analyses reveal distinct local and systemic responses. Sci. Rep. 2017, 7, 40453. [Google Scholar] [CrossRef]
- Van Moerkercke, A.; Steensma, P.; Schweizer, F.; Pollier, J.; Gariboldi, I.; Payne, R.; Vanden Bossche, R.; Miettinen, K.; Espoz, J.; Purnama, P.C.; et al. The bHLH transcription factor BIS1 controls the iridoid branch of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Proc. Natl. Acad. Sci. USA 2015, 112, 8130–8135. [Google Scholar] [CrossRef]
- Van Moerkercke, A.; Steensma, P.; Gariboldi, I.; Espoz, J.; Purnama, P.C.; Schweizer, F.; Miettinen, K.; Vanden Bossche, R.; De Clercq, R.; Memelink, J.; et al. The basic helix-loop-helix transcription factor BIS2 is essential for monoterpenoid indole alkaloid production in the medicinal plant Catharanthus roseus. Plant J. Cell Mol. Biol. 2016, 88, 3–12. [Google Scholar] [CrossRef]
- Zhang, H.; Hedhili, S.; Montiel, G.; Zhang, Y.; Chatel, G.; Pré, M.; Gantet, P.; Memelink, J. The basic helix-loop-helix transcription factor CrMYC2 controls the jasmonate-responsive expression of the ORCA genes that regulate alkaloid biosynthesis in Catharanthus roseus. Plant J. Cell Mol. Biol. 2011, 67, 61–71. [Google Scholar] [CrossRef]
- Menke, F.L.; Champion, A.; Kijne, J.W.; Memelink, J. A novel jasmonate- and elicitor-responsive element in the periwinkle secondary metabolite biosynthetic gene Str interacts with a jasmonate- and elicitor-inducible AP2-domain transcription factor, ORCA2. EMBO J. 1999, 18, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- Van der Fits, L.; Memelink, J. ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science 2000, 289, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Singh, S.K.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2017, 213, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Patra, B.; Pattanaik, S.; Wang, Y.; Yuan, L. GATA and Phytochrome Interacting Factor Transcription Factors Regulate Light-Induced Vindoline Biosynthesis in Catharanthus roseus. Plant Physiol. 2019, 180, 1336–1350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, M.; Wen, W.; Yu, R. Biosynthesis and regulation of terpenoid indole alkaloids in Catharanthus roseus. Pharmacogn. Rev. 2015, 9, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. Epigenetics—A historical perspective. In Plant Epigenetics Coming of Age for Breeding Applications; Elsevier: Amsterdam, The Netherlands, 2018; Volume 88, pp. 1–19. [Google Scholar]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Reviews. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Vidalis, A.; Živković, D.; Wardenaar, R.; Roquis, D.; Tellier, A.; Johannes, F. Methylome evolution in plants. Genome Biol. 2016, 17, 264. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Takuno, S.; Ran, J.H.; Gaut, B.S. Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2016, 2, 15222. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.Q.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef]
- Kooke, R.; Morgado, L.; Becker, F.; van Eekelen, H.; Hazarika, R.; Zheng, Q.; de Vos, R.C.H.; Johannes, F.; Keurentjes, J.J.B. Epigenetic mapping of the Arabidopsis metabolome reveals mediators of the epigenotype-phenotype map. Genome Res. 2019, 29, 96–106. [Google Scholar] [CrossRef]
