Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections


Abstract
1. Introduction
2. Rearrangement of Intracellular Organelles during Viral Infections
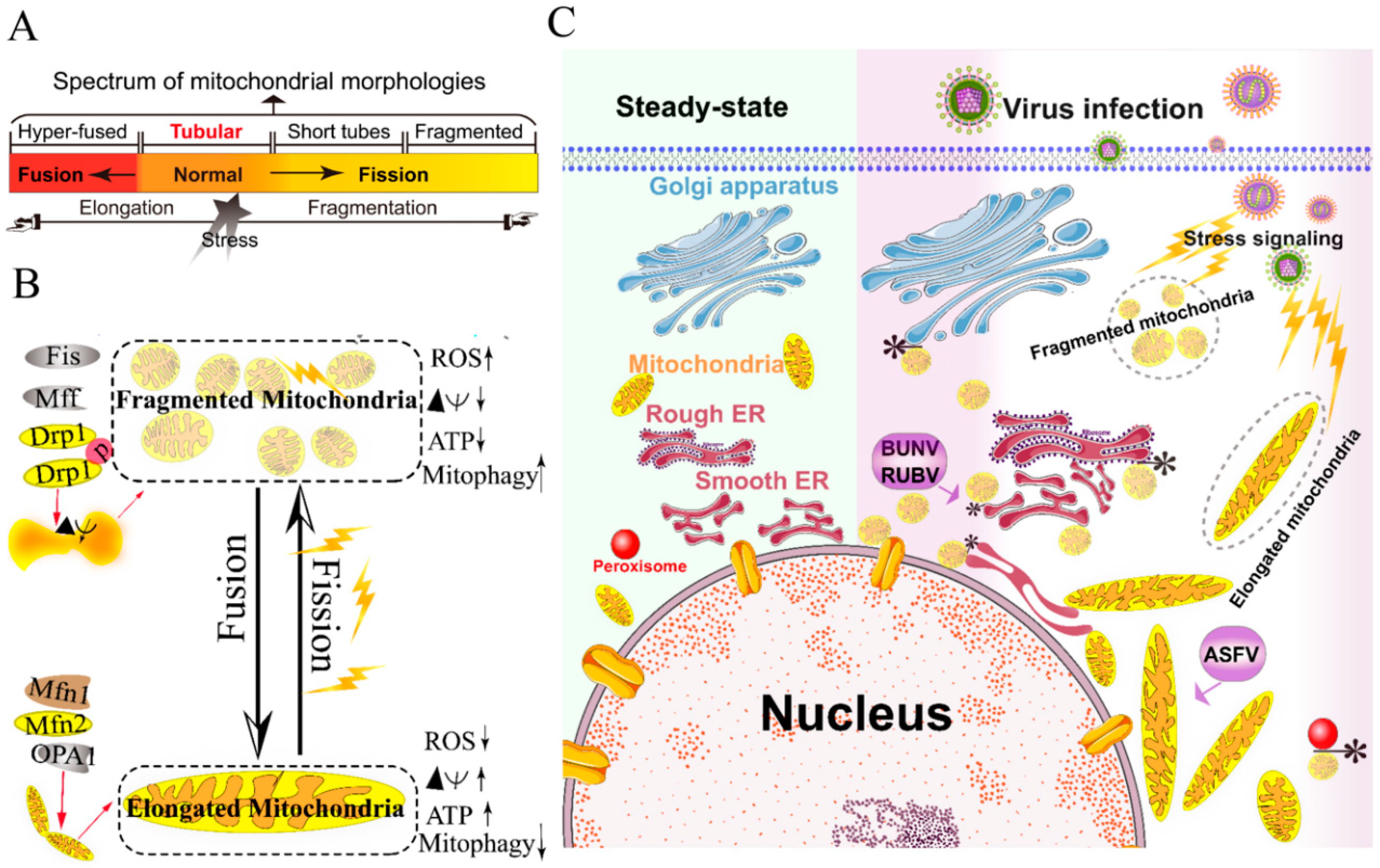
2.1. Remodeling of the Mitochondria for Viral Replication
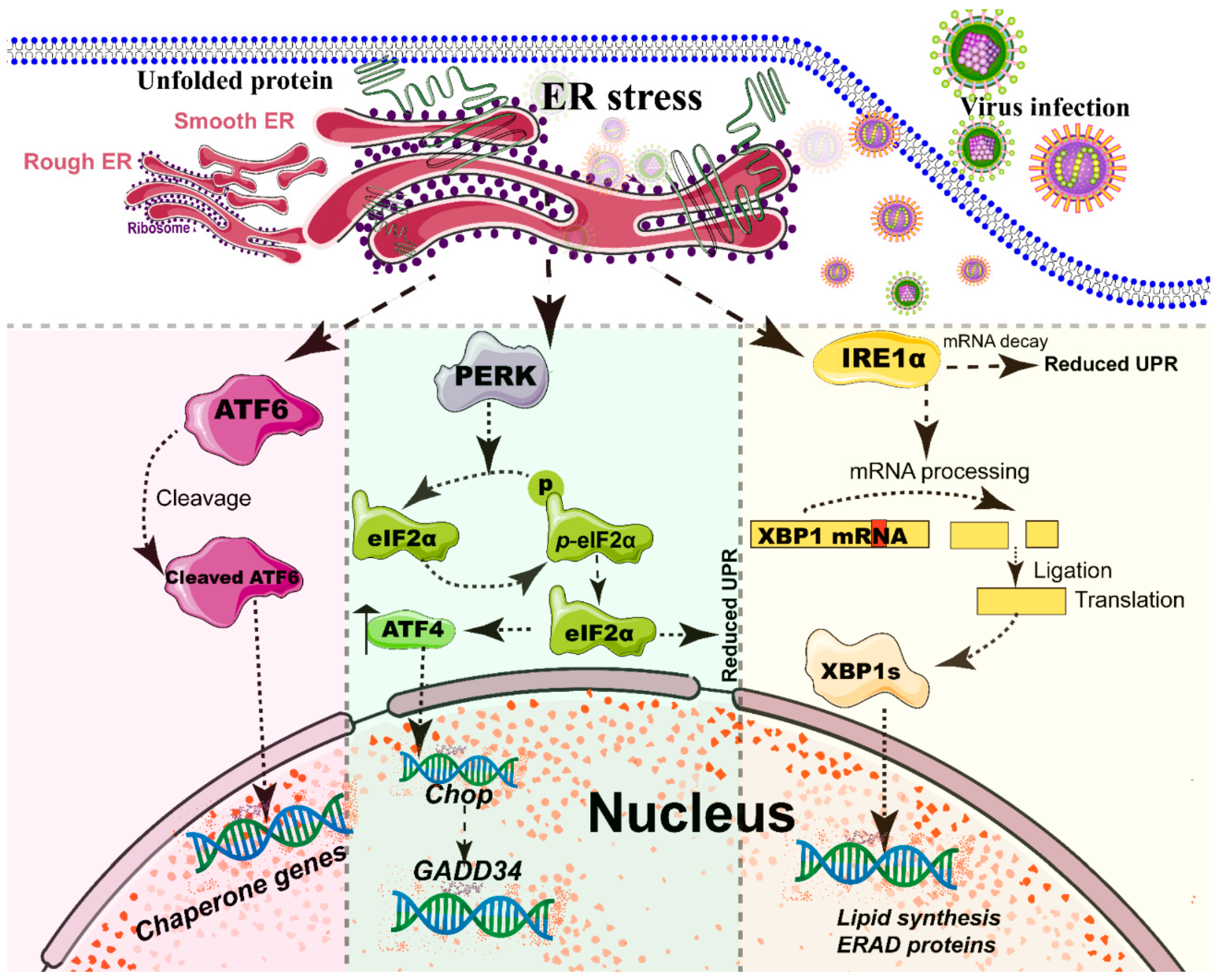
2.2. Rearrangement of ER and Unfolded Protein Response (UPR) during Viral Infection
2.3. Rearrangement of Peroxisome for Infectious Progeny
2.4. Hijacking of Golgi Apparatus for Infectious Progeny
2.5. Role of the Lysosome and Endosome in Viral Infections
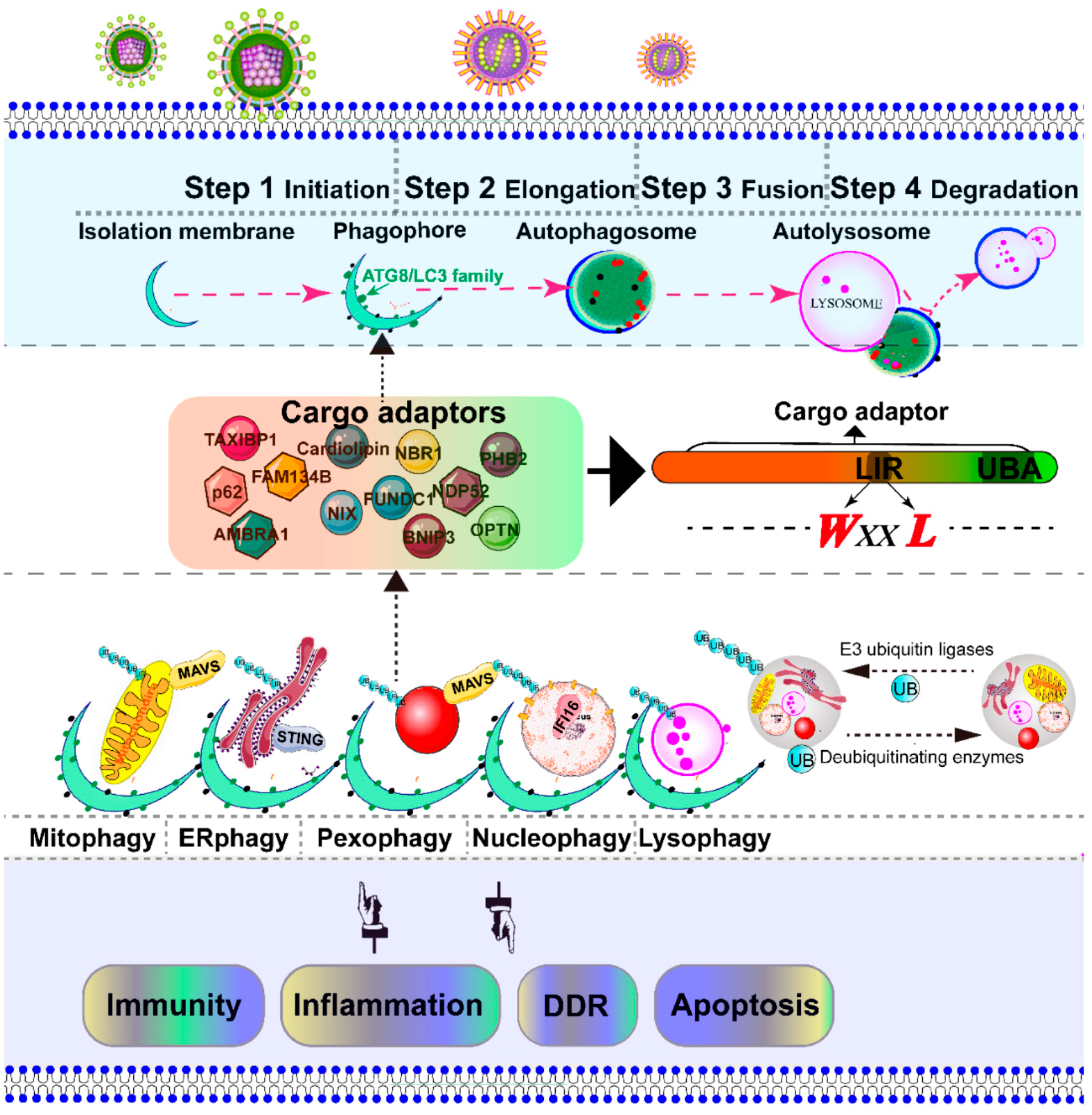
3. Degradation of Intracellular Organelles by Virus-Triggered Selective Autophagy
3.1. Overview of Mitophagy and Pexophagy Signaling
3.2. Virus Triggers Mitophagy and Pexophagy to Suppress MAVS-Dependent RLR Signaling
3.3. ER-Phagy (reticulophagy) Trigged by Viral Infection
3.4. Lysophagy and Nucleophagy Trigged by Viral Infection
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| AMBRA1 | Autophagy and beclin-1 regulator 1 |
| ASFV | African swine fever virus |
| BUNV | Bunyamwera virus |
| BNIP3L/NIX | BCL2 interacting protein 3 like |
| BNIP3 | BCL2 interacting protein 3 |
| cGAS | Cyclic GMP-AMP synthase |
| CVB | Coxsackievirus B |
| CHIKV | Chikungunya virus |
| CALCOCO2/NDP52 | Calcium binding and coiled-coil domain 2/Antigen nuclear dot 52 kDa protein |
| COP | Coat protein complex |
| CV | Coxsackieviruses |
| DENV | Dengue virus |
| DRP-1 | Dynamic relative GTPase protein |
| ERGIC | ER-Golgi-intermediate compartment |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| EAV | Equine arteritis virus |
| FHV | Flock house virus |
| FUNDC1 | FUN14 domain containing 1 |
| FAM134B | Family with sequence similarity 134, member B |
| HIV | Human immune deficiency |
| HPIV3 | Human parainfluenza virus typ3 |
| HCV | Hepatitis C virus |
| HSV-1 | Human herpes simplex virus-1 |
| HBV | Hepatitis B virus |
| IAV | Influenza A virus |
| IMM | Inner mitochondrial membranes |
| IBV | Avian coronavirus infectious bronchitis virus |
| IAV | Influenza A virus |
| IRE1α | Inositol requiring enzyme 1α |
| JEV | Japanese encephalitis virus |
| LC3 | Light chain 3 |
| LIR | LC3-interacting region |
| MHV | Murine hepatitis virus |
| Mfns | Mitofusin GTPases |
| Mfn1 | Mitofusin 1 |
| Mfn2 | Mitofusin 2 |
| Mff | Mitochondrial fission factor |
| MAM | Mitochondrial-associated-membrane |
| MCMV | Murine cytomegalovirus |
| MV | Measles virus |
| MAPL | Mitochondrial-anchored protein ligase |
| MDVs | Mitochondrial-derived vesicles |
| MAVS | Mitochondrial antiviral signaling protein |
