The Roles of GRKs in Hemostasis and Thrombosis



{kind=link}
{kind=link}
Abstract
1. Introduction
2. GRKs in Platelets
3. The Role of GRKs and Arrestins during Platelet Activation
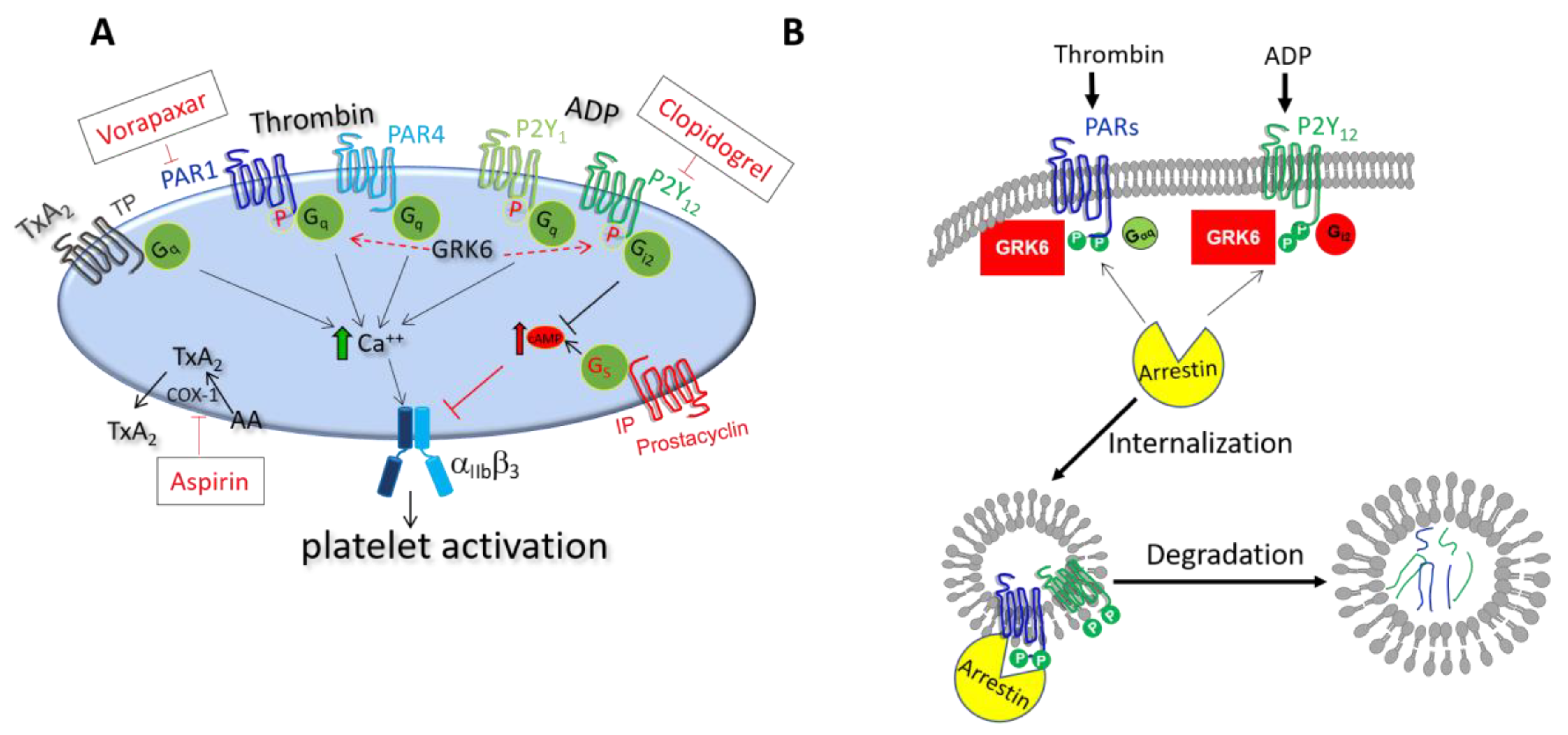
3.1. Role of GRK6 during Platelet Activation
3.1.1. Regulation of Thrombin Receptor Signaling by GRK6
3.1.2. Regulation of TxA2 Receptor Signaling by GRK6
3.1.3. Regulation of ADP Receptor Signaling by GRK6
3.2. Role of Arrestins during Platelet Activation
3.3. Regulation of GRKs in Platelets
4. The Function of GRKs during the Hemostatic Plug Formation
4.1. Regulation of GPCR Signaling at Site of Vascular Injury
4.2. GPCR Desensitization during Platelet Activation: An Old Question to Revisit
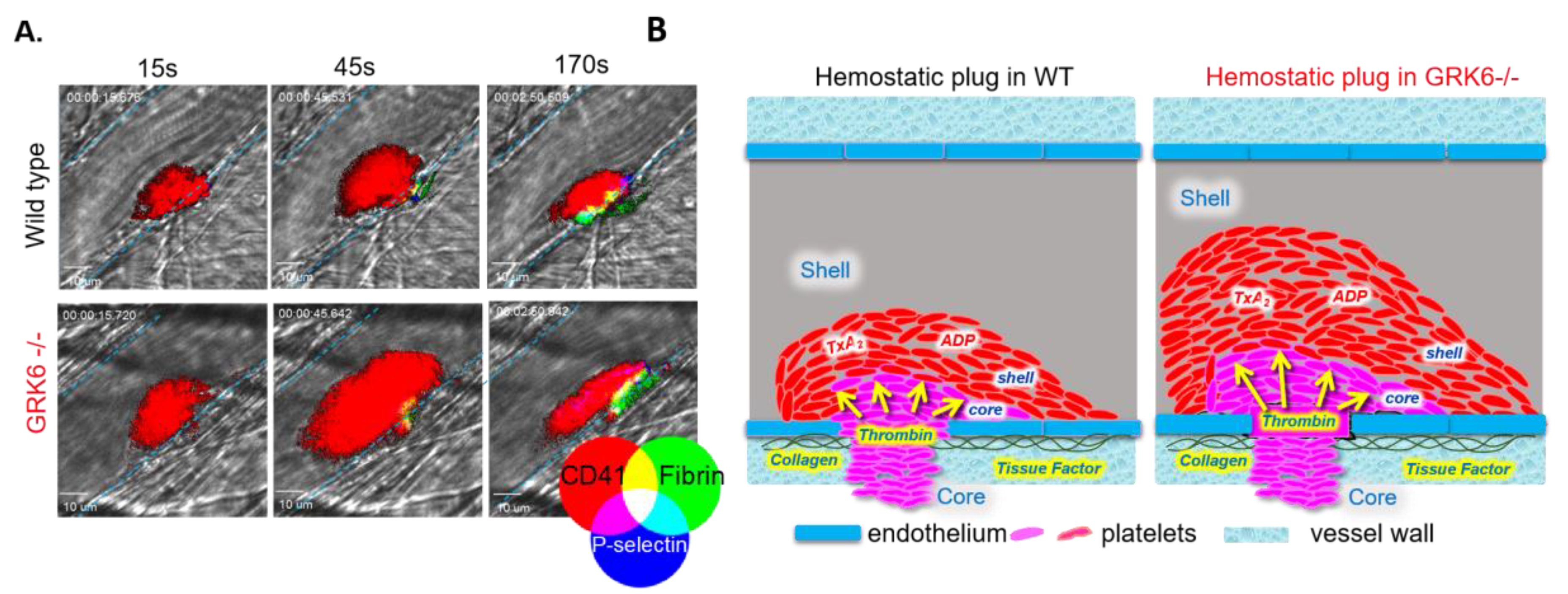
4.3. The In Vivo Consequences of Deletion of GRK6 at Site of Vascular Injury
5. GRKs in Human Pathology-Related Platelet Dysfunction
5.1. Antiplatelet Drugs Targeting at GPCRs and Their Regulators
5.2. GRKs Polymorphisms and Their Role in Cardiovascular Disease
5.3. COVID-19 and Thrombotic Events: A Future Direction for Studying GRKs?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| RGS | regulator of G protein signaling |
| GRK | G protein-coupled receptor kinase |
| GTPase | intrinsic guanosine triphosphatase |
| PAR | protease-activated receptor |
| GP1b | glycoprotein 1b |
| GPVI | glycoprotein VI |
| VWF | von Willebrand factor |
| ADP | adenosine 5′-diphosphate |
| ATP | adenosine triphosphate |
| COX | cyclooxygenase |
| TXA2 | thromboxane A2 |
| IP | prostacyclin receptor |
| PECAM | platelet endothelial cell adhesion molecule |
| iPSC | induced pluripotent stem cell |
| MEG-01 | megakaryoblastic cells |
| GWAS | genome-wide association studies |
| eQTL | expression quantitative trait loci |
| Ang II | angiotensin II |
| VTE | venous thromboembolism |
| ACE | angiotensin-converting enzyme |
| DVT | deep vein thrombosis |
| PE | pulmonary embolism |
| MPV | mean platelet volume |
| PDW | platelet volume distribution width |
| aPTT | activated partial thromboplastin time |
| PKC | protein kinase C |
| PP1 | protein phosphatase-1 |
| SPL | spinophilin |
| MAPK | mitogen-activated protein kinase |
| RAS | renin-angiotensin-system |
| WT | wild-type |
References
- Thon, J.N.; Italiano, J.E. Platelet formation. Semin. Hematol. 2010, 47, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Thon, J.N.; Montalvo, A.; Patel-Hett, S.; Devine, M.T.; Richardson, J.L.; Ehrlicher, A.; Larson, M.K.; Hoffmeister, K.; Hartwig, J.H.; Italiano, J.E., Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. J. Cell Biol. 2010, 191, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Guichard, J.; Masse, J.M.; Debili, N.; Cramer, E.M. Of mice and men: Comparison of the ultrastructure of megakaryocytes and platelets. Exp. Hematol. 2001, 29, 1295–1302. [Google Scholar] [CrossRef]
