Abstract
Structural information of biological macromolecules is crucial and necessary to deliver predictions about the effects of mutations—whether polymorphic or deleterious (i.e., disease causing), wherein, thermodynamic parameters, namely, folding and binding free energies potentially serve as effective biomarkers. It may be emphasized that the effect of a mutation depends on various factors, including the type of protein (globular, membrane or intrinsically disordered protein) and the structural context in which it occurs. Such information may positively aid drug-design. Furthermore, due to the intrinsic plasticity of proteins, even mutations involving radical change of the structural and physico–chemical properties of the amino acids (native vs. mutant) can still have minimal effects on protein thermodynamics. However, if a mutation causes significant perturbation by either folding or binding free energies, it is quite likely to be deleterious. Mitigating such effects is a promising alternative to the traditional approaches of designing inhibitors. This can be done by structure-based in silico screening of small molecules for which binding to the dysfunctional protein restores its wild type thermodynamics. In this review we emphasize the effects of mutations on two important biophysical properties, stability and binding affinity, and how structures can be used for structure-based drug design to mitigate the effects of disease-causing variants on the above biophysical properties.
1. Effect of Mutations on Stability and Binding
The study of the effect of amino acid mutations within proteins has been a traditional chapter in protein science. Earlier studies were applied site-directed mutagenesis to assess the importance of an amino acid for stability and function of the corresponding protein. Nowadays, the focus has shifted to understanding the effects caused by genetic variants, namely, non-synonymous single nucleoside polymorphisms (nsSNP), with respect to disease predisposition. The phage-T4 lysozyme, for example, has served as one of the most well-studied systems with regard to mutations [1]. These studies were facilitated by the availability of X-ray structures (native and mutants) allowing for structural investigations of the effects on protein packing, stability, and activity. At the same time, lysozyme mutants in human have also been studied to characterize the molecular mechanism of diseases, such as hereditary systemic amyloidosis [2]. Similarly, the barnase–barstar protein-inhibitor complex was subjected to extensive mutagenesis to reveal the role of various residues on binding affinity [3,4,5]. This high-resolution complex has served as a model system to study protein–protein recognition by single and double mutant cycles [4,5]. Such studies have also served to rationalize optimization theories in the binding energetics [5,6] generally applicable to protein–protein recognition. Recent advances in the study of genetic (DNA) variants in the same system have also explored their influence in the manifestation of differential immunogenicity, and this very property has then been applied in bio-therapeutics, for example, by constructing heterodimeric barnase–barstar DNA vaccine molecules [7], ground-breaking in the development of novel DNA vaccines. Effectively, numerous works in molecular biophysics were and are focusing on understanding the effects of mutations on protein stability and binding. Below we review the relevant concepts and works associated with the two most fundamental biophysical events in protein science, folding and binding.
To begin with the effects of amino acid substitutions on protein folding, we emphasize that the same substitution may have different effects when occurring in globular, membrane or intrinsically disordered proteins. It is perhaps good to reiterate the fact that globular proteins are characterized by the presence of densely packed interiors (hydrophobic core) with packing densities (0.7 to 0.8) resembling that of crystalline solids [8] and, that, interior packing is known to be one of the most dominant forces in protein folding [9], also related to the stability, dynamics and the de novo design of the foldable globules. The dense interior packing within globular proteins is known to be achieved by a nucleation–condensation of ‘packing motifs’ [10], concomitant to the rapid collapse of hydrophobic residues in an aqueous environment. On the other hand, helix packing in integral membrane proteins [11] inserted within the lipid bilayer does not involve the ‘hydrophobic effect’ and yet, scales to an equivalent magnitude of packing to that of the globular proteins [12]. The polar vs. hydrophobic environment presented in the two cases demands differential amino acid compositions in the two types of proteins to achieve an equivalent degree of packing in both. Interestingly, small hydrophobic (Gly, Ala) [11,12] and small hydroxyl-containing (Ser, Thr) [12] amino acids have been found to contribute the most in tight packing of helices in membrane proteins as opposed to large hydrophobic and aromatic residues [10,13] in globular protein interiors. Apart from the tight packing of helices, membrane proteins are also known to involve a distinct pattern of charges [14,15] embedded in their sequence to remain stable and active within the amphiphilic lipid bilayer. In dramatic contrast, in the case of intrinsically disordered proteins (IDPs), the interior packing is practically negligible [16], since (unlike globular proteins) the few hydrophobic residues in them are so placed that it forbids the possibility of a hydrophobic collapse to attain a stable fold with a well-packed core. This, in fact, enables them to retain their characteristic disorder or dynamic flexibility using existing conformational ensembles rather than a single stably folded global minima structure similar to either globular [9] or membrane proteins [17]. The major component in retaining this dynamic flexibility in IDPs is electrostatic interactions [18] involving hydrogen bonds, salt-bridges, charge–dipole, and dipole–dipole interactions. Hence, when subjected to mutational studies, the sites to perform the mutations are chosen based on the knowledge-based prediction of the expected differential effect in folding, stability, and dynamics for the three major class of proteins. For example, salt-bridge mutations have served to constitute one of the prime chapters in understanding the modus operandi in IDPs [19,20] while the study of hydrophobic core mutations has traditionally served to probe interior packing within globular proteins [21], which will be discussed in more detail in the next section. Electrostatics also serves as an indispensable component in the folding and stability of globular proteins [22]. For membrane proteins, mutations have been chosen mostly based on structure–function relationships [23], such as oligomerization [24], thermostability [25], etc., involving both packing and electrostatics. There have also been instances of strategic point mutations (e.g., involving proline and/or glycine the well-known helix-breakers) introducing kinks (Figure 1) and wedging on transmembrane helix–helix interfaces [26]. Apart from the specific emphasis on individual structure–functional attributes of these different classes of proteins, mutational studies have also been attempted as a mean to trace their evolutionary origin (or common ancestor), particularly relevant in the context of the ‘globular-disordered interface’ [27,28] in proteins.
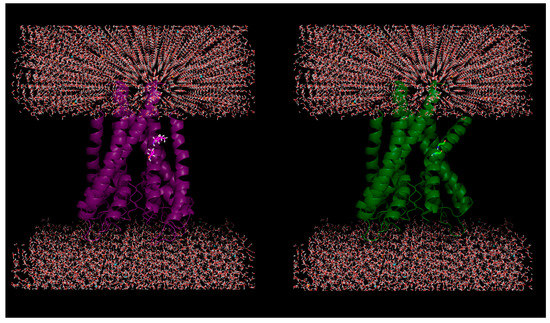
Figure 1.
Plausible effect of mutations in membrane proteins: Helical kink is introduced due to in silico mutations of two successive residues (100-Ile, 101-Thr) to glycine (helix breaker) in a KcsA potassium channel protein (PDB ID: 1J95).
2. Mutation and its Compensation: Structural Plasticity and Conformational Relaxation
Plasticity in the context of protein conformations [29,30,31] describes their adaptability in response to applied external forces (for example, by introducing mutations). This is a key physical property for protein dynamics, wherein, it serves to facilitate protein evolution and other protein functions, such as allostery and self-assembly [32]. Structural studies have further shown how conformational relaxation of both main- and side-chain atoms could compensate the deleterious effects of mutations, thereby, preserving the overall fold [33,34]. The random mutation of the 12 out of 13 core residues of ribonuclease barnase is an example where 23% of the mutants retained their enzymatic activity in vivo [35]. Other similar studies followed, and the idea of introducing strategic multiple mutations was eventually extended into the realm of de novo design of proteins [36]. Parallel (α/β)8—TIM barrel [37,38] served as an exemplary early model system, wherein, the specificity in side-chain packing as well as the pattern of hydrophobicities, both were detected to play their part. However, from all such studies, it was unmistakable that conformational plasticity is an inherent feature in proteins, resulting in structural relaxation to reduce the effect of mutations, particularly applicable in the context of multiple core mutations (Figure 2) in foldable globules [39], which is also relevant for IDPs [40,41].