- Leung, J.; Gaudin, V. Who Rules the Cell? An Epi-Tale of Histone, DNA, RNA, and the Metabolic Deep State. Front. Plant Sci. 2020, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Li, W.F.; Ning, G.X.; Mao, J.; Guo, Z.G.; Zhou, Q.; Chen, B.H. Whole-genome DNA methylation patterns and complex associations with gene expression associated with anthocyanin biosynthesis in apple fruit skin. Planta 2019, 250, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Xiao, P.G. Deep in shadows: Epigenetic and epigenomic regulations of medicinal plants. Chin. Herbal Med. 2018. [Google Scholar] [CrossRef]
- Yang, D.; Huang, Z.; Jin, W.; Xia, P.; Jia, Q.; Yang, Z.; Hou, Z.; Zhang, H.; Ji, W.; Han, R. DNA methylation: A new regulator of phenolic acids biosynthesis in Salvia miltiorrhiza. Ind. Crops Prod. 2018, 124, 402–411. [Google Scholar] [CrossRef]
- Bulut, B.; Aydinli, Z.; Türktaş-Erken, M. MSAP analysis reveals diverse epigenetic statuses in opium poppy varieties with different benzyisoquinoline alkaloid content. Turk. J. Biol. Turk Biyol. Derg. Sci. Tech. Res. Counc. Turk. 2020, 44, 103–109. [Google Scholar] [CrossRef]
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenetics Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Song, Q.X.; Lu, X.; Li, Q.T.; Chen, H.; Hu, X.Y.; Ma, B.; Zhang, W.K.; Chen, S.Y.; Zhang, J.S. Genome-wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef]
- Ausin, I.; Feng, S.; Yu, C.; Liu, W.; Kuo, H.Y.; Jacobsen, E.L.; Zhai, J.; Gallego-Bartolome, J.; Wang, L.; Egertsdotter, U.; et al. DNA methylome of the 20-gigabase Norway spruce genome. Proc. Natl. Acad. Sci. USA 2016, 113, E8106–E8113. [Google Scholar] [CrossRef]
- Roessler, K.; Takuno, S.; Gaut, B.S. CG Methylation Covaries with Differential Gene Expression between Leaf and Floral Bud Tissues of Brachypodium distachyon. PLoS ONE 2016, 11, e0150002. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.T.S.; Inzé, D.; Nelissen, H. Differential methylation during maize leaf growth targets developmentally regulated genes. Plant Physiol. 2014, 164, 1350–1364. [Google Scholar] [CrossRef]
- Vining, K.J.; Pomraning, K.R.; Wilhelm, L.J.; Priest, H.D.; Pellegrini, M.; Mockler, T.C.; Freitag, M.; Strauss, S.H. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: Tissue-level variation and relationship to gene expression. BMC Genom. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- González, B.; Vera, P. Folate Metabolism Interferes with Plant Immunity through 1C Methionine Synthase-Directed Genome-wide DNA Methylation Enhancement. Mol. Plant 2019, 12, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Lindermayr, C.; Rudolf, E.E.; Durner, J.; Groth, M. Interactions between metabolism and chromatin in plant models. Molecular Metabolism 2020, 38, 100951. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Placette, C.; Le Gac, A.L.; Chauveau, D.; Segura, V.; Delaunay, A.; Lesage-Descauses, M.C.; Hummel, I.; Cohen, D.; Jesson, B.; Le Thiec, D.; et al. Changes in the epigenome and transcriptome of the poplar shoot apical meristem in response to water availability affect preferentially hormone pathways. J. Exp. Bot. 2018, 69, 537–551. [Google Scholar] [CrossRef]