- Clemetson, K.J.; Clemetson, J.M. Platelet collagen receptors. Thromb. Haemost. 2001, 86, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Bryckaert, M.; Rosa, J.P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell Mol. Life Sci. 2015, 72, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 210, 59–85. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Farrell, D.H.; Weisel, J.W.; Bennett, J.S. The Platelet Integrin alphaIIbbeta3 Differentially Interacts with Fibrin Versus Fibrinogen. J. Biol. Chem. 2016, 291, 7858–7867. [Google Scholar] [CrossRef]
- Whiteheart, S.W. Platelet granules: Surprise packages. Blood 2011, 118, 1190–1191. [Google Scholar] [CrossRef]
- Needleman, P.; Moncada, S.; Bunting, S.; Vane, J.R.; Hamberg, M.; Samuelsson, B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 1976, 261, 558–560. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Hausdorff, W.P.; Caron, M.G.; Lefkowitz, R.J. Turning off the signal: Desensitization of beta-adrenergic receptor function. FASEB J. 1990, 4, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef]
- Ferguson, S.S.; Downey, W.E., 3rd; Colapietro, A.M.; Barak, L.S.; Menard, L.; Caron, M.G. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 1996, 271, 363–366. [Google Scholar] [CrossRef]
- Gao, B.; Mumby, S.; Gilman, A.G. The G protein beta 2 complementary DNA encodes the beta 35 subunit. J. Biol. Chem. 1987, 262, 17254–17257. [Google Scholar]
- Stewart, A.; Fisher, R.A. Introduction: G Protein-coupled Receptors and RGS Proteins. Prog. Mol. Biol. Transl. Sci. 2015, 133, 1–11. [Google Scholar] [CrossRef]
- Sterne-Marr, R.; Tesmer, J.J.; Day, P.W.; Stracquatanio, R.P.; Cilente, J.A.; O’Connor, K.E.; Pronin, A.N.; Benovic, J.L.; Wedegaertner, P.B. G protein-coupled receptor Kinase 2/G alpha q/11 interaction. A novel surface on a regulator of G protein signaling homology domain for binding G alpha subunits. J. Biol. Chem. 2003, 278, 6050–6058. [Google Scholar] [CrossRef]
- Tesmer, V.M.; Kawano, T.; Shankaranarayanan, A.; Kozasa, T.; Tesmer, J.J. Snapshot of activated G proteins at the membrane: The Galphaq-GRK2-Gbetagamma complex. Science 2005, 310, 1686–1690. [Google Scholar] [CrossRef]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Schumbrutzki, C.; Wortelkamp, S.; Sickmann, A.; Zahedi, R.P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteom. 2012, 75, 1454–1462. [Google Scholar] [CrossRef]
- Zeiler, M.; Moser, M.; Mann, M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol. Cell Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef]