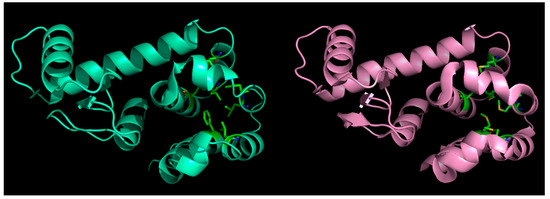
Figure 2.
Effect of mutations in globular proteins: seven core (hydrophobic) residues mutated to methionine (left panel: native, right: mutant) in phage T4-lysozyme and yet, the fold is preserved without almost any marked distortions. This happens because of ‘structural relaxation’ in proteins due to their inherent conformational plasticity (adaptability to changes).
3. Mutations in IDPs as Compared to Globular and Membrane Proteins
Although one of the hallmarks of IDPs is to harbor a high degree of structural plasticity, this may not always guarantee compensation of the deleterious effects caused by certain missense mutations. We should recall that many human diseases, such as cancer, diabetes, neurodegenerative and cardiovascular disorders, are associated with IDPs. Interestingly, similar mutational prototypes have generally been found to be more damaging in IDPs than in globular proteins [42,43,44], which is somewhat paradoxical, given the fact that IDPs have a substantially greater degree of structural plasticity and, therefore, are expected to have a corresponding greater potential to compensate for the mutational damage than that of globular proteins. However, the step-wise molecular and cellular consequences of a certain mutational prototype is hierarchical, multi-factored and complex. For example, amyloidosis may be defined as the formation of amyloid fibrils in protein polymers consisting of identical monomeric units which are the macromolecular end-effects responsible for many neurodegenerative disorders (e.g., Parkison’s, Alzheimer’s) which, in turn, is a consequence of β-aggregation. Again, β-aggregation may be accounted for by hydrophobicity and/or β-sheet propensity of a protein region [43]. Comparative studies in α- and γ-synuclein have revealed increased aggregation in the former with a higher propensity for β-sheets, which further suggests that an increased α-helical propensity in the amyloid-forming region may protect against γ-synuclein aggregation [45]. Interestingly, IDPs have been found to be more prone to amyloidosis in spite of having a much lower aggregation propensity (having only one third of aggregation nucleating regions) compared to globular (and membrane) proteins [43]. This high aggregation propensity also explains the considerable amount of structural frustration in globular proteins [43]. However, it may not be straightforward to draw a correlation between the solution conformations of amyloidogenic proteins and their pathogenicity. Though, lately, biophysical characterization of misfolded states and their aggregation mechanisms have gained considerable attention [46], aggregation pathways remain complex (whether pathogenic or not), often involving peculiarities of protein misfolding and characterized by remarkable polymorphism, wherein, the final product may consist of soluble oligomers, fibrils as well as amorphous aggregates [47].
As a matter of fact, the ‘disease-associated missense mutations’ in IDPs are also found in a higher prevalence with greater functional impact [48] than the ‘neutral polymorphisms’ [49]. More importantly, the IDP-disease-mutations are found to be associated with the ‘disorder-to-order transitions’ [41] at a far greater frequency than the polymorphic ones [46]. The cancerous mutations in p53 [48,50] in its DNA-binding domain are classic examples of IDP-disease-mutations, wherein, dramatic destabilization of the domain renders it disordered at physiological conditions [51]. Overall, there are many investigations associated with mutational studies on IDPs revealing their molecular evolution [27] and pathological features [20]. Traditionally, the mutations can be viewed as mostly ‘hereditary’ [20], chosen on the basis of geographic and ethnic variations, pedigree of individual families with a history of a certain (say, the Alzheimer’s) disease. At the molecular level, one of the major insights revealed by these mutational studies has been the influential role of salt-bridges in mediating the ‘mutation-induced rigidity’ associated with enhanced aggregation of the candidate IDP, which has also found support by recent molecular dynamic (MD) studies exploring the nitty-gritty and transient nature of salt-bridge dynamics (Figure 3) in IDPs [19].
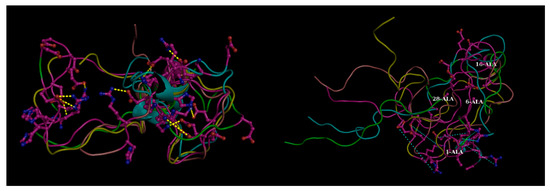
Figure 3.
Effect of mutations in disordered proteins. Four transient (flitting) salt bridge forming charged residues (1-Asp, 6-His, 16-Lys, 28-Lys) mutated to alanine in beta amyloid (Aβ42) resulting in the dismantling in salt-bridges globally throughout the structural ensemble (Left Panel: Mutant compared to the Right: Native). These transient salt-bridges continuously keep altering their partners throughout the whole simulation trajectory supporting different conformations at different time points and thereby supporting a conformational ensemble (illustrated in Figure 3. of Reference [19]). The yellow dashed lines in the left panel (native) show the salt-bridges found individually in the five randomly chosen conformers (within 4 Å) while the same connections are shown by thinner cyan dashed lines in the right panel to portray the absence of these ionic interactions (far greater than 4 Å). Molecular Dynamics simulation trajectories collected from Reference [19]. Briefly, explicit-water molecular dynamic (MD) simulation was performed with AMBER 12 at T = 300 K using the ff99SB force field with periodic boundary conditions and TIP3P water model. Figure reconstructed in Pymol.
Another effective and important way to classify mutations may be based on the actual consequence of a mutation as to whether it purely disrupts the structural integrity of a protein [52] or affects protein functions. For example, proximal residues may co-evolve together in a protein fold to preserve global stability, while point mutations (including insertion–deletions) may potentially fine-tune protein function, modifying functional sites and protein interactions [53]. Again, functional mutations may be proximal or direct to the catalytic/active site [54,55] as well as distal (allosteric and regulatory). The effect of distal mutations have been found to propagate throughout the whole protein fold affecting both its dynamics and catalysis, wherein low-frequency torsional oscillations [56,57] appear to play a pivotal role. Mutational hot-spots [58] have been identified (e.g., in human monoacylglycerol lipase, human DNA polymerase β) based on such long-range communication hubs in protein conformational dynamics [59,60,61]. Such information may also potentially facilitate developing novel ligands with therapeutic value [59].
