PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells

{kind=link}
{kind=link}
Abstract
1. Introduction
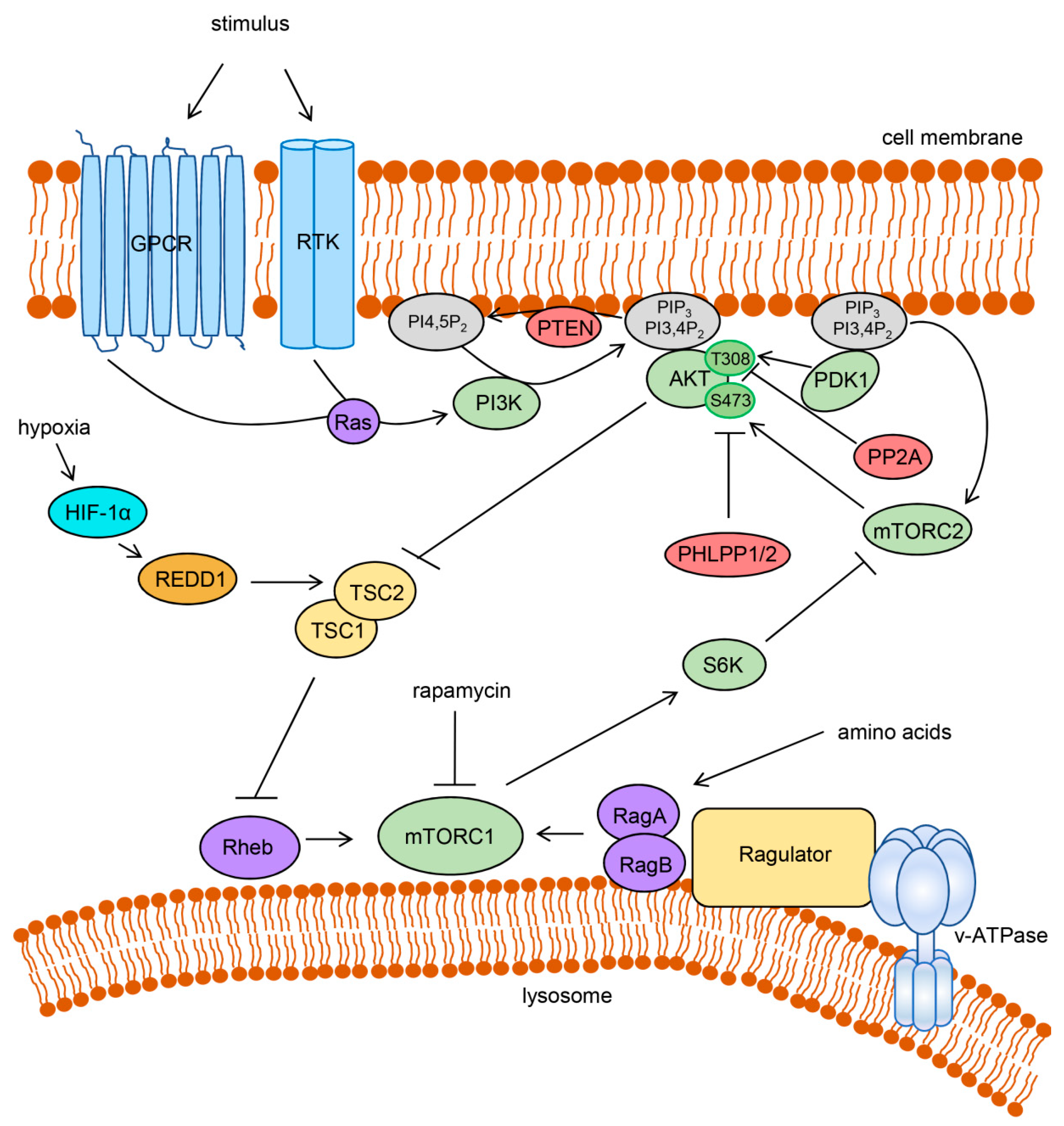
2. The PI3K/AKT/mTOR Pathway and Cancer
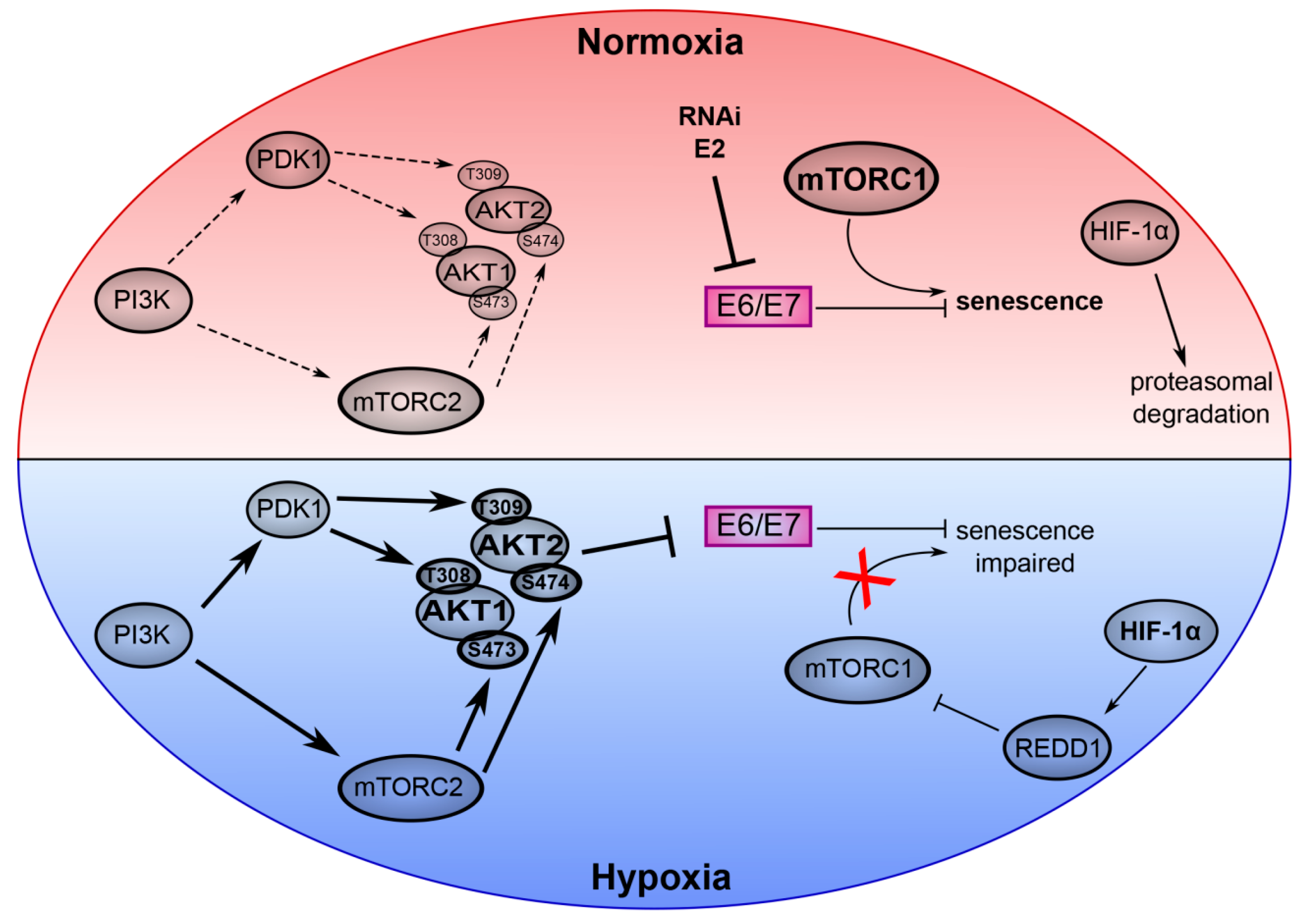
3. mTOR Signaling and Senescence in HPV-Positive Cancer Cells
4. AKT Signaling in Hypoxic E6/E7 Repression
5. Role of the PI3K/AKT/mTOR Pathway in HPV-Positive Tumors in the Clinic
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjose, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.; Dillner, J. Clinical trials of human papillomavirus vaccines and beyond. Nat. Rev. Clin. Oncol. 2013, 10, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 2018, 36, 4768–4773. [Google Scholar] [CrossRef] [PubMed]
- Castle, P.E.; Maza, M. Prophylactic HPV vaccination: Past, present, and future. Epidemiol. Infect. 2016, 144, 449–468. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Müller, M. Next generation prophylactic human papillomavirus vaccines. Lancet Oncol. 2015, 16, e217–e225. [Google Scholar] [CrossRef]
- Bruni, L.; Diaz, M.; Barrionuevo-Rosas, L.; Herrero, R.; Bray, F.; Bosch, F.X.; de Sanjose, S.; Castellsague, X. Global estimates of human papillomavirus vaccination coverage by region and income level: A pooled analysis. Lancet Glob. Health 2016, 4, e453–e463. [Google Scholar] [CrossRef]
- Vaupel, P. Pathophysiology of Solid Tumors. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies; Molls, M., Vaupel, P., Nieder, C., Anscher, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 51–92. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Guerrieri, M.E.; Greco, C. Tissue biomarkers as prognostic variables of cervical cancer. Crit. Rev. Oncol. Hematol. 2013, 86, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Mändl, J.; Adrian, S.; Kuhn, B.J.; Hoppe-Seyler, F. Virus/Host Cell Crosstalk in Hypoxic HPV-Positive Cancer Cells. Viruses 2017, 9, 174. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rösl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Durst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E990–e998. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Beitner-Johnson, D.; Rust, R.T.; Hsieh, T.C.; Millhorn, D.E. Hypoxia activates Akt and induces phosphorylation of GSK-3 in PC12 cells. Cell. Signal. 2001, 13, 23–27. [Google Scholar] [CrossRef]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [CrossRef]