| NDV | Newcastle disease virus |
| NBR1 | Next to BRCA1 gene 1 protein |
| OMM | Out mitochondrial membranes |
| OPA1 | Optic atrophy 1 |
| OPTN | Optineurin |
| PERK | Double-stranded RNA-activated protein kinase-like kinase |
| PRRSV | Porcine reproductive and respiratory syndrome virus |
| PHB2 | Prohibitin 2 |
| PEX | Peroxin |
| PV | Polioviruses |
| PINK1 | PTEN-induced putative kinase protein 1 |
| RUBV | Rubella virus |
| ROS | Reactive oxygen species |
| Ref | Reference |
| RIG-I | Retinoic acid-inducible gene I protein |
| STING | Stimulator of interferon genes |
| SIV | Sindbis virus |
| SFV | Semliki forest virus |
| SARS | Severe acute respiratory syndrome |
| TBSV | Tomato bushy stunt virus |
| TBEV | Tick-borne encephalitis virus |
| TAXIBP1 | Tax1 binding protein 1 |
| UPR | Unfolded proteins response |
| UPRmt | Unfolded protein response (UPR) of mitochondria |
References
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Helenius, A. Virus entry at a glance. J. Cell Sci. 2013, 126, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. BioMed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ni, H.M.; Guo, F.; Ding, Y.; Shi, Y.H.; Lahiri, P.; Frohlich, L.F.; Rulicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 Protein Is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015, 11, e1005174. [Google Scholar] [CrossRef]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2013, 9, 1256–1257. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef]
- Lazarow, P.B. Viruses exploiting peroxisomes. Curr. Opin. Microbiol. 2011, 14, 458–469. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e08828. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial fission: Regulation and ER connection. Mol. Cells 2014, 37, 89–94. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Castello, A.; Quintas, A.; Sanchez, E.G.; Sabina, P.; Nogal, M.; Carrasco, L.; Revilla, Y. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 2009, 5, e1000562. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia virus requires glutamine but not glucose for efficient replication. J. Virol. 2014, 88, 4366–4374. [Google Scholar] [CrossRef]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef]
- Fontana, J.; Lopez-Montero, N.; Elliott, R.M.; Fernandez, J.J.; Risco, C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell Microbiol. 2008, 10, 2012–2028. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Onomoto, K.; Takamatsu, S.; Jogi, M.; Takemura, A.; Morimoto, S.; Julkunen, I.; Namiki, H.; Yoneyama, M.; Fujita, T. Virus-infection or 5’ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010, 6, e1001012. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 336. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.E.; Wang, S.; Liu, T.Y.; Rapoport, T.A. Reconstitution of the tubular endoplasmic reticulum network with purified components. Nature 2017, 543, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Verchot, J. How does the stressed out ER find relief during virus infection? Curr. Opin. Virol. 2016, 17, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- Guo, F.J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell. Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Moon, S.L.; Barnhart, M.D.; Wilusz, J. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr. Opin. Microbiol. 2012, 15, 500–505. [Google Scholar] [CrossRef]
- Tenorio, R.; Fernandez de Castro, I.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Mainou, B.A.; Dermody, T.S.; Risco, C. Reovirus sigmaNS and muNS Proteins Remodel the Endoplasmic Reticulum to Build Replication Neo-Organelles. mBio 2018, 9, e01253-18. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Risco, C.; Rodriguez, J.R.; Lopez-Iglesias, C.; Carrascosa, J.L.; Esteban, M.; Rodriguez, D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002, 76, 1839–1855. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Moss, B. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 2003, 77, 11754–11766. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Barcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Garcia-Escudero, R.; Salas, M.L.; Andres, G. African swine fever virus structural protein p54 is essential for the recruitment of envelope precursors to assembly sites. J. Virol. 2004, 78, 4299–4313. [Google Scholar] [CrossRef]
- Gao, P.; Chai, Y.; Song, J.; Liu, T.; Chen, P.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLoS Pathog. 2019, 15, e1008169. [Google Scholar] [CrossRef]
- Ren, S.; Rehman, Z.U.; Shi, M.; Yang, B.; Liu, P.; Yin, Y.; Qu, Y.; Meng, C.; Yang, Z.; Gao, X.; et al. Hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus cooperatively disturb fusion–fission homeostasis to enhance mitochondrial function by activating the unfolded protein response of endoplasmic reticulum and mitochondrial stress. Vet. Res. 2019, 50, 37. [Google Scholar]
- Targett-Adams, P.; Boulant, S.; Douglas, M.W.; McLauchlan, J. Lipid metabolism and HCV infection. Viruses 2010, 2, 1195–1217. [Google Scholar] [CrossRef]
- Cobbold, C.; Brookes, S.M.; Wileman, T. Biochemical requirements of virus wrapping by the endoplasmic reticulum: Involvement of ATP and endoplasmic reticulum calcium store during envelopment of African swine fever virus. J. Virol. 2000, 74, 2151–2160. [Google Scholar] [CrossRef]