4. Probing the Role of Mutations in Diseases: Tracking Changes in Thermodynamic Parameters
Changes in folding and binding free energies (∆∆G) are the standard thermodynamic measures to probe the effect of mutations on protein stability and binding [50]. It has been demonstrated that for assessing the effect, one needs to take into account the relative change in ∆∆G with respect to the ∆GWT rather than considering ∆∆G alone [62]. Changes in ∆∆G were used to characterize sequence and structural patterns on human disease-causing amino acid variants [63,64]. Particular attention was paid to mutations involving a reversal of biophysical characteristics of the wild type residue(s). For example, salt-bridge mutations have been found to be typically disease-causing as demonstrated in the case of hyper-aldosteronism, wherein the mere removal of the charge (while keeping intact the side chain geometry) on a single strategic amino acid site (Glu → Gln) [65], and thereby effectively dismantling a critical salt-bridge, was found to be nitpicking. Salt-bridge mutations in IDPs have also been found to be deleterious with enhanced aggregation of the proteins (e.g., in Alzheimer’s and Parkinson’s Diseases) [20]. Recent MD simulation studies on IDPs have explored a plausible interpretation of the corresponding molecular events, wherein a considerable reduction in the conformational variation was found in Aβ42 upon dismantling both high persistence as well as transient salt-bridges [19].
Several computational approaches have been developed to predict folding and binding free energy changes (∆∆G) as a mean to link them with pathogenicity of mutations. These approaches vary from sequence-based [66,67], to structure-based [68,69,70], depending on the input [63,64]. Methodologies vary from empirical approaches [71,72], first-principle approaches [73], combination of knowledge-based terms and physics [74,75,76] to machine learning approaches [77,78]. It should be emphasized that for effective drug discovery, one needs to know not only the thermodynamic effects of mutation but also the 3D structure of the target biomolecule(s). Notably, for amyoidogenic proteins, kinetics may take over thermodynamics as aggregation is often found to be kinetically driven [79]. It is also important to consider that a protein is never isolated in a cellular context, rather, all cellular biochemical processes take place in heterogeneous, highly volume occupied, crowded environments [80,81], wherein stabilization of a particular protein [82] may occur by complex formation with specific partner molecules. Taking this into account, most biophysical experimental assays, as well as computational methods, may be seen as reductionist approaches, wherein free energy calculations may strongly be biased and would, therefore, require corrections by appropriate normalization factors [83], also taking into account convergence and sampling [84]. It is, therefore, of utmost importance to, at least, perform cross-validations of the thermodynamic parameters calculated between experimental (calorimetric and/or other indirect spectroscopic techniques) and structure driven computational approaches [85] wherever applicable and possible. In addition, to that end, there have been studies vividly addressing the thermodynamic consequences of excluded volume and macromolecular crowding, both, in vitro and in vivo using labeled tracer macromolecules [80]. Strategies have also been proposed to extend quantitative analyses of crowding from simple model systems to systems with increasing complexity up to the labels of intact cells [80].
5. Statistical Classification of Mutations Based on Their Degree of Harmfulness
The effect of some mutations is more pronounced [86] than others. To that end, statistical studies [64,87] have broadly classified the nsSNPs into two major categories: (i) polymorphic (or harmless) and (ii) disease variants. The influential causal factors considered in such statistical studies are genetic variations, frequency of occurrence, and statistical measure(s) of the degree of harmfulness [64,88]. The object of the exercise was to find empirical correlations between the variation type and the degree of harmfulness, if any. To that end, the entire combinatorial space of 380 possible amino acid mutations (20 amino acids each can be replaced by one out of the other 19 makes it 380) (that can occur from a set of 20 naturally occurring amino acids) was explored, and the frequency of each mutation in the corresponding database was recorded. To overcome any possible database-bias, the calculations were repeated as a means to cross-validate the results on updated database(s). Major observations were that in the HumVar dataset [89], 108 out of 380 possible mutations were never found, while, contrastingly, the top 26 most frequent variants made up as much as 46% of the whole dataset. As a matter of fact, only about one quarter (only 87 out of 380) of the variants were found to belong to the “harmless” category [64] in the same database. As a cross-validation, when the analysis was repeated in an expanded dataset of more than three-fold increased size, a jump was observed in the ‘polymorphic-to-disease variant’ ratio from 0.74 to 1.54 in the new compared to the older dataset. Such indifference resulting from database bias inherent to all these statistical/knowledge-based approaches actually speak in favor of using ∆∆G as a more reliable and preferred probe to be applied on a case-to-case basis to make predictions about the effect of a particular mutation in relation to pathogenicity.
6. Mitigating and Clustering the Effects of Disease-Causing Genetic Variants in Relation to Drug Design
With the rapid development of computer techniques, computer-aided approaches have been widely applied in aiding early-stage drug discovery both in industrial as well as in academic projects [90,91,92,93]. By discovering the potential compounds that target and affect the function of specific proteins, biological processes can be modulated to mitigate or eliminate the disease-causing effects [90,92]. Advances in human genome projects have provided a large plethora of target proteins for drug discovery projects [94,95]. Meanwhile, breakthroughs in structural biology have offered in-depth structural information of more and more targets and elucidated the disease mechanisms at the molecular level [96,97,98,99]. Such advances have further stimulated the application of computational approaches to integrate the available structural information, functional mechanism, and physico–chemical properties related to drug discovery [91,100]. Drug discovery traditionally is a time- and energy-consuming process and it would be difficult to imagine that the process can be reduced to the time-span of (say) cancer illness of a single patient. Then again, discovery of compounds to mitigate or eliminate the disease-causing effects induced by a specific amino acid mutation is the main goal of personalized medicines [101]. In other words, benefiting from an individual’s genomic information (by means of comparing to the sequence consensus of the standard human genome), followed by the identification of drug-like compounds such as screening of FDA approved drugs over a particular novel mutation may potentially provide precise treatment to target specific disease-associated mutations on these individuals. In addition, the individual’s genomic information can be of great help to include or exclude patients most appropriate for clinical trails at the final stage of drug development, which would not only increase the safety of the patients but also accelerate the drug testing process.
In a drug-design methodology, targeting specific disease-causing mutations and elucidation of the mutational effects together is of great importance, especially for the approaches requiring structural information of the target protein. Free energy calculation methods are used to determine the dominant effects of mutations, whether affecting protein stability, protein binding or both. With the in-depth analysis of the effect of mutations at the molecular-level, the disease-causing mutations in the target proteins can further be clustered by their major effects such as destabilizing mutation, catalytic mutations, mutations affecting dimerization or protein conformations [102,103,104]. Such types of classification can help designing drugs for certain groups of mutations with similar effects and is, thus, applicable to a broader spectrum of diagnosis and therapy.
7. Structure-Based Approach in Drug Design
Structure-based drug design (SBDD) is the computational approach that relies on knowledge of 3D structures (Figure 4) of the biological targets to identify or design the potential chemical structure suitable for clinical tests [100,105]. With the explosion of genomic, functional, and structural information in recent decades, the majority of biological targets with 3D structure have been identified and stimulated the applications of structure-based approaches in the current design pipeline. SBDD is popular for virtual screening to filter the drug-like compounds from a large library of small molecules, including widely applied approaches, such as docking and structure-based pharmacophore design [105]. While the established high-throughput screening (HTS) [106] allows for automatic testing of a wide range of compounds (up to millions), the low success rate and high cost together limit its applications. Alternatively, one can use computational approaches to reduce the number of compounds subjected to testing [105,106], wherein docking and structure-based pharmacophore design are the two most popular approaches, targeting deleterious mutations.

Figure 4.
Schematic presentation of the drug discovery process to mitigate the effects of disease-causing mutations.