- Feng, S.J.; Liu, X.S.; Tao, H.; Tan, S.K.; Chu, S.S.; Oono, Y.; Zhang, X.D.; Chen, J.; Yang, Z.M. Variation of DNA methylation patterns associated with gene expression in rice (Oryza sativa) exposed to cadmium. Plant Cell Environ. 2016, 39, 2629–2649. [Google Scholar] [CrossRef]
- N’Diaye, A.; Byrns, B.; Cory, A.T.; Nilsen, K.T.; Walkowiak, S.; Sharpe, A.; Robinson, S.J.; Pozniak, C.J. Machine learning analyses of methylation profiles uncovers tissue-specific gene expression patterns in wheat. Plant Genome 2020. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA methylation in context: From genomes to gene expression in plants. Biochim. Et Biophys. Acta Gene Regul. Mech. 2017, 1860, 149–156. [Google Scholar] [CrossRef]
- Zhang, Y.; Wendte, J.M.; Ji, L.; Schmitz, R.J. Natural variation in DNA methylation homeostasis and the emergence of epialleles. Proc. Natl. Acad. Sci. USA 2020, 117, 4874–4884. [Google Scholar] [CrossRef]
- Al-Harrasi, I.; Al-Yahyai, R.; Yaish, M.W. Differential DNA methylation and transcription profiles in date palm roots exposed to salinity. PLoS ONE 2018, 13, e0191492. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, Y.; Chang, X.; Jing, S.; Zhang, Q.; You, C.; Yuan, H.; Wang, H. DNA methylome analysis provides evidence that the expansion of the tea genome is linked to TE bursts. Plant Biotechnol. J. 2019, 17, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Zhang, Y.; Wendte, J.M.; Zhang, X.; Schmitz, R.J. Evolutionary and experimental loss of gene body methylation and its consequence to gene expression. G3 Bethesda Md. 2019, 9, 2441–2445. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Laenen, B.; Takuno, S.; Slotte, T. Single-cell expression noise and gene-body methylation in Arabidopsis thaliana. Heredity 2019, 123, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef]
- Ngernprasirtsiri, J.; Kobayashi, H.; Akazawa, T. Transcriptional regulation and DNA methylation of nuclear genes for photosynthesis in nongreen plant cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7919–7923. [Google Scholar] [CrossRef]
- Ma, K.; Song, Y.; Jiang, X.; Zhang, Z.; Li, B.; Zhang, D. Photosynthetic response to genome methylation affects the growth of Chinese white poplar. Tree Genet. Genomes 2012, 8, 1407–1421. [Google Scholar] [CrossRef]
- Perrin, A.; Daccord, N.; Roquis, D.; Celton, J.M.; Vergne, E.; Bucher, E. Divergent DNA methylation signatures of juvenile seedlings, grafts and adult apple trees. Epigenomes 2020, 4, 4. [Google Scholar] [CrossRef]
- Attaran, E.; Major, I.T.; Cruz, J.A.; Rosa, B.A.; Koo, A.J.K.; Chen, J.; Kramer, D.M.; He, S.Y.; Howe, G.A. Temporal dynamics of growth and photosynthesis suppression in response to jasmonate signaling. Plant Physiol. 2014, 165, 1302–1314. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Ngom, B.; Mamati, E.; Goudiaby, M.F.; Kimatu, J.; Sarr, I.; Diouf, D.; Kane, N.A. Methylation analysis revealed salicylic acid affects pearl millet defense through external cytosine DNA demethylation. J. Plant Interact. 2018, 13, 288–293. [Google Scholar] [CrossRef]