- Liu, X.; Palaniyandi, S.; Zhu, I.; Tang, J.; Li, W.; Wu, X.; Ochsner, S.P.; Pauza, C.D.; Cohen, J.I.; Zhu, X. Human cytomegalovirus evades antibody-mediated immunity through endoplasmic reticulum-associated degradation of the FcRn receptor. Nat. Commun. 2019, 10, 3020. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Ko, D.H.; Shin, N.; Pyo, C.W.; Choi, S.Y. Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza A virus by regulating the host redox state. Free Radic. Biol. Med. 2019, 135, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1alpha protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, e00887-19. [Google Scholar] [CrossRef]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch. Virol. 2016, 161, 699–703. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Achazi, K.; Niedrig, M. Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 2013, 178, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.H.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.; Monick, M.M. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 2012, 287, 4679–4689. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C. eIF2alpha-CHOP-BCl-2/JNK and IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Disease 2019, 10, 891. [Google Scholar] [CrossRef]
- Stahl, S.; Burkhart, J.M.; Hinte, F.; Tirosh, B.; Mohr, H.; Zahedi, R.P.; Sickmann, A.; Ruzsics, Z.; Budt, M.; Brune, W. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 2013, 9, e1003544. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine cytomegalovirus targets transcription factor ATF4 to exploit the unfolded-protein response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef]
- Zhang, P.; Su, C.; Jiang, Z.; Zheng, C. Herpes Simplex Virus 1 UL41 Protein Suppresses the IRE1/XBP1 Signal Pathway of the Unfolded Protein Response via Its RNase Activity. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma (1) 34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef]
- Mulvey, M.; Arias, C.; Mohr, I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 2007, 81, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Till, A.; Lakhani, R.; Burnett, S.F.; Subramani, S. Pexophagy: The selective degradation of peroxisomes. Int. J. Cell Biol. 2012, 2012, 512721. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Yamashita, S.-I.; Fujiki, Y. Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 984–991. [Google Scholar] [CrossRef]
- Delille, H.K.; Schrader, M. Targeting of hFis1 to peroxisomes is mediated by Pex19p. J. Biol. Chem. 2008, 283, 31107–31115. [Google Scholar] [CrossRef]