7.1. Docking
Docking is one of the most common approaches for compound screening, and the basic idea is to use scoring functions to evaluate the fitness of the target protein in complex with the docked compound [92]. Currently, vast docking programs have been developed to perform fast docking calculations with a wide array of protocols and scoring functions, such as Dock6 [107], Autodock Vina [108], Glide [109], Surflex [110], and many others. Such approaches require the structure of the target protein to be either experimentally solved or computationally modeled. As mentioned above, one should do intensive modeling to generate the best representative structure or set of structures to be subjected to docking [91,111]. In the past, SBDD has been widely applied in mitigating the effects of mutations related to many common diseases. Examples include the p53 protein, which is the so-called “guardian protein” in cancer, functioning as a tumor-suppressor [112]. Again, only, some mutations in p53 result in the malfunctioning of the protein and increase the risk of cancers [113]. In cancer patients, mutations destabilizing the DNA binding to p53 are frequently observed and rescuing the native function(s) in the ‘mutant p53 protein’ is one central objective in current cancer research [114,115]. In the past, it has been shown that the binding of small molecules can stabilize the DNA binding domain and rescue mutant functions [110]. Recent work modeled the wild-type and several mutants [116] to elucidate the mechanism of p53 reactivation [116]. A novel transiently open L1/L3 pocket was identified and indicated the exposure of Cys-124 in the formation of such cavity [116]. Such finding is crucial as Cys-124 has been suggested to be the covalent docking site for known alkylating p53 stabilizers [117] while compounds can be docked onto this pocket to search for other potential stabilizers. As a matter of fact, 1,324 compounds from the NCI/DTP Open Chemical Repository Diversity Set II were docked onto the generated ensemble structures of R273H cancer mutant out of which 45 compounds were selected for biological assay [116]. Finally, one compound, stictic acid (NSC-87511) (Figure 5) was experimentally validated to be an efficient reactivation compound for mutant p53 [116].
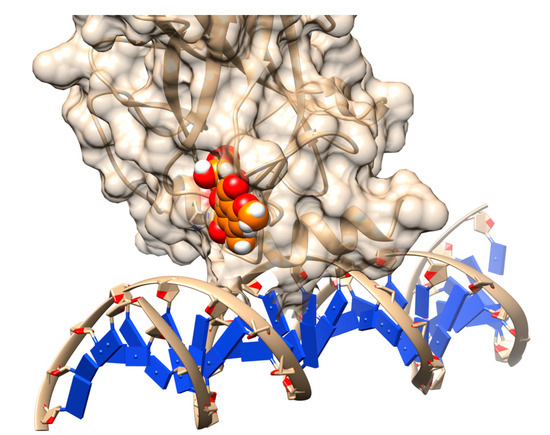
Figure 5.
Rescuing the activity of mutant p53 by binding stictic acid into the open L1/L3 pocket. The representative scheme is generated using the structure of p53 core domain complex with DNA (PDB: 1TSR). Atoms in sphere representation belong to stictic acid in a given docked pose (hetero atoms colored as per the default coloring scheme of Chimera). The DNA bases are represented as blue squares.
Besides cancer research, docking based screening has also been used in rare diseases. Snyder–Robinson Syndrome (SRS) is a rare X-linked mental disease, caused by the malfunctioning of an important human enzyme, the spermine synthase [118]. Spermine synthase functions as homo-dimer and mutations affecting the dimerization such as G56S are shown to abolish the enzyme activity to result in the disease [96,118]. Recent work has targeted identification of dimer stabilizers by binding to the mutant homo-dimer interface [119]. Integrated large commercial compound libraries were used for this docking-based virtual screening with the representative structures of the dimer [119]. The best-ranked 51 compounds were then subjected to experimental screening out of which three top-ranked compounds (also known as ‘leads’) have been shown to enhance the catalytic activity up to 30% [119,120].
7.2. Structure-Based Pharmacophore Design
Pharmacophore models can be used to make an ensemble of abstract steric and electronic features representing macromolecular (target protein) interactions with drug-like small molecules [121,122]. In other words, three-dimensional arrangements of these features such as hydrophobic centroids, aromatic rings and hydrogen bonds are representation of the binding mode between the ligand and the target [122,123]. Pharmacophores are generated from common features of active ligands, which are identified by aligning or superimposing the conformers of either ligand-target complexes or known active molecules [123]. Multiple degenerate atomic models can potentially be output from pharmacophore modeling programs requiring further optimization and validation to select the best one. Pharmacophore models are commonly used for virtual screening of active small molecules from large compound databases [121,122,123]. Such approaches can be more efficient than docking for certain targets, especially when a large number of existing known active compounds are available [124,125,126].
Pharmacophore models have also been used to identify active molecules to mitigate the effects of mutations in many diseases [108,109,110,111]. For cases where a sufficient number of active molecules are previously known for generating high-quality pharmacophore models, pharmacophore proves to be a powerful tool for drug ‘lead’ identification [106]. Recent work has applied structure-based pharmacophore analysis to identify the novel ROS-1 inhibitors to curb the drug resistance problem caused by mutations [127]. Proto-oncogene receptor tyrosine kinase ROS-1 is ectopicly and oncogenicly expressed in many cancers, mainly in non-small cell lung cancer (NSCLC) [127]. ROS-1 is highly homologous with the kinase domain of anaplastic lymphoma kinase (ALK) and FDA approved ALK inhibitors such as Crizotinib are experimentally validated as therapeutics against ROS-1 driven tumors [127]. However, these commercial ROS-1 inhibitors lack a broad spectrum of activity due to the growing resistance from ROS-1 mutations, primarily G2032R [128]. Following on, a pharmacophore model was built using the complex structure of both wildtype (WT) and mutant ROS-1 with previously known inhibitors to identify more general inhibitors against both WT and mutant [129,130]. Pharmacophore-based virtual screening was then performed to selected candidates from commercial databases with further filtering and scoring analysis. Five hits were eventually identified with good binding affinities to both WT and mutant [130].
Thus, pharmacophore essentially defines the interaction framework among the active ligands, and their specific targets [121,122] and the corresponding models can also be built with libraries of active ligands alone, in the absence of the 3D structure of the target—an approach known as ligand based pharmacophore. The models, therein, can then be trained for discrimination between active and inactive molecules [121]. In fact, this serves as the prime reason of widespread use of pharmacophore models in virtual screening especially when lacking the target structure. In addition, as the pharmacophore model represents the binding (or interaction map) of ‘active compounds-target interaction’, it provides a plausible relationship between the structure and the ligand activity and could help to elucidate the underlying biochemical mechanism to further guide the design of the novel active compounds [122]. For example, by exploring the different pharmacological properties, recent studies have seemed to improve the potency of existing pharmacophore and designed novel epidermal growth factor receptor (EGFR) inhibitor potentially inhibited by primary mutants (L858R, del9) and drug-resistant mutants, such as L858R/T790M [128].
8. Ligand-Based Approaches in Drug Design
In the lack of structural information of the target protein(s), the aforementioned structure-based approaches may not be suitable for drug design. As an alternative, ligand-based drug design (LBDD) can be applied to aid such cases [131,132,133,134]. Ligand-based methods only focus on the analysis of physico–chemical properties of known ligands that interact with the target of interests. Most popular approaches, however, are the quantitative structure–activity relationship (QSAR) models and the ligand-based pharmacophore modeling [134]. In terms of drug design, targeting the mutant proteins, LBDD could be efficient for novel mutations whose effects have not yet been investigated.