- Hardy, A.R.; Conley, P.B.; Luo, J.; Benovic, J.L.; Poole, A.W.; Mundell, S.J. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Blood 2005, 105, 3552–3560. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gupta, S.; Cooper, M.; DeHelian, D.; Zhao, X.; Naik, M.U.; Wurtzel, J.G.T.; Stalker, T.J.; Goldfinger, L.E.; Benovic, J.; et al. GRK6 regulates the hemostatic response to injury through its rate-limiting effects on GPCR signaling in platelets. Blood Adv. 2020, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.H.; Park, K.M.; Kim, S. Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. Int. J. Mol. Sci. 2020, 21, 3932. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Bergmeier, W. Negative regulators of platelet activation and adhesion. J. Thromb. Haemost. JTH 2018, 16, 220–230. [Google Scholar] [CrossRef]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996, 51, 319–351; discussion 352–353. [Google Scholar]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef]
- Komolov, K.E.; Benovic, J.L. G protein-coupled receptor kinases: Past, present and future. Cell Signal 2018, 41, 17–24. [Google Scholar] [CrossRef]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Potz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef]
- Solari, F.A.; Mattheij, N.J.; Burkhart, J.M.; Swieringa, F.; Collins, P.W.; Cosemans, J.M.; Sickmann, A.; Heemskerk, J.W.; Zahedi, R.P. Combined Quantification of the Global Proteome, Phosphoproteome, and Proteolytic Cleavage to Characterize Altered Platelet Functions in the Human Scott Syndrome. Mol. Cell Proteom. 2016, 15, 3154–3169. [Google Scholar] [CrossRef]
- Traynham, C.J.; Hullmann, J.; Koch, W.J. Canonical and non-canonical actions of GRK5 in the heart. J. Mol. Cell Cardiol. 2016, 92, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.M.; Koch, W.J. Noncanonical Roles of G Protein-coupled Receptor Kinases in Cardiovascular Signaling. J. Cardiovasc. Pharmacol. 2017, 70, 129–141. [Google Scholar] [CrossRef]
- Mayor, F., Jr.; Cruces-Sande, M.; Arcones, A.C.; Vila-Bedmar, R.; Briones, A.M.; Salaices, M.; Murga, C. G protein-coupled receptor kinase 2 (GRK2) as an integrative signalling node in the regulation of cardiovascular function and metabolic homeostasis. Cell Signal 2017, 41, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Gomez, J. Molecular cloning and expression of GRK6. A new member of the G protein-coupled receptor kinase family. J. Biol. Chem. 1993, 268, 19521–19527. [Google Scholar] [PubMed]
- Han, X.; Nieman, M.T. PAR4 (Protease-Activated Receptor 4): PARticularly Important 4 Antiplatelet Therapy. Arter. Thromb. Vasc. Biol. 2018, 38, 287–289. [Google Scholar] [CrossRef]
- Posma, J.J.; Grover, S.P.; Hisada, Y.; Owens, A.P., 3rd; Antoniak, S.; Spronk, H.M.; Mackman, N. Roles of Coagulation Proteases and PARs (Protease-Activated Receptors) in Mouse Models of Inflammatory Diseases. Arter. Thromb. Vasc. Biol. 2019, 39, 13–24. [Google Scholar] [CrossRef]