- Stegeman, H.; Kaanders, J.H.; Wheeler, D.L.; van der Kogel, A.J.; Verheijen, M.M.; Waaijer, S.J.; Iida, M.; Grenman, R.; Span, P.N.; Bussink, J. Activation of AKT by hypoxia: A potential target for hypoxic tumors of the head and neck. BMC Cancer 2012, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Bossler, F.; Kuhn, B.J.; Günther, T.; Kraemer, S.J.; Khalkar, P.; Adrian, S.; Lohrey, C.; Holzer, A.; Shimobayashi, M.; Dürst, M.; et al. Repression of Human Papillomavirus Oncogene Expression under Hypoxia Is Mediated by PI3K/mTORC2/AKT Signaling. mBio 2019, 10, e02323-18. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Yang, E.; Lee, C.J.; Lee, H.W.; DiMaio, D.; Hwang, E.S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983. [Google Scholar] [CrossRef]
- Hall, A.H.; Alexander, K.A. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J. Virol. 2003, 77, 6066–6069. [Google Scholar] [CrossRef]
- Magaldi, T.G.; Almstead, L.L.; Bellone, S.; Prevatt, E.G.; Santin, A.D.; DiMaio, D. Primary human cervical carcinoma cells require human papillomavirus E6 and E7 expression for ongoing proliferation. Virology 2012, 422, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- DeFilippis, R.A.; Goodwin, E.C.; Wu, L.; DiMaio, D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J. Virol. 2003, 77, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.I.; Francis, D.A.; Karpova, A.Y.; Dowhanick, J.J.; Benson, J.D.; Howley, P.M. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 2000, 19, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle 2018, 1–11. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Natarajan, V.; Demidenko, Z.N.; Burdelya, L.G.; Gudkov, A.V.; Blagosklonny, M.V. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 13314–13318. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-type-dependent regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2 and LKB1 in response to hypoxia. Mol. Cell. Biol. 2011, 31, 1870–1884. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Contreras-Paredes, A.; De la Cruz-Hernandez, E.; Martinez-Ramirez, I.; Duenas-Gonzalez, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, O.; Yang, Z.Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef]