- Schrader, M. Shared components of mitochondrial and peroxisomal division. Biochim. Biophys. Acta 2006, 1763, 531–541. [Google Scholar] [CrossRef]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef]
- Mohan, K.V.; Som, I.; Atreya, C.D. Identification of a type 1 peroxisomal targeting signal in a viral protein and demonstration of its targeting to the organelle. J. Virol. 2002, 76, 2543–2547. [Google Scholar] [CrossRef][Green Version]
- Aguado, B.; Vinuela, E.; Alcami, A. African swine fever virus fatty acid acylated proteins. Virology 1991, 185, 942–945. [Google Scholar] [CrossRef]
- McCartney, A.W.; Greenwood, J.S.; Fabian, M.R.; White, K.A.; Mullen, R.T. Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 2005, 17, 3513–3531. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk, M.; Pathak, K.B.; Sharma, M.; Nagy, P.D. Exploiting alternative subcellular location for replication: Tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 2007, 362, 320–330. [Google Scholar] [CrossRef] [PubMed]
- James Morre, D.; Mollenhauer, H.H. Microscopic morphology and the origins of the membrane maturation model of Golgi apparatus function. Int. Rev. Cytol. 2007, 262, 191–218. [Google Scholar]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherdt, M.; Schekman, R. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Rabouille, C.; Klumperman, J. Opinion: The maturing role of COPI vesicles in intra-Golgi transport. Nat. Rev. Mol. Cell Biol. 2005, 6, 812–817. [Google Scholar] [CrossRef]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2007, 81, 558–567. [Google Scholar] [CrossRef]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [CrossRef]
- Dodd, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beato, R.; Salas, M.L.; Vinuela, E.; Salas, J. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 1992, 188, 637–649. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Colocalization of Transcription and Translation within Cytoplasmic Poxvirus Factories Coordinates Viral Expression and Subjugates Host Functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J.; Moscona, A. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef]
- Bost, A.G.; Prentice, E.; Denison, M.R. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology 2001, 285, 21–29. [Google Scholar] [CrossRef][Green Version]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef]