The basic assumption in ligand-based drug design is that small molecules with similar shape and biophysical properties will likewise interact with the same target receptor [123,131]. By identifying the fingerprints of known active ligands and constructing LBDD models, large databases can be screened to retrieve the novel compounds as potential leads for the target of interest [134]. QSAR is a widely applied LBDD approach, which utilizes mathematical models to correlate the physio–chemical properties of compounds to their experimentally measured bio-activity. Generally, QSAR methodology identifies the molecular descriptors associated with properties of the ligands and further uses mathematical models to discover correlations between the molecular descriptors and their biological activity. Finally, these QSAR models are tested and validated for the predicted biological activity of the compounds. As it stands, the current state-of-the-art is to apply the QSAR models widely in computer-aided drug design, targeting the mutant protein(s). One major success is the discovery of the potential corrector for cystic fibrosis (CF) mutations, namely, F508del in cystic fibrosis transmembrane conductance regulator gene (CFTR) [132,133,134]. F508del is the most frequent CF causing mutation, which leads to the improper folding of the protein and its degradation [132]. Subsequent to the identification, QSAR analysis has further been applied to guide the synthesis of novel compounds to treat CF by improving the trafficking of the mutant CFTR (the CF corrector) [133]. Recent works have collected all compounds known to improve the F508del trafficking and then applied QSAR analysis to decipher the critical chemical descriptors for the potential F508del correctors [133]. A novel predictive model was then constructed with these descriptors to provide further guidelines to the design and optimization of the novel corrector [134]. Again, the combination of ligand and structure-based approaches is expected to add significantly more to the current state-of-the-art [135,136]. Such combinations can either be sequential, parallel or hybrid, integrated contextually into a drug discovery pipeline, and, have already shown much promise [136].
A more sophisticated case would be to consider targeting proteins that lack both 3D structures and known active ligands and, therefore, will not have sufficient information to build robust pharmacophore and QSAR models. On such instances, one may switch on to sequence-based ligand predictor approaches such as meta-structure [137]. The basic idea behind developing meta-structure is the transformation of 3D structural information into the topological space via calculating the residue interaction networks from a database [137,138]. The residues and the corresponding neighborhood relationships are represented by nodes and edges. The predictor is trained against sets of representative protein 3D structure to derive statistical topological information for all possible amino acid pairs and, thus, can be subsequently used to perform predictions solely based on primary sequences [137]. Based on the sequence analysis, the quantitative information about the local secondary structure and residue compactness for each residue can be acquired to describe the intricate interaction networks in the topological space for the target protein [137]. Such meta-structure features can further be applied in drug development especially for target proteins that lack 3D structural information. For example, inspired by the protein-structure similarity clustering (PSSC) approach in structure-based drug development [139], the meta-structure similarities in ligand binding site can be used to cluster proteins with similar ligand-binding properties. Thus, the meta-structure features of any one member of the cluster would serve as a valuable starting point for ligand development of other members in the cluster [137].
9. Aiding Drug Design by the Knowledge of Mutations on Globular, Membrane and Disordered Proteins
Understanding of molecular mechanism of disease-associated mutations can directly be applied to drug design [91,101]. Structural biology has been instrumental in such understanding, effectively contributing to early drug discovery [140], and, also elucidating the impacts of disease-associated mutations and drug resistance in cancers and infectious diseases. Information regarding the differential effects of mutations on globular, membrane and disordered proteins may serve beneficially to select and apply the most appropriate and effective strategy to design potential drug-like molecules for each individual case. The majority of diseases are directly associated with the alterations of binding stability or folding stability of mutated proteins [63]—probed by binding or folding free energies. Such information also indicates to what extent the mutations are disrupting the protein interactions or structural integrity providing important guidelines towards the design of stabilizers and/or inhibitors to mitigate or eliminate the deleterious effects of mutations.
In addition, having 3D structures of target proteins is of great advantage to be used in free energy calculations coupled with MD simulations to extensively investigate the underlying structural mechanism (e.g., disruption of the hydrophobic core or loss of hydrogen bonding) of the mutational effects on binding or folding. As discussed in an earlier section, such information has been successfully used to identify the correct drug-like molecules targeting the mutations related to Snyder–Robinson Syndrome (SRS). SRS is caused by the malfunctioning of the human enzyme, spermine synthase (a globular protein), wherein the known existing deleterious mutations affect the native protein functionality by a wide range of molecular mechanisms, such as dedimerization, destabilization of the monomer, and disruption of the catalytic core [96,119,120,141].
Mutational resistance towards drugs also limits the lifetime of many successful drugs. As an alternative to the design of novel drug-like molecules to overcome such resistance, strategies, such as ensemble-based protein design [142], have been developed to be administered early in the development process to predict and overcome the effects of possible mutational resistance (e.g., in dihydrofolate reductase of Staphylococcus aureus). Such design protocol has a dual attribute, namely, positive design to maintain catalytic function and negative design to interfere with binding of a lead inhibitor simultaneously.
Alteration in protein conformation and dynamics are also closely related to a significant number of human diseases [143,144,145]. Computational approaches, such as MD or Mote Carlo (MC) simulations, are powerful tools to study protein dynamics. Mutations can alter protein dynamics in various ways, such as altering local flexibility, transition in conformational states, allosteric regulations etc. Exploring allosteric regulations may serve as potential alternatives for the cases where the native binding pocket is deemed to be too difficult to bind with small molecules [91]. Solving experimental structures of destabilizing mutants is often found difficult, particularly for membrane proteins due to their inherent insolubility and instability [146,147] and in such cases, molecular modeling of mutant structures can give some guidelines about the mutational effects. Such alternative structures are frequently subjected to docking of compound libraries in virtual screening—a methodology known as “ensemble-based drug design” [148,149]. Especially for IDPs where there is a definite lack of ordered structures, molecular modeling and MD simulation have together been widely applied to retrieve the representative structural ensemble in structure-based drug design [145,150].
On the other hand, high-throughput screening and rational drug design have considerably aided drugging membrane protein interactions as they are accessible on the cell surface and can directly alter cellular signaling [151]. This, in fact, is the key reason why the majority of therapeutics target membrane proteins. Techniques, such as alanine scanning, have also served to identify stabilizing mutations in the computational design of membrane proteins as well as in drug development [152]. To that end, frameworks, such as RosettaMP, have been developed to provide a general membrane representation that interfaces with scoring, conformational sampling, and mutation routines offering great ease and flexibility to integrate them into new design protocols [153]. Peptide architectonics [154] have been a relatively new addition in the subject, wherein, the idea is to select for sub-sequences of a native peptide, selectively toxic towards the pathogenic membrane proteins alone. As an alternative to drugging (as there is often a lack of structural information for transmembrane proteins), engineering of protein therapeutics [147] has also been attempted to membrane protein targets, though, its full potential is yet to be explored.
10. Conclusions
Macromolecular structural analyses may potentially be used to aid probing of genetic variants linked with disease. Such studies are usually complemented by a wide range of biophysical solution assays and computational modeling. Research along these directions has also opened up avenues towards developing diagnostic tools and plausible therapeutics. In such a context, it is of foremost importance to conceptualize (i) how traditionally mutational effects on protein stability and binding have been probed and (ii) the basis of the differential effects of mutations to different classes of proteins (globular, membrane, and disordered proteins) based on conformational relaxation, structural plasticity, compensation, and other physico–chemical factors. In the second half of the paper, we took the opportunity to discuss how this knowledge-base of the effect of mutations in globular, membrane, and disordered proteins may potentially aid drug design. As a probing technique, we particularly highlighted the importance of tracking changes in thermodynamic parameters (∆GWT) and also took the opportunity to discuss the limitations of knowledge-based approaches such as the statistical classification of mutations based on their degree of harmfulness. The review particularly highlighted the emergence of the ever-so-promising recent approach to computationally mitigate the effects of disease-causing genetic variants, alternative to the traditional approaches in designing inhibitors. A wide array of structure-based approaches in drug design including docking, structure-based pharmacophore design, and ligand-based approaches have been vividly discussed along with their proper context of applicability, as to whether they are to be aided in presence or absence of the experimental coordinates of the target protein and/or known ligands.