- Dummler, B.; Hemmings, B.A. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef]
- Rodon, J.; Tabernero, J. Improving the Armamentarium of PI3K Inhibitors with Isoform-Selective Agents: A New Light in the Darkness. Cancer Discov. 2017, 7, 666–669. [Google Scholar] [CrossRef]
- McIntyre, J.B.; Wu, J.S.; Craighead, P.S.; Phan, T.; Kobel, M.; Lees-Miller, S.P.; Ghatage, P.; Magliocco, A.M.; Doll, C.M. PIK3CA mutational status and overall survival in patients with cervical cancer treated with radical chemoradiotherapy. Gynecol. Oncol. 2013, 128, 409–414. [Google Scholar] [CrossRef]
- Wright, A.A.; Howitt, B.E.; Myers, A.P.; Dahlberg, S.E.; Palescandolo, E.; Van Hummelen, P.; MacConaill, L.E.; Shoni, M.; Wagle, N.; Jones, R.T.; et al. Oncogenic mutations in cervical cancer: Genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 2013, 119, 3776–3783. [Google Scholar] [CrossRef]
- Lachkar, B.; Minaguchi, T.; Akiyama, A.; Liu, S.; Zhang, S.; Xu, C.; Shikama, A.; Tasaka, N.; Sakurai, M.; Nakao, S.; et al. Prognostic significance of PIK3CA mutation in stage IIB to IVA cervical cancers treated by concurrent chemoradiotherapy with weekly cisplatin. Medicine (Baltimore) 2018, 97, e11392. [Google Scholar] [CrossRef]
- Schwarz, J.K.; Payton, J.E.; Rashmi, R.; Xiang, T.; Jia, Y.; Huettner, P.; Rogers, B.E.; Yang, Q.; Watson, M.; Rader, J.S.; et al. Pathway-specific analysis of gene expression data identifies the PI3K/Akt pathway as a novel therapeutic target in cervical cancer. Clin. Cancer Res. 2012, 18, 1464–1471. [Google Scholar] [CrossRef]
- Faried, L.S.; Faried, A.; Kanuma, T.; Sano, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Predictive and prognostic role of activated mammalian target of rapamycin in cervical cancer treated with cisplatin-based neoadjuvant chemotherapy. Oncol. Rep. 2006, 16, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Faried, L.S.; Faried, A.; Kanuma, T.; Aoki, H.; Sano, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Expression of an activated mammalian target of rapamycin in adenocarcinoma of the cervix: A potential biomarker and molecular target therapy. Mol. Carcinog. 2008, 47, 446–457. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wei, S.J.; Lin, Y.C.; Lung, J.C.; Chang, T.C.; Whang-Peng, J.; Liu, J.M.; Yang, D.M.; Yang, W.K.; Shen, C.Y. PIK3CA as an oncogene in cervical cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef]
