Thyroid Hormone Protects from Fasting-Induced Skeletal Muscle Atrophy by Promoting Metabolic Adaptation

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results
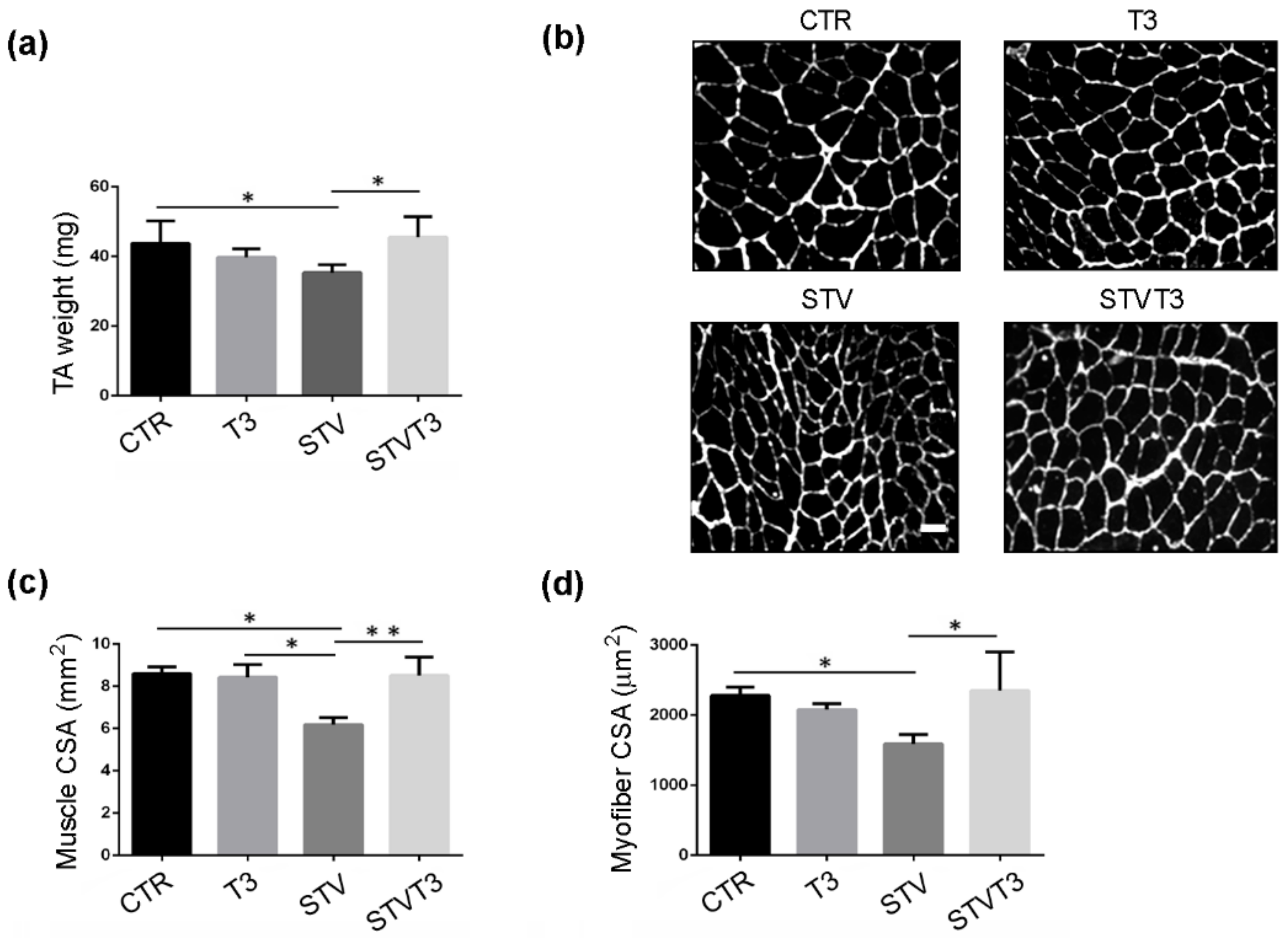
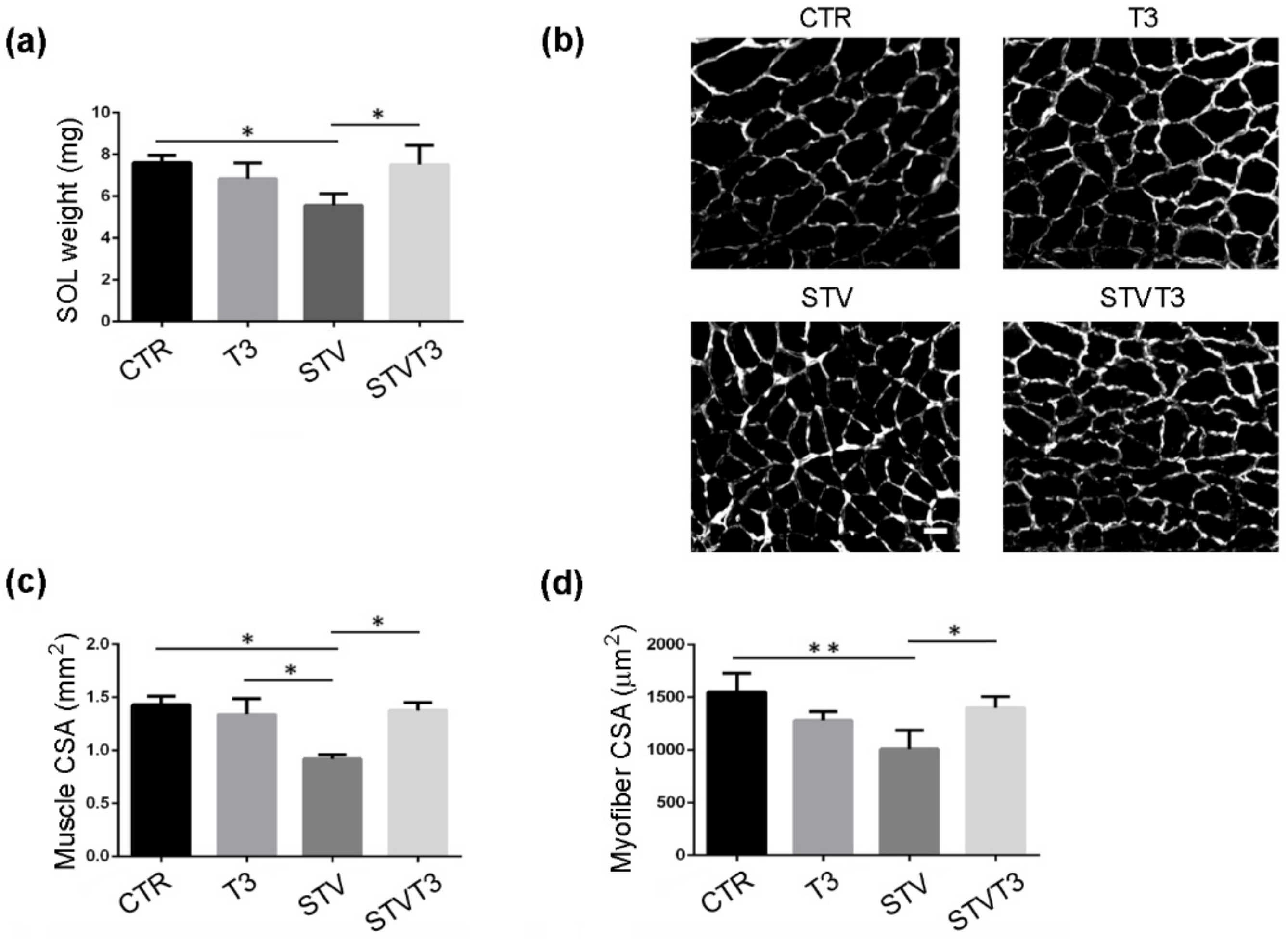
2.1. T3 Counteracts Starvation-Induced Skeletal Muscle Wasting