Author Contributions
E.A. conceived the problem. S.B. and Y.P. did the literature survey and wrote the paper with help from E.A. S.B. organized the manuscript and extensively edited it during the revisions. All authors read and approved the final manuscript.
Funding
The work was supported by a grant from NIH, grant number 1R01GM125639.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hawkins, P.N.; Booth, D.R.; Vigushin, D.M.; Tennent, G.A.; Soutar, A.K.; Totty, N.; Nguyen, O.; Blake, C.C.F.; Terry, C.J.; et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993, 362, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Hartley, R.W. Directed mutagenesis and barnase-barstar recognition. Biochemistry 1993, 32, 5978–5984. [Google Scholar] [CrossRef] [PubMed]
- Buckle, A.M.; Schreiber, G.; Fersht, A.R. Protein-protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry 1994, 33, 8878–8889. [Google Scholar] [CrossRef]
- Schreiber, G.; Fersht, A.R. Energetics of protein-protein interactions: Analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995, 248, 478–486. [Google Scholar] [CrossRef]
- Wang, T.; Tomic, S.; Gabdoulline, R.R.; Wade, R.C. How optimal are the binding energetics of barnase and barstar? Biophys. J. 2004, 87, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Spång, H.C.L.; Braathen, R.; Bogen, B. Heterodimeric Barnase-Barstar Vaccine Molecules: Influence of One versus Two Targeting Units Specific for Antigen Presenting Cells. PLoS ONE 2012, 7, e45393. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M. The interpretation of protein structures: Total volume, group volume distributions and packing density. J. Mol. Biol. 1974, 82, 1–14. [Google Scholar] [CrossRef]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Mapping the distribution of packing topologies within protein interiors shows predominant preference for specific packing motifs. BMC Bioinf. 2011, 12, 195. [Google Scholar] [CrossRef]
- Javadpour, M.M.; Eilers, M.; Groesbeek, M.; Smith, S.O. Helix packing in polytopic membrane proteins: Role of glycine in transmembrane helix association. Biophys. J. 1999, 77, 1609–1618. [Google Scholar] [CrossRef]
- Eilers, M.; Shekar, S.C.; Shieh, T.; Smith, S.O.; Fleming, P.J. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 5796–5801. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Sen, M.; Bhattacharya, D.; Saha, P. The jigsaw puzzle model: Search for conformational specificity in protein interiors. J. Mol. Biol. 2003, 333, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Charneski, C.A.; Hurst, L.D. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol. Biol. Evol. 2014, 31, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.A.; Tipper, D.J. The Role of Charged Residues in Determining Transmembrane Protein Insertion Orientation in Yeast. J. Biol. Chem. 1996, 271, 24625–24633. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Skach, W.R. Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 2009, 16, 606–612. [Google Scholar] [CrossRef]
- Nakamura, H. Roles of electrostatic interaction in proteins. Q. Rev. Biophys. 1996, 29, 1–90. [Google Scholar] [CrossRef]
- Basu, S.; Biswas, P. Salt-bridge dynamics in intrinsically disordered proteins: A trade-off between electrostatic interactions and structural flexibility. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 624–641. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Gassner, N.C.; Baase, W.A.; Matthews, B.W. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc. Natl. Acad. Sci. USA 1996, 93, 12155–12158. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Applications of complementarity plot in error detection and structure validation of proteins. Indian J. Biochem. Biophys. 2014, 51, 188–200. [Google Scholar] [PubMed]
- Liang, J.; Naveed, H.; Jimenez-Morales, D.; Adamian, L.; Lin, M. Computational studies of membrane proteins: Models and predictions for biological understanding. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Fung, H.K.; Rajgaria, R.; Filizola, M.; Weinstein, H.; Floudas, C.A. Mutations Affecting the Oligomerization Interface of G-Protein-Coupled Receptors Revealed by a Novel De Novo Protein Design Framework. Biophys. J. 2008, 94, 2470–2481. [Google Scholar] [CrossRef]
- Zhou, Y.; Bowie, J.U. Building a Thermostable Membrane Protein. J. Biol. Chem. 2000, 275, 6975–6979. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Situ, A.J.; Ulmer, T.S. Structural and thermodynamic basis of proline-induced transmembrane complex stabilization. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sepulveda, E.; Hartmann, M.D.; Kogenaru, M.; Ursinus, A.; Sulz, E.; Albrecht, R.; Coles, M.; Martin, J.; Lupas, A.N. Origin of a folded repeat protein from an intrinsically disordered ancestor. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Baruah, A.; Biswas, P. Globular–disorder transition in proteins: A compromise between hydrophobic and electrostatic interactions? Phys. Chem. Chem. Phys. 2016, 18, 23207–23214. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Mas, G.; Hiller, S. Conformational plasticity of molecular chaperones involved in periplasmic and outer membrane protein folding. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef]
- Ikura, M.; Ames, J.B. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: Two ways to promote multifunctionality. Proc. Natl. Acad. Sci. USA 2006, 103, 1159–1164. [Google Scholar] [CrossRef]
- Bastolla, U.; Porto, M.; Roman, H.E. The emerging dynamic view of proteins: Protein plasticity in allostery, evolution and self-assembly. Biochim. Biophys. Acta 2013, 1834, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Buckle, A.M.; Cramer, P.; Fersht, A.R. Structural and energetic responses to cavity-creating mutations in hydrophobic cores: Observation of a buried water molecule and the hydrophilic nature of such hydrophobic cavities. Biochemistry 1996, 35, 4298–4305. [Google Scholar] [CrossRef]
- Eriksson, A.E.; Baase, W.A.; Zhang, X.J.; Heinz, D.W.; Blaber, M.; Baldwin, E.P.; Matthews, B.W. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [Google Scholar] [CrossRef]
- Axe, D.D.; Foster, N.W.; Fersht, A.R. Active barnase variants with completely random hydrophobic cores. Proc. Natl. Acad. Sci. USA 1996, 93, 5590–5594. [Google Scholar] [CrossRef] [PubMed]
- Dahiyat, B.I.; Sarisky, C.A.; Mayo, S.L. De novo protein design: Towards fully automated sequence selection. J. Mol. Biol. 1997, 273, 789–796. [Google Scholar] [CrossRef]
- Goraj, K.; Renard, A.; Martial, J.A. Synthesis, purification and initial structural characterization of octarellin, a de novo polypeptide modelled on the alpha/beta-barrel proteins. Protein Eng. 1990, 3, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Offredi, F.; Dubail, F.; Kischel, P.; Sarinski, K.; Stern, A.S.; Van de Weerdt, C.; Hoch, J.C.; Prosperi, C.; François, J.M.; Mayo, S.L.; et al. De novo backbone and sequence design of an idealized alpha/beta-barrel protein: Evidence of stable tertiary structure. J. Mol. Biol. 2003, 325, 163–174. [Google Scholar] [CrossRef]