- Koch, M.; Vorwerk, S.; Masur, C.; Sharifi-Sirchi, G.; Olivieri, N.; Schlaich, N.L. A role for a flavin-containing mono-oxygenase in resistance against microbial pathogens in Arabidopsis. Plant J. Cell Mol. Biol. 2006, 47, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, A.; Möller, B.; Liu, W.; Kientz, M.; Flipse, J.; Rademacher, E.H.; Schmid, M.; Jürgens, G.; Weijers, D. MONOPTEROS controls embryonic root initiation by regulating a mobile transcription factor. Nature 2010, 464, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Tognetti, V.B.; Van Aken, O.; Morreel, K.; Vandenbroucke, K.; van de Cotte, B.; De Clercq, I.; Chiwocha, S.; Fenske, R.; Prinsen, E.; Boerjan, W.; et al. Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell 2010, 22, 2660–2679. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, C.; Zhu, J.K.; Yang, Z. Epigenetic modifications and plant hormone action. Mol. Plant 2016, 9, 57–70. [Google Scholar] [CrossRef]
- Kim, J.; Kang, S.H.; Park, S.G.; Yang, T.J.; Lee, Y.; Kim, O.T.; Chung, O.; Lee, J.; Choi, J.P.; Kwon, S.J.; et al. Whole-genome, transcriptome, and methylome analyses provide insights into the evolution of platycoside biosynthesis in Platycodon grandiflorus, a medicinal plant. Hortic. Res. 2020, 7, 112. [Google Scholar] [CrossRef]
- Colinas, M.; Goossens, A. Combinatorial transcriptional control of plant specialized metabolism. Trends Plant Sci. 2018, 23, 324–336. [Google Scholar] [CrossRef]
- Van der Fits, L.; Memelink, J. The jasmonate-inducible AP2/ERF-domain transcription factor ORCA3 activates gene expression via interaction with a jasmonate-responsive promoter element. Plant J. Cell Mol. Biol. 2001, 25, 43–53. [Google Scholar] [CrossRef]
- Kanaoka, M.M.; Pillitteri, L.J.; Fujii, H.; Yoshida, Y.; Bogenschutz, N.L.; Takabayashi, J.; Zhu, J.K.; Torii, K.U. SCREAM/ICE1 and SCREAM2 specify three cell-state transitional steps leading to arabidopsis stomatal differentiation. Plant Cell 2008, 20, 1775–1785. [Google Scholar] [CrossRef]
- Billingsley, J.M.; DeNicola, A.B.; Barber, J.S.; Tang, M.C.; Horecka, J.; Chu, A.; Garg, N.K.; Tang, Y. Engineering the biocatalytic selectivity of iridoid production in Saccharomyces cerevisiae. Metab. Eng. 2017, 44, 117–125. [Google Scholar] [CrossRef]
- Dugé de Bernonville, T.; Foureau, E.; Parage, C.; Lanoue, A.; Clastre, M.; Londono, M.A.; Oudin, A.; Houillé, B.; Papon, N.; Besseau, S.; et al. Characterization of a second secologanin synthase isoform producing both secologanin and secoxyloganin allows enhanced de novo assembly of a Catharanthus roseus transcriptome. BMC Genom. 2015, 16, 619. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.M.E.; Xu, D.; Foureau, E.; Teto Carqueijeiro, M.I.S.; Oudin, A.; Bernonville, T.D.D.; Novak, V.; Burow, M.; Olsen, C.E.; Jones, D.M.; et al. An NPF transporter exports a central monoterpene indole alkaloid intermediate from the vacuole. Nat. Plants 2017, 3, 16208. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Safonova, O.; De Luca, V. Completion of the canonical pathway for assembly of anticancer drugs vincristine/vinblastine in Catharanthus roseus. Plant J. Cell Mol. Biol. 2019, 97, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Daviaud, C.; Renault, V.; Mauger, F.; Deleuze, J.F.; Tost, J. Whole-Genome Bisulfite Sequencing Using the Ovation®Ultralow Methyl-Seq Protocol. Methods Mol. Biol. 2018, 1708, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Ghangal, R.; Sharma, R.; Sinha, A.K.; Jain, M. Transcriptome analysis of Catharanthus roseus for gene discovery and expression profiling. PLoS ONE 2014, 9, e103583. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Grehl, C.; Wagner, M.; Lemnian, I.; Glaser, B.; Grosse, I. Performance of Mapping Approaches for Whole-Genome Bisulfite Sequencing Data in Crop Plants. Front. Plant Sci. 2020, 11, 176. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2020. Available online: http://www.R-project.org/ (accessed on 20 August 2020).
- Chen, Y.; Pal, B.; Visvader, J.E.; Smyth, G.K. Differential methylation analysis of reduced representation bisulfite sequencing experiments using edgeR. F1000Research 2017, 6, 2055. [Google Scholar] [CrossRef]
- Wu, H.; Xu, T.; Feng, H.; Chen, L.; Li, B.; Yao, B.; Qin, Z.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Dowle, M.; Srinivasan, A. Data.Table: Extension of ‘data.frame’. 2019. Available online: http://CRAN.R-project.org/package=data.table (accessed on 20 August 2020).
- Wickham, H. ggplot2-Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- De Vries, A.; Ripley, B.D. ggdendro: Create Dendrograms and Tree Diagrams Using ’ggplot2’. 2016. Available online: http://CRAN.R-project.org/package=ggdendro (accessed on 20 August 2020).
- Slowikowski, K. ggrepel: Automatically Position Non-Overlapping Text Labels with ’ggplot2’. 2019. Available online: http://CRAN.R-project.org/package=ggrepel (accessed on 20 August 2020).
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: http://CRAN.R-project.org/package=pheatmap (accessed on 20 August 2020).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. 2006, 1695, 1–9. [Google Scholar]
- Maury, S.; Sow, M.D.; Le Gac, A.L.; Genitoni, J.; Lafon-Placette, C.; Mozgova, I. Phytohormone and chromatin crosstalk: The missing link for developmental plasticity? Front. Plant Sci. 2019, 10, 395. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Best Hit in Uniprot Database (blastx) | % id | e-Value | Comparison | LogFC | Methylation Difference |
|---|---|---|---|---|---|---|
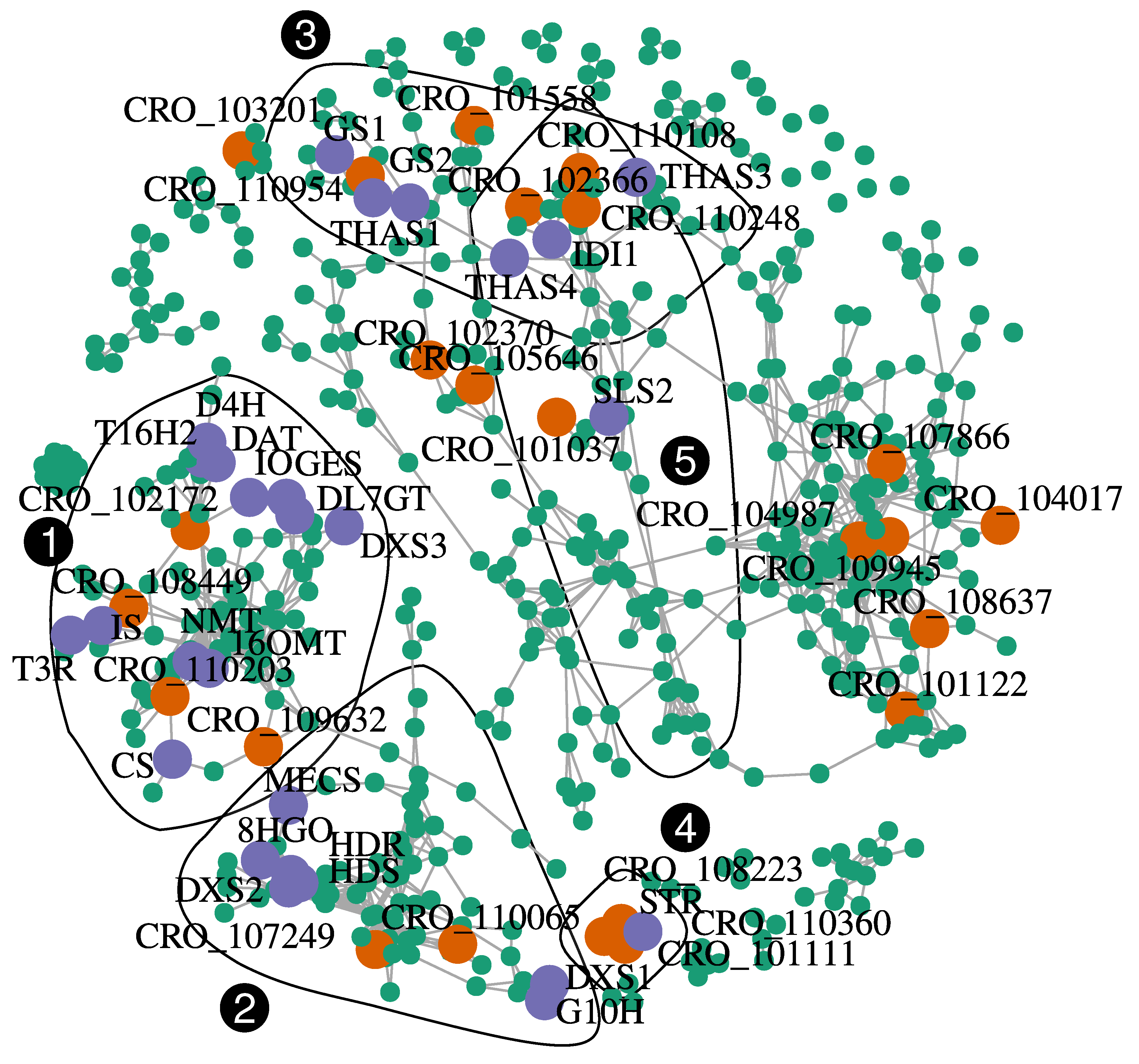
| Community 1 (GES, IS, IO, and DL7GT, T16H2, 16OMT, T3R, NMT, D4H, and DAT, vindoline-related community) | ||||||
| CRO_T102172 | DOF56_ARATH, Dof zinc finger protein DOF5.6 (AtDOF5.6) | 71.77 | 4 | Distal vs. Aroot | 2.54 | 0.31 |
| CRO_T109632 | BH048_ARATH, Transcription factor bHLH48 (AtbHLH48) | 40.55 | 3 | Root vs. Basal | −2.27 | 0.29 |
| CRO_T110203 | WOX1_ARATH, WUSCHEL-related homeobox 1 | 56.95 | 4 | Root vs. Yleafcat | −9.16 | 0.22 |
| Community 2 (DXS1, DXS2, HDS, HDR, MECS, G10H, and 8HGO; terpene precursor community) | ||||||
| CRO_T107249 | UNE10_ARATH, Transcription factor UNE10 (AtbHLH16) (bHLH 16) | 52.71 | 5 | Root vs. Mleaf | −8.76 | 0.19 |
| CRO_T107392 | MYB06_ANTMA, Myb-related protein 306 | 61.43 | 3 | Root vs. Flower | −6.72 | −0.29 |
| CRO_T110065 | UNE10_ARATH, Transcription factor UNE10 (AtbHLH16) (bHLH 16) | 46.13 | 2 | Yleafcat vs. Aroot | 2.44 | 0.24 |
| Community 3 (GS1, GS2, THAS1, THAS3, and THAS4, strictosidine aglycone acting enzymes) | ||||||
| CRO_T110954 | BH113_ARATH, Transcription factor bHLH113 (AtbHLH113) | 42.74 | 1 | Yleafcat vs. Aroot | −1.85 | 0.18 |