- Bertelsen, B.I.; Steine, S.J.; Sandvei, R.; Molven, A.; Laerum, O.D. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: Frequent PIK3CA amplification and AKT phosphorylation. Int. J. Cancer 2006, 118, 1877–1883. [Google Scholar] [CrossRef]
- Henken, F.E.; Banerjee, N.S.; Snijders, P.J.; Meijer, C.J.; De-Castro Arce, J.; Rösl, F.; Broker, T.R.; Chow, L.T.; Steenbergen, R.D. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Mol. Cancer 2011, 10, 71. [Google Scholar] [CrossRef]
- Bahrami, A.; Hasanzadeh, M.; Hassanian, S.M.; ShahidSales, S.; Ghayour-Mobarhan, M.; Ferns, G.A.; Avan, A. The Potential Value of the PI3K/Akt/mTOR Signaling Pathway for Assessing Prognosis in Cervical Cancer and as a Target for Therapy. J. Cell. Biochem. 2017, 118, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Tsimberidou, A.M.; Garrido-Laguna, I.; Wang, X.; Luthra, R.; Hong, D.S.; Naing, A.; Falchook, G.S.; Moroney, J.W.; Piha-Paul, S.A.; et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol. Cancer Ther. 2011, 10, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Berman, T.A.; Schiller, J.T. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef]
- Wang, Z.; Valera, J.C.; Zhao, X.; Chen, Q.; Gutkind, J.S. mTOR co-targeting strategies for head and neck cancer therapy. Cancer Metastasis Rev. 2017, 36, 491–502. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature, 2015; 517, 576–582. [Google Scholar] [CrossRef]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Day, T.A.; Shirai, K.; O’Brien, P.E.; Matheus, M.G.; Godwin, K.; Sood, A.J.; Kompelli, A.; Vick, J.A.; Martin, D.; Vitale-Cross, L.; et al. Inhibition of mTOR Signaling and Clinical Activity of Rapamycin in Head and Neck Cancer in a Window of Opportunity Trial. Clin. Cancer Res. 2019, 25, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Shirai, K.; Day, T.A.; Szabo, E.; Waes, C.V.; O’Brien, P.E.; Matheus, M.G.; Godwin, K.; Sood, A.J.; Vick, J.A.; Martin, D.; et al. A pilot, single arm, prospective trial using neoadjuvant rapamycin prior to definitive therapy in head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 6071. [Google Scholar] [CrossRef]