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016, 97, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.; Marshall, J.A.; Bowden, D.S.; Vardaxis, N.; Meanger, J.; Lee, J.Y. Rubella virus replication complexes are virus-modified lysosomes. Virology 1998, 240, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Schwartz, M.D.; Ahlquist, P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 2001, 75, 11664–11676. [Google Scholar] [CrossRef] [PubMed]
- Ertel, K.J.; Benefield, D.; Castano-Diez, D.; Pennington, J.G.; Horswill, M.; den Boon, J.A.; Otegui, M.S.; Ahlquist, P. Cryo-electron tomography reveals novel features of a viral RNA replication compartment. eLife 2017, 6, e25940. [Google Scholar] [CrossRef]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest Virus RNA Replication Complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Shi, X.; Kohl, A.; Leonard, V.H.; Li, P.; McLees, A.; Elliott, R.M. Requirement of the N-terminal region of orthobunyavirus nonstructural protein NSm for virus assembly and morphogenesis. J. Virol. 2006, 80, 8089–8099. [Google Scholar] [CrossRef]
- Gomez, C.; Hope, T.J. The ins and outs of HIV replication. Cell. Microbiol. 2005, 7, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Tout, I.; Gomes, M.; Ainouze, M.; Marotel, M.; Pecoul, T.; Durantel, D.; Vaccarella, S.; Dubois, B.; Loustaud-Ratti, V.; Walzer, T.; et al. Hepatitis B Virus Blocks the CRE/CREB Complex and Prevents TLR9 Transcription and Function in Human B Cells. J. Immunol. 2018, 201, 2331–2344. [Google Scholar] [CrossRef]
- Lai, J.H.; Wang, M.Y.; Huang, C.Y.; Wu, C.H.; Hung, L.F.; Yang, C.Y.; Ke, P.Y.; Luo, S.F.; Liu, S.J.; Ho, L.J. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef]
- Yamashita, S.; Abe, K.; Tatemichi, Y.; Fujiki, Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014, 10, 1549–1564. [Google Scholar] [CrossRef]
- Hara-Kuge, S.; Fujiki, Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp. Cell Res. 2008, 314, 3531–3541. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547.e4. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; Leveille, S.; Danthi, P.; Hiscott, J.; Sherry, B.; Dermody, T.S. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar] [CrossRef]
- Yu, C.Y.; Chiang, R.L.; Chang, T.H.; Liao, C.L.; Lin, Y.L. The interferon stimulator mitochondrial antiviral signaling protein facilitates cell death by disrupting the mitochondrial membrane potential and by activating caspases. J. Virol. 2010, 84, 2421–2431. [Google Scholar] [CrossRef]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Foy, E.; Li, K.; Sumpter, R., Jr.; Loo, Y.M.; Johnson, C.L.; Wang, C.; Fish, P.M.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 2986–2991. [Google Scholar] [CrossRef]