- Shapiro, M.J.; Weiss, E.J.; Faruqi, T.R.; Coughlin, S.R. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J. Biol. Chem. 2000, 275, 25216–25221. [Google Scholar] [CrossRef]
- Covic, L.; Gresser, A.L.; Kuliopulos, A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000, 39, 5458–5467. [Google Scholar] [CrossRef]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Zheng, Y.W.; Sulciner, D.J.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404, 609–613. [Google Scholar] [CrossRef]
- Nakahata, N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharm. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef]
- Parent, J.L.; Labrecque, P.; Orsini, M.J.; Benovic, J.L. Internalization of the TXA2 receptor alpha and beta isoforms. Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J. Biol. Chem. 1999, 274, 8941–8948. [Google Scholar] [CrossRef] [PubMed]
- Murugappan, S.; Shankar, H.; Kunapuli, S.P. Platelet receptors for adenine nucleotides and thromboxane A2. Semin. Thromb. Hemost. 2004, 30, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Penn, R.B.; Pronin, A.N.; Benovic, J.L. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc. Med. 2000, 10, 81–89. [Google Scholar] [CrossRef]
- Paing, M.M.; Johnston, C.A.; Siderovski, D.P.; Trejo, J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell Biol. 2006, 26, 3231–3242. [Google Scholar] [CrossRef]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Wilden, U.; Hall, S.W.; Kuhn, H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc. Natl. Acad. Sci. USA 1986, 83, 1174–1178. [Google Scholar] [CrossRef]
- Krasel, C.; Bunemann, M.; Lorenz, K.; Lohse, M.J. Beta-arrestin binding to the beta2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 2005, 280, 9528–9535. [Google Scholar] [CrossRef]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [PubMed]
- Murakami, A.; Yajima, T.; Sakuma, H.; McLaren, M.J.; Inana, G. X-arrestin: A new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993, 334, 203–209. [Google Scholar] [CrossRef]
- Schaff, M.; Receveur, N.; Bourdon, C.; Ohlmann, P.; Lanza, F.; Gachet, C.; Mangin, P.H. beta-arrestin-1 participates in thrombosis and regulates integrin aIIbbeta3 signalling without affecting P2Y receptors desensitisation and function. Thromb. Haemost. 2012, 107, 735–748. [Google Scholar] [CrossRef]
- Li, D.; D’Angelo, L.; Chavez, M.; Woulfe, D.S. Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J. Biol. Chem. 2011, 286, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.L.; Zhao, X.; Hill, R.; Mundell, S.J. Arrestin-3 differentially regulates platelet GPCR subsets. Platelets 2019, 7, 1–5. [Google Scholar] [CrossRef]
- Sato, P.Y.; Chuprun, J.K.; Schwartz, M.; Koch, W.J. The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev. 2015, 95, 377–404. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef]