| Community 4 (ORCA3 and STR; control of strictosidine accumulation) | ||||||
| CRO_T101111 | BH030_ARATH, Transcription factor bHLH30 (AtbHLH30) | 40.18 | 5 | Root vs. Basal | 2.92 | −0.28 |
| CRO_T108223 | PTI6_SOLLC, Pathogenesis-related genes transcriptional activator PTI6 | 59.51 | 3 | Root vs. Yleafcat | 5.66 | 0.26 |
| Community 5 (SLS2 and IDI1) | ||||||
| CRO_T101037 | PCF5_ORYSJ, Transcription factor PCF5 | 85.92 | 4 | Flower vs. Yleafcat | 3.72 | −0.29 |
| CRO_T102366 | BH079_ARATH, Transcription factor bHLH79 (AtbHLH79) (bHLH 79) | 57.19 | 2 | Flower vs. Aroot | 5.46 | −0.24 |
| CRO_T110108 | YAB1_ARATH, Axial regulator YABBY 1 (Fl-54) | 65.12 | 3 | Mleafcat vs. Yleafcat | −2.41 | −0.32 |
| CRO_T110248 | ICE1_ARATH, Transcription factor ICE1 (AtbHLH116) | 60.62 | 2 | Flower vs. Mleafcat | 1.74 | −0.26 |
| Sample | Accession | Reference | Project Accession Number |
|---|---|---|---|
| Root WGBS RNA-seq | SRR11932631 SRR1271858 | This study [76] | PRJNA635601 PRJNA246273 |
| Adventitious root WGBS RNA-seq | SRR11932632 ERR2112587 | This study [10] | PRJNA635601 PRJEB22378 |
| Young Leaf WGBS RNA-seq | SRR11932629 ERR1512377 | This study [14] | PRJNA635601 PRJEB14626 |
| Mature Leaf WGBS RNA-seq | SRR11932637 ERR1512375 | This study [14] | PRJNA635601 PRJEB14626 |
| Mature leaf 24 h post folivory WGBS RNA-seq | SRR11932636 ERR1512373 | This study [14] | PRJNA635601 PRJEB14626 |
| Young leaf from folivory subjected plants WGBS RNA-seq | SRR11932635 ERR1512376 | This study [14] | PRJNA635601 PRJEB14626 |
| Flower WGBS RNA-seq | SRR11932630 ERR4235477 | This study This study | PRJNA635601 PRJEB38854 |
| Young Leaf basal part WGBS RNA-seq | SRR11932634 ERR4235478 | This study This study | PRJNA635601 PRJEB38854 |
| Young Leaf distal part WGBS RNA-seq | SRR11932633 ERR4235479 | This study This study | PRJNA635601 PRJEB38854 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugé de Bernonville, T.; Maury, S.; Delaunay, A.; Daviaud, C.; Chaparro, C.; Tost, J.; O’Connor, S.E.; Courdavault, V. Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation. Int. J. Mol. Sci. 2020, 21, 6028. https://doi.org/10.3390/ijms21176028
Dugé de Bernonville T, Maury S, Delaunay A, Daviaud C, Chaparro C, Tost J, O’Connor SE, Courdavault V. Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation. International Journal of Molecular Sciences. 2020; 21(17):6028. https://doi.org/10.3390/ijms21176028
Chicago/Turabian StyleDugé de Bernonville, Thomas, Stéphane Maury, Alain Delaunay, Christian Daviaud, Cristian Chaparro, Jörg Tost, Sarah Ellen O’Connor, and Vincent Courdavault. 2020. "Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation" International Journal of Molecular Sciences 21, no. 17: 6028. https://doi.org/10.3390/ijms21176028
APA StyleDugé de Bernonville, T., Maury, S., Delaunay, A., Daviaud, C., Chaparro, C., Tost, J., O’Connor, S. E., & Courdavault, V. (2020). Developmental Methylome of the Medicinal Plant Catharanthus roseus Unravels the Tissue-Specific Control of the Monoterpene Indole Alkaloid Pathway by DNA Methylation. International Journal of Molecular Sciences, 21(17), 6028. https://doi.org/10.3390/ijms21176028