- Loo, Y.M.; Owen, D.M.; Li, K.; Erickson, A.K.; Johnson, C.L.; Fish, P.M.; Carney, D.S.; Wang, T.; Ishida, H.; Yoneyama, M.; et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 6001–6006. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Tomar, D.; Singh, R.; Singh, A.K.; Pandya, C.D.; Singh, R. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 2012, 1823, 316–326. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Munro, S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell 2010, 21, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses 2015, 7, 6316–6345. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Viruses | Family | Genome Structure | Virion Structure | Viral Protein | ATF6 | PERK | IRE1α | Ref |
|---|---|---|---|---|---|---|---|---|
| PRRSV | Arteriviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | √ | √ | [87] |
| IBV | Coronaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | × | √ | [97] |
| DENV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | × | √ | √ | [98] |
| JEV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | NS4B | √ | √ | √ | [99,100,101] |
| TBEV | Flaviridae | Linear, ssRNA(+) | Enveloped; Rounded | ? | √ | × | √ | [102] |
| HCV | Flaviridae | Linear, ssRNA(+) | Enveloped; Spherical | ? | √ | × | × | [103] |
| IAV | Orthomyxoviridae | Segmented, ssRNA (-) | Enveloped; Rounded | ? | × | √ | [104] | |
| NDV | Paramyxoviridae | Linear, ssRNA(-) | Enveloped; Spherical | F and HN | √ | √ | √ | [88,105] |
| MCMV | Herpesviridae | Linear, ds DNA | Enveloped; Spherical | ? | ? | √ | × | [106,107] |
| HSV-1 | Herpesviridae | Linear, dsDNA | Enveloped; Spherical | UL41/ICP0/γ134.5 | √ | √ | × | [108,109,110,111] |
| HBV | Hepadnaviridae | Circular dsDNA | Enveloped; Spherical | ? | √ | × | √ | [112] |
| Family | Viruses | Genome Structure | Virion Structure | Replication Site | Ref | Assembly Site | Ref |
|---|---|---|---|---|---|---|---|
| Asfarviridae | ASFV | Liner, dsDNA | Enveloped, spherical | Nuclear and cytoplasmic | [49,132,133] | ER | [86,90] |
| Poxviridae | VV | Liner, dsDNA | Enveloped, brick-shaped | cytoplasmic | [134] | ERGIC, ER | [82,83] |
| Arteriviridae/Arteriviruses | EAV, PRRSV | Liner, ssRNA (+) | Enveloped, spherical | Cytoplasmic double membranes | [84,135] | ER | [84] |
| Coronaviridae/Coronavirus | SARS/MHV | Liner, ssRNA (+) | Enveloped, spherical | Cytoplasmic double membranes | [136] | ERGIC, Golgi, ER | [137,138] |
| Flaviviridae/Hepacivirus | HCV | Linear, ssRNA(+) | Enveloped, spherical | Cytoplasmic | [139] | Autophagosome | [140,141] |
| Metonaviridae/Rubivirus | RUBV | Liner, ssRNA (+) | Enveloped, spherical isometric capsid? | Cytoplasmic | [56] | Golgi, Lysosome | [56,142] |