- Pronin, A.N.; Satpaev, D.K.; Slepak, V.Z.; Benovic, J.L. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem. 1997, 272, 18273–18280. [Google Scholar] [CrossRef]
- Andrews, R.K.; Munday, A.D.; Mitchell, C.A.; Berndt, M.C. Interaction of calmodulin with the cytoplasmic domain of the platelet membrane glycoprotein Ib-IX-V complex. Blood 2001, 98, 681–687. [Google Scholar] [CrossRef]
- Gardiner, E.E.; Arthur, J.F.; Berndt, M.C.; Andrews, R.K. Role of calmodulin in platelet receptor function. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2005, 3, 283–287. [Google Scholar] [CrossRef]
- Wong, M.X.; Harbour, S.N.; Wee, J.L.; Lau, L.M.; Andrews, R.K.; Jackson, D.E. Proteolytic cleavage of platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) is regulated by a calmodulin-binding motif. FEBS Lett. 2004, 568, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [PubMed]
- Beyett, T.S.; Fraley, A.E.; Labudde, E.; Patra, D.; Coleman, R.C.; Eguchi, A.; Glukhova, A.; Chen, Q.; Williams, R.M.; Koch, W.J.; et al. Perturbation of the interactions of calmodulin with GRK5 using a natural product chemical probe. Proc. Natl. Acad. Sci. USA 2019, 116, 15895–15900. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.T.; Paolucci, L.; De Blasi, A. Inhibition of G protein-coupled receptor kinase subtypes by Ca2+/calmodulin. J. Biol. Chem. 1996, 271, 28691–28696. [Google Scholar] [CrossRef] [PubMed]
- Hullmann, J.E.; Grisanti, L.A.; Makarewich, C.A.; Gao, E.; Gold, J.I.; Chuprun, J.K.; Tilley, D.G.; Houser, S.R.; Koch, W.J. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ. Res. 2014, 115, 976–985. [Google Scholar] [CrossRef]
- Signarvic, R.S.; Cierniewska, A.; Stalker, T.J.; Fong, K.P.; Chatterjee, M.S.; Hess, P.R.; Ma, P.; Diamond, S.L.; Neubig, R.R.; Brass, L.F. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood 2010, 116, 6092–6100. [Google Scholar] [CrossRef]
- Ma, P.; Foote, D.C.; Sinnamon, A.J.; Brass, L.F. Dissociation of SHP-1 from spinophilin during platelet activation exposes an inhibitory binding site for protein phosphatase-1 (PP1). PLoS ONE 2015, 10, e0119496. [Google Scholar] [CrossRef]
- Ma, P.; Ou, K.; Sinnamon, A.J.; Jiang, H.; Siderovski, D.P.; Brass, L.F. Modulating platelet reactivity through control of RGS18 availability. Blood 2015, 126, 2611–2620. [Google Scholar] [CrossRef]
- Ma, P.; Gupta, S.; Sampietro, S.; DeHelian, D.; Tutwiler, V.; Tang, A.; Stalker, T.J.; Brass, L.F. RGS10 shapes the hemostatic response to injury through its differential effects on intracellular signaling by platelet agonists. Blood Adv. 2018, 2, 2145–2155. [Google Scholar] [CrossRef]
- Hensch, N.R.; Karim, Z.A.; Druey, K.M.; Tansey, M.G.; Khasawneh, F.T. RGS10 Negatively Regulates Platelet Activation and Thrombogenesis. PLoS ONE 2016, 11, e0165984. [Google Scholar] [CrossRef]