- Teng, S.; Madej, T.; Panchenko, A.; Alexov, E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 2009, 96, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Kalmar, L.; Tompa, P.; Han, K.-H.; Selenko, P.; Dunker, A.K.; Daughdrill, G.W.; Uversky, V.N. The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 2013, 1, e24360. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Söderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Globular and disordered—The non-identical twins in protein-protein interactions. Front. Mol. Biosci. 2015, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004, 342, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, J.S.; Miles, A.J.; Araujo, A.P.U.; Wallace, B.A. Differential dehydration effects on globular proteins and intrinsically disordered proteins during film formation. Protein Sci. 2017, 26, 718–726. [Google Scholar] [CrossRef]
- Marsh, J.A.; Singh, V.K.; Jia, Z.; Forman-Kay, J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 2006, 15, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 2010, 277, 2940–2953. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Iakoucheva, L.M. Disease mutations in disordered regions–exception to the rule? Mol. Biosyst. 2012, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Mechanic, L.E.; Marrogi, A.J.; Welsh, J.A.; Bowman, E.D.; Khan, M.A.; Enewold, L.; Zheng, Y.-L.; Chanock, S.; Shields, P.G.; Harris, C.C. Polymorphisms in XPD and TP53 and mutation in human lung cancer. Carcinogenesis 2005, 26, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue mutations and their impact on protein structure and function: Detecting beneficial and pathogenic changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Daudé, D.; Topham, C.M.; Remaud-Siméon, M.; André, I. Probing impact of active site residue mutations on stability and activity of Neisseria polysaccharea amylosucrase. Protein Sci. 2013, 22, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Gerton, J.L.; Ohgi, S.; Olsen, M.; DeRisi, J.; Brown, P.O. Effects of Mutations in Residues near the Active Site of Human Immunodeficiency Virus Type 1 Integrase on Specific Enzyme-Substrate Interactions. J. Virol. 1998, 72, 5046–5055. [Google Scholar]
- Woods, K.N.; Pfeffer, J.; Dutta, A.; Klein-Seetharaman, J. Vibrational resonance, allostery, and activation in rhodopsin-like G protein-coupled receptors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Luk, L.Y.P.; Javier Ruiz-Pernía, J.; Dawson, W.M.; Roca, M.; Loveridge, E.J.; Glowacki, D.R.; Harvey, J.N.; Mulholland, A.J.; Tuñón, I.; Moliner, V.; et al. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. USA 2013, 110, 16344–16349. [Google Scholar] [CrossRef]
- Dixit, A.; Yi, L.; Gowthaman, R.; Torkamani, A.; Schork, N.J.; Verkhivker, G.M. Sequence and Structure Signatures of Cancer Mutation Hotspots in Protein Kinases. PLoS ONE 2009, 4, e7485. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Rajarshi, G.; Karageorgos, I.; Zvonok, N.; Gallagher, E.S.; Huang, H.; Vemuri, K.; Hudgens, J.W.; Ma, X.; Nasr, M.L.; et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Sci. Rep. 2018, 8, 1719. [Google Scholar] [CrossRef]
- Klvaňa, M.; Murphy, D.L.; Jeřábek, P.; Goodman, M.F.; Warshel, A.; Sweasy, J.B.; Florián, J. Catalytic Effects of Mutations of Distant Protein Residues in Human DNA Polymerase β: Theory and Experiment. Biochemistry 2012, 51, 8829–8843. [Google Scholar] [CrossRef]
- Souza, V.P.; Ikegami, C.M.; Arantes, G.M.; Marana, S.R. Mutations close to a hub residue affect the distant active site of a GH1 β-glucosidase. PLoS ONE 2018, 13, e0198696. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.-C.R.; Stratakis, C.A.; Williams, T.A.; et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics 2004, 20 (Suppl. 1), i63–i68. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM–A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef]
- Blanco, J.D.; Radusky, L.; Climente-González, H.; Serrano, L. FoldX accurate structural protein-DNA binding prediction using PADA1 (Protein Assisted DNA Assembly 1). Nucleic Acids Res. 2018, 46, 3852–3863. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012, 28, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Getov, I.; Petukh, M.; Alexov, E. SAAFEC: Predicting the Effect of Single Point Mutations on Protein Folding Free Energy Using a Knowledge-Modified MM/PBSA Approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Dai, L.; Alexov, E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int. J. Mol. Sci. 2016, 17, 547. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Wei, G.-W. Analysis and prediction of protein folding energy changes upon mutation by element specific persistent homology. Bioinformatics 2017, 33, 3549–3557. [Google Scholar] [CrossRef]
- Cang, Z.; Wei, G.-W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLOS Comput. Biol. 2017, 13, e1005690. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Shu, W.; Devlin, G.L.; Meehan, S.; Auer, S.; Dobson, C.M.; Welland, M.E. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA 2007, 104, 10016–10021. [Google Scholar] [CrossRef]
- Rivas, G.; Minton, A.P. Macromolecular crowding in vitro, in vivo, and in between. Trends Biochem. Sci. 2016, 41, 970–981. [Google Scholar] [CrossRef]
- Lee, H.-T.; Kilburn, D.; Behrouzi, R.; Briber, R.M.; Woodson, S.A. Molecular crowding overcomes the destabilizing effects of mutations in a bacterial ribozyme. Nucleic Acids Res. 2015, 43, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Senske, M.; Törk, L.; Born, B.; Havenith, M.; Herrmann, C.; Ebbinghaus, S. Protein Stabilization by Macromolecular Crowding through Enthalpy Rather Than Entropy. J. Am. Chem. Soc. 2014, 136, 9036–9041. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Hwang, H.; Pierce, B.G.; Weng, Z. Prediction of protein–protein binding free energies. Protein Sci. 2012, 21, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Domański, J.; Hedger, G.; Best, R.B.; Stansfeld, P.J.; Sansom, M.S.P. Convergence and Sampling in Determining Free Energy Landscapes for Membrane Protein Association. J. Phys. Chem. B 2017, 121, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, N.M.; Fenley, A.T.; Gilson, M.K. Computational Calorimetry: High-Precision Calculation of Host–Guest Binding Thermodynamics. J. Chem. Theory Comput. 2015, 11, 4377–4394. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Mutations: Types and Causes. Molecular Cell Biology 4th Edition 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21578/ (accessed on 22 December 2018).
- Gao, M.; Zhou, H.; Skolnick, J. Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure 2015, 23, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Luigi Martelli, P. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Michel, J. Current and emerging opportunities for molecular simulations in structure-based drug design. Phys. Chem. Chem. Phys. 2014, 16, 4465–4477. [Google Scholar] [CrossRef]
- Hung, C.-L.; Chen, C.-C. Computational approaches for drug discovery. Drug Dev. Res. 2014, 75, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in Structure-Based Rational Drug Design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, M.P.; Samara, G.; Hurwitz, M.; Passaro, E. Human Genome Project. Am. J. Surg. 1993, 165, 258–264. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073.