| Nodaviridae | FHV | Linear, ssRNA(+) | Non-envelop, icosahedral | Cytoplasmic | [143,144] | OMM | [143,144] |
| Togaviridae/Alphavirus | SFV | Liner, ssRNA (+) | Enveloped, spherical and icosahedral | Cytoplasmic | [145,146] | Endosome/Lysosome | [145,146,147] |
| Tombusviridae | TBSV | Linear, ssRNA(+) | Non-envelop, icosahedral | Cytoplasmic | [123] | Peroxisome, ER | [122,123] |
| Picornaviridae/Enterovirus | CV/PV | Linear, ssRNA (+) | Non-envelop, spherical | Cytoplasmic | [148] | ER | [85,148] |
| Orthomyxoviridae | IAV | Segmented, ssRNA (−) | Enveloped; Rounded | Nuclear | [149] | ER, Golgi | [149] |
| Peribunyaviridae/Bunyavirus | BUNV | Segmented, ssRNA (−) | Enveloped, spherical | cytoplasmic | [57] | Golgi | [57,150] |
| Retroviridae/Lentivirus | HIV | Linear, ssRNA (+) | Enveloped, Spherical | Nuclear | [151] | ER or Golgi | [151] |
| Name | Mitophagy | ER-Phagy | Pexophagy | Nucleophagy | Lysophagy | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cargo Proteins | Mitochondria | Ref | ER | Ref | Peroxisome | Ref | Nucleus | Ref | Lysosome | Ref |
| p62/SQSTM1 | √ | [10] | √ | [12] | √ | [175,176,180] | ? | - | √ | [208,209] |
| NDP52 | √ | [173,210] | ? | - | ? | - | ? | - | ? | - |
| NBR1 | √ | [14] | ? | - | √ | [175,177,181] | ? | - | ? | - |
| OPTN | √ | [15,16,173,210] | ? | - | ? | - | ? | - | ? | - |
| AMBRA1 | √ | [13] | ? | - | ? | - | ? | - | ? | - |
| BNIP3L/NIX | √ | [19,20] | ? | - | ? | - | ? | - | ? | - |
| BNIP3 | √ | [21,211] | √ | [21] | ? | - | ? | - | ? | - |
| TAX1BP1 | √ | [15,17] | ? | - | ? | - | ? | - | ? | - |
| FUNDC1 | √ | [23] | ? | - | ? | - | ? | - | ? | - |
| Cardiolipin | √ | [24] | ? | - | ? | - | ? | - | ? | - |
| Prohibitin2 | √ | [22] | ? | ? | - | ? | - | - | ||
| FAM134B | ? | - | √ | [25] | ? | - | ? | - | ? | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, S.; Ding, C.; Sun, Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. Int. J. Mol. Sci. 2020, 21, 3689. https://doi.org/10.3390/ijms21103689
Ren S, Ding C, Sun Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. International Journal of Molecular Sciences. 2020; 21(10):3689. https://doi.org/10.3390/ijms21103689
Chicago/Turabian StyleRen, Shanhui, Chan Ding, and Yingjie Sun. 2020. "Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections" International Journal of Molecular Sciences 21, no. 10: 3689. https://doi.org/10.3390/ijms21103689
APA StyleRen, S., Ding, C., & Sun, Y. (2020). Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. International Journal of Molecular Sciences, 21(10), 3689. https://doi.org/10.3390/ijms21103689