- Ma, P.; Cierniewska, A.; Signarvic, R.; Cieslak, M.; Kong, H.; Sinnamon, A.J.; Neubig, R.R.; Newman, D.K.; Stalker, T.J.; Brass, L.F. A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood 2012, 119, 1935–1945. [Google Scholar] [CrossRef]
- Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. USA 1997, 94, 9956–9961. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Obaishi, H.; Satoh, A.; Wada, M.; Mandai, K.; Satoh, K.; Nishioka, H.; Matsuura, Y.; Mizoguchi, A.; Takai, Y. Neurabin: A novel neural tissue-specific actin filament-binding protein involved in neurite formation. J. Cell Biol. 1997, 139, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; di Tommaso, A.; Metaye, T.; Ladeveze, V. Spinophilin: From partners to functions. Biochimie 2006, 88, 1099–1113. [Google Scholar] [CrossRef]
- Stalker, T.J.; Welsh, J.D.; Brass, L.F. Shaping the platelet response to vascular injury. Curr. Opin. Hematol. 2014, 21, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.D.; Poventud-Fuentes, I.; Sampietro, S.; Diamond, S.L.; Stalker, T.J.; Brass, L.F. Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature. J. Thromb. Haemost. 2017, 15, 526–537. [Google Scholar] [CrossRef]
- Shen, J.; Sampietro, S.; Wu, J.; Tang, J.; Gupta, S.; Matzko, C.N.; Tang, C.; Yu, Y.; Brass, L.F.; Zhu, L.; et al. Coordination of platelet agonist signaling during the hemostatic response in vivo. Blood Adv. 2017, 1, 2767–2775. [Google Scholar] [CrossRef]
- Stalker, T.J. Mouse laser injury models: Variations on a theme. Platelets 2020, 31, 423–431. [Google Scholar] [CrossRef]
- Brass, L.F.; Newman, D.K.; Wannemacher, K.M.; Zhu, L.; Stalker, T.J. Signal transduction during platelet plug formation. In Platelets, 3rd ed.; Michelson, A.D., Ed.; Elsevier: New York, NY, USA, 2013; pp. 367–398. [Google Scholar]
- Vandendries, E.R.; Hamilton, J.R.; Coughlin, S.R.; Furie, B.; Furie, B.C. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc. Natl. Acad. Sci. USA 2007, 104, 288–292. [Google Scholar] [CrossRef]
- Brass, L.F. Homologous desensitization of HEL cell thrombin receptors. Distinguishable roles for proteolysis and phosphorylation. J. Biol. Chem. 1992, 267, 6044–6050. [Google Scholar]
- Molino, M.; Bainton, D.F.; Hoxie, J.A.; Coughlin, S.R.; Brass, L.F. Thrombin receptors on human platelets. Initial localization and subsequent redistribution during platelet activation. J. Biol. Chem. 1997, 272, 6011–6017. [Google Scholar] [CrossRef]
- French, S.L.; Hamilton, J.R. Protease-activated receptor 4: From structure to function and back again. Br. J. Pharmacol. 2016, 173, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.; Ishii, K.; Coughlin, S.R.; Kobilka, B.K. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. J. Biol. Chem. 1994, 269, 27719–27726. [Google Scholar]