- Peng, Y.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the Effects of Missense Mutations Causing Snyder-Robinson Syndrome on the Stability and Dimerization of Spermine Synthase. Int. J. Mol. Sci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, Z.; Peng, Y.; Godar, S.; Getov, I.; Teng, S.; Alper, J.; Alexov, E. Forces and Disease: Electrostatic force differences caused by mutations in kinesin motor domains can distinguish between disease-causing and non-disease-causing mutations. Sci. Rep. 2017, 7, 8237. [Google Scholar] [CrossRef] [PubMed]
- Spellicy, C.J.; Norris, J.; Bend, R.; Bupp, C.; Mester, P.; Reynolds, T.; Dean, J.; Peng, Y.; Alexov, E.; Schwartz, C.E.; et al. Key apoptotic genes APAF1 and CASP9 implicated in recurrent folate-resistant neural tube defects. Eur. J. Hum. Genet. 2018, 26, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, K.; Niranjan, T.; Selvan, N.; Teo, C.F.; May, M.; Patel, S.; Weatherly, B.; Skinner, C.; Opitz, J.; Carey, J.; et al. Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability. J. Biol. Chem. 2017, 292, 8948–8963. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Hong, C.-J.; Lin, Y.-T.; Chang, W.-H.; Huang, H.-T.; Liao, J.-Y.; Chang, Y.-J.; Hsieh, Y.-F.; Cheng, C.-Y.; Liu, H.-C.; et al. Amyloid-beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS ONE 2012, 7, e35807. [Google Scholar] [CrossRef] [PubMed]
- Alexov, E. Advances in Human Biology: Combining Genetics and Molecular Biophysics to Pave the Way for Personalized Diagnostics and Medicine. Available online: https://www.hindawi.com/journals/ab/2014/471836/ (accessed on 8 January 2019).
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef]
- Peng, Y.; Myers, R.; Zhang, W.; Alexov, E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Peng, Y.; Suryadi, J.; Yang, Y.; Kucukkal, T.G.; Cao, W.; Alexov, E. Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. Int. J. Mol. Sci. 2015, 16, 27270–27287. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Nair, P.C.; Miners, J.O. Molecular dynamics simulations: From structure function relationships to drug discovery. In Silico Pharmacol 2014, 2, 4. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Wesley Hatfield, G.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Michael, A.J. Spermine synthase. Cell. Mol. Life Sci. 2010, 67, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Martiny, V.; Lagorce, D.; Ikeguchi, Y.; Alexov, E.; Miteva, M.A. Rational Design of Small-Molecule Stabilizers of Spermine Synthase Dimer by Virtual Screening and Free Energy-Based Approach. PLoS ONE 2014, 9, e110884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel approach for efficient pharmacophore-based virtual screening: Method and applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef]
- Lee, C.-H.; Huang, H.-C.; Juan, H.-F. Reviewing ligand-based rational drug design: The search for an ATP synthase inhibitor. Int. J. Mol. Sci. 2011, 12, 5304–5318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, H.; Zhang, Q.; Bao, X.; Yu, K.; Luo, X.; Zhu, W.; Jiang, H. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Silakari, O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018, 79, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Springsteel, M.F.; Galietta, L.J.V.; Ma, T.; By, K.; Berger, G.O.; Yang, H.; Dicus, C.W.; Choung, W.; Quan, C.; Shelat, A.A.; et al. Benzoflavone activators of the cystic fibrosis transmembrane conductance regulator: Towards a pharmacophore model for the nucleotide-binding domain. Bioorg. Med. Chem. 2003, 11, 4113–4120. [Google Scholar] [CrossRef]
- Pathak, D.; Chadha, N.; Silakari, O. Identification of non-resistant ROS-1 inhibitors using structure based pharmacophore analysis. J. Mol. Graph. Model. 2016, 70, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, X.; Wu, H.; Zou, F.; Yan, X.-E.; Chen, C.; Hu, C.; Yu, K.; Wang, W.; Zhao, P.; et al. Discovery of (R)-1-(3-(4-Amino-3-(3-chloro-4-(pyridin-2-ylmethoxy)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (CHMFL-EGFR-202) as a Novel Irreversible EGFR Mutant Kinase Inhibitor with a Distinct Binding Mode. J. Med. Chem. 2017, 60, 2944–2962. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.-L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired Resistance to Crizotinib from a Mutation in CD74–ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Noy, E.; Senderowitz, H. Combating cystic fibrosis: In search for CF transmembrane conductance regulator (CFTR) modulators. ChemMedChem 2011, 6, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.J.V.; et al. Synthesis and biological evaluation of novel thiazole- VX-809 hybrid derivatives as F508del correctors by QSAR-based filtering tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Konrat, R. The protein meta-structure: A novel concept for chemical and molecular biology. Cell. Mol. Life Sci. 2009, 66, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, Y.; Pons, M.; Konrat, R. Meta-structure correlation in protein space unveils different selection rules for folded and intrinsically disordered proteins. Mol. Biosyst. 2012, 8, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Waldmann, H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discov. Today 2005, 10, 471–483. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ascher, D.B.; Thomas, S.E.; Blundell, T.L. Genomes, structural biology and drug discovery: Combating the impacts of mutations in genetic disease and antibiotic resistance. Biochem. Soc. Trans. 2017, 45, 303–311. [Google Scholar] [CrossRef]
- Zhang, Z.; Norris, J.; Schwartz, C.; Alexov, E. In Silico and In Vitro Investigations of the Mutability of Disease-Causing Missense Mutation Sites in Spermine Synthase. PLoS ONE 2011, 6, e20373. [Google Scholar] [CrossRef]
- Frey, K.M.; Georgiev, I.; Donald, B.R.; Anderson, A.C. Predicting resistance mutations using protein design algorithms. Proc. Natl. Acad. Sci. USA 2010, 107, 13707–13712. [Google Scholar] [CrossRef]
- Gilchrist, S.; Gilbert, N.; Perry, P.; Östlund, C.; Worman, H.J.; Bickmore, W.A. Altered protein dynamics of disease-associated lamin A mutants. BMC Cell. Biol. 2004, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; De Felice, F.G. PABMB Lecture. Protein dynamics, folding and misfolding: From basic physical chemistry to human conformational diseases. FEBS Lett. 2001, 498, 129–134. [Google Scholar] [CrossRef]
- Cheng, L.S.; Amaro, R.E.; Xu, D.; Li, W.W.; Arzberger, P.W.; McCammon, J.A. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J. Med. Chem. 2008, 51, 3878–3894. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, C.; Michel, H. Crystallization of membrane proteins. Curr. Opin. Struct. Biol. 1997, 7, 697–701. [Google Scholar] [CrossRef]
- Lluis, M.W.; Godfroy, J.I.; Yin, H. Protein engineering methods applied to membrane protein targets. Protein Eng. Des. Sel. 2013, 26, 91–100. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef]
- Yu, C.; Niu, X.; Jin, F.; Liu, Z.; Jin, C.; Lai, L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 22298. [Google Scholar] [CrossRef]
- Chen, C.Y.-C.; Tou, W.I. How to design a drug for the disordered proteins? Drug Discov. Today 2013, 18, 910–915. [Google Scholar] [CrossRef]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef]
- Perez-Aguilar, J.M.; Saven, J.G. Computational Design of Membrane Proteins. Structure 2012, 20, 5–14. [Google Scholar] [CrossRef]
- Alford, R.F.; Koehler Leman, J.; Weitzner, B.D.; Duran, A.M.; Tilley, D.C.; Elazar, A.; Gray, J.J. An Integrated Framework Advancing Membrane Protein Modeling and Design. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Patra, H.K.; Islam, M.; Basu, S.; Griffith, M. Peptide Architectonics for Biotherapeutics. Indian Patent Application No. 201741036721; Filed on 16 October 2017,
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).