- Sun, Y.; Coppe, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Stern, P.L.; van der Burg, S.H.; Hampson, I.N.; Broker, T.R.; Fiander, A.; Lacey, C.J.; Kitchener, H.C.; Einstein, M.H. Therapy of human papillomavirus-related disease. Vaccine 2012, 30 (Suppl. 5), F71–F82. [Google Scholar] [CrossRef]
- Skeate, J.G.; Woodham, A.W.; Einstein, M.H.; Da Silva, D.M.; Kast, W.M. Current therapeutic vaccination and immunotherapy strategies for HPV-related diseases. Hum. Vaccin. Immunother. 2016, 12, 1418–1429. [Google Scholar] [CrossRef]
- Smola, S. Immunopathogenesis of HPV-Associated Cancers and Prospects for Immunotherapy. Viruses 2017, 9, 254. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.-H.; Peng, W.; Milton, D.; Amaria, R.N.; Glitza, I.C.; Hwu, W.-J.; Patel, S.P.; Wong, M.K.K.; Woodman, S.E.; Yee, C.; et al. Phase I/II study of the PI3Kβ inhibitor GSK2636771 in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. J. Clin. Oncol. 2018, 36, TPS9596. [Google Scholar] [CrossRef]
- Tolcher, A.; Hong, D.; Sullivan, R.; Mier, J.; Shapiro, G.; Chmielowski, B.; Ribas, A.; Postow, M.; Pearlberg, J.; Brail, L.; et al. Abstract CT089: IPI-549-01 - A Phase 1/1b, first-in-human study of IPI-549, a PI3K-γ inhibitor, as monotherapy and in combination with nivolumab in patients with advanced solid tumors. Cancer Res. 2017, 77, CT089. [Google Scholar] [CrossRef]
- Hellner, K.; Münger, K. Human papillomaviruses as therapeutic targets in human cancer. J. Clin. Oncol. 2011, 29, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bossler, F.; Hoppe-Seyler, K.; Hoppe-Seyler, F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2188. https://doi.org/10.3390/ijms20092188
Bossler F, Hoppe-Seyler K, Hoppe-Seyler F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. International Journal of Molecular Sciences. 2019; 20(9):2188. https://doi.org/10.3390/ijms20092188
Chicago/Turabian StyleBossler, Felicitas, Karin Hoppe-Seyler, and Felix Hoppe-Seyler. 2019. "PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells" International Journal of Molecular Sciences 20, no. 9: 2188. https://doi.org/10.3390/ijms20092188
APA StyleBossler, F., Hoppe-Seyler, K., & Hoppe-Seyler, F. (2019). PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. International Journal of Molecular Sciences, 20(9), 2188. https://doi.org/10.3390/ijms20092188