- Packham, M.A.; Mustard, J.F. Platelet aggregation and adenosine diphosphate/adenosine triphosphate receptors: A historical perspective. Semin. Thromb. Hemost. 2005, 31, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Baurand, A.; Eckly, A.; Hechler, B.; Kauffenstein, G.; Galzi, J.L.; Cazenave, J.P.; Leon, C.; Gachet, C. Differential regulation and relocalization of the platelet P2Y receptors after activation: A way to avoid loss of hemostatic properties? Mol. Pharmacol. 2005, 67, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Ziegler, N.; Reiner, S.; Krasel, C.; Lohse, M.J. Agonist-selective, receptor-specific interaction of human P2Y receptors with beta-arrestin-1 and -2. J. Biol. Chem. 2008, 283, 30933–30941. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Shattil, S.J.; Ferry, N.; Rozansky, D.; Insel, P.A. Desensitization of epinephrine-initiated platelet aggregation does not alter binding to the alpha 2-adrenergic receptor or receptor coupling to adenylate cyclase. Mol. Pharmacol. 1986, 29, 1–6. [Google Scholar] [PubMed]
- Mackman, N.; Bergmeier, W.; Stouffer, G.A.; Weitz, J.I. Therapeutic strategies for thrombosis: New targets and approaches. Nat. Rev. Drug Discov. 2020, 19, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, J.; Gresham, K.; Koch, W.J. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol. 2019, 16, 612–622. [Google Scholar] [CrossRef]
- Eagle, K.A.; Ginsburg, G.S.; Musunuru, K.; Aird, W.C.; Balaban, R.S.; Bennett, S.K.; Blumenthal, R.S.; Coughlin, S.R.; Davidson, K.W.; Frohlich, E.D.; et al. Identifying patients at high risk of a cardiovascular event in the near future: Current status and future directions: Report of a national heart, lung, and blood institute working group. Circulation 2010, 121, 1447–1454. [Google Scholar] [CrossRef]
- Bray, P.F.; Mathias, R.A.; Faraday, N.; Yanek, L.R.; Fallin, M.D.; Herrera-Galeano, J.E.; Wilson, A.F.; Becker, L.C.; Becker, D.M. Heritability of platelet function in families with premature coronary artery disease. J. Thromb. Haemost. 2007, 5, 1617–1623. [Google Scholar] [CrossRef]
- Edelstein, L.C.; Simon, L.M.; Lindsay, C.R.; Kong, X.; Teruel-Montoya, R.; Tourdot, B.E.; Chen, E.S.; Ma, L.; Coughlin, S.; Nieman, M.; et al. Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood 2014, 124, 3450–3458. [Google Scholar] [CrossRef]
- Warren, D.M.; Soria, J.M.; Souto, J.C.; Comuzzie, A.; Fontcuberta, J.; Blangero, J.; MacCluer, J.W.; Almasy, L. Heritability of hemostasis phenotypes and their correlation with type 2 diabetes status in Mexican Americans. Hum. Biol. 2005, 77, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, T.R.; Lowe, G.D.; Lawlor, D.A.; Casas, J.P.; Day, I.N. A gene-centric analysis of activated partial thromboplastin time and activated protein C resistance using the HumanCVD focused genotyping array. Eur. J. Hum. Genet. 2013, 21, 779–783. [Google Scholar] [CrossRef]
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R.; et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef]
- Lobmeyer, M.T.; Wang, L.; Zineh, I.; Turner, S.T.; Gums, J.G.; Chapman, A.B.; Cooper-DeHoff, R.M.; Beitelshees, A.L.; Bailey, K.R.; Boerwinkle, E.; et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharm. Genom. 2011, 21, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Trimarco, V.; Di Marino, S.; Marino, M.; Iaccarino, G.; Trimarco, B. L41Q polymorphism of the G protein coupled receptor kinase 5 is associated with left ventricular apical ballooning syndrome. Eur. J. Heart Fail. 2010, 12, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429. [Google Scholar] [CrossRef]
- Lindstrom, S.; Wang, L.; Smith, E.N.; Gordon, W.; van Hylckama Vlieg, A.; de Andrade, M.; Brody, J.A.; Pattee, J.W.; Haessler, J.; Brumpton, B.M.; et al. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood 2019, 134, 1645–1657. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Han, X.; Hofmann, L.; de la Fuente, M.; Alexander, N.; Palczewski, K.; Invent Consortium, I.; Nieman, M.T. PAR4 activation involves extracellular loop-3 and transmembrane residue Thr153. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Tal, S.; Spectre, G.; Kornowski, R.; Perl, L. Venous Thromboembolism Complicated with COVID-19: What Do We Know So Far? Acta Haematol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood 2020. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Yang, Q. Antiplatelet Therapy After Percutaneous Coronary Intervention in Patients With COVID-19: Implications From Clinical Features to Pathologic Findings. Circulation 2020, 141, 1736–1738. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. Proteinase-activated receptor 1 (PAR1): A target for repurposing in the treatment of COVID-19? Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Oro, C.; Qian, H.; Thomas, W.G. Type 1 angiotensin receptor pharmacology: Signaling beyond G proteins. Pharmacol. Ther. 2007, 113, 210–226. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zhao, X.; Cooper, M.; Ma, P. The Roles of GRKs in Hemostasis and Thrombosis. Int. J. Mol. Sci. 2020, 21, 5345. https://doi.org/10.3390/ijms21155345
Chen X, Zhao X, Cooper M, Ma P. The Roles of GRKs in Hemostasis and Thrombosis. International Journal of Molecular Sciences. 2020; 21(15):5345. https://doi.org/10.3390/ijms21155345
Chicago/Turabian StyleChen, Xi, Xuefei Zhao, Matthew Cooper, and Peisong Ma. 2020. "The Roles of GRKs in Hemostasis and Thrombosis" International Journal of Molecular Sciences 21, no. 15: 5345. https://doi.org/10.3390/ijms21155345
APA StyleChen, X., Zhao, X., Cooper, M., & Ma, P. (2020). The Roles of GRKs in Hemostasis and Thrombosis. International Journal of Molecular Sciences, 21(15), 5345. https://doi.org/10.3390/ijms21155345