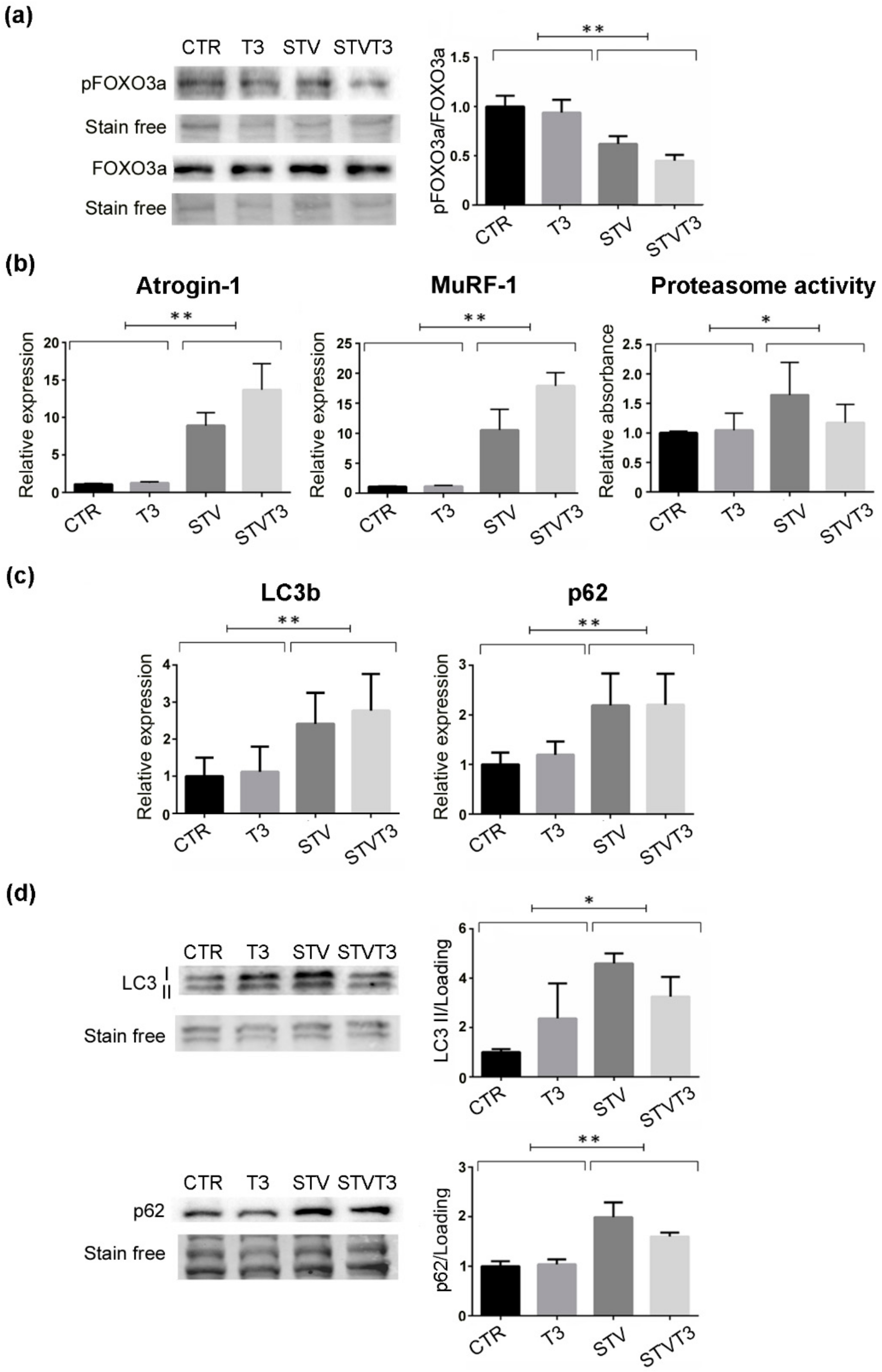
2.2. Thyroid Hormone Does Not Modulate the Catabolic Pathways Induced by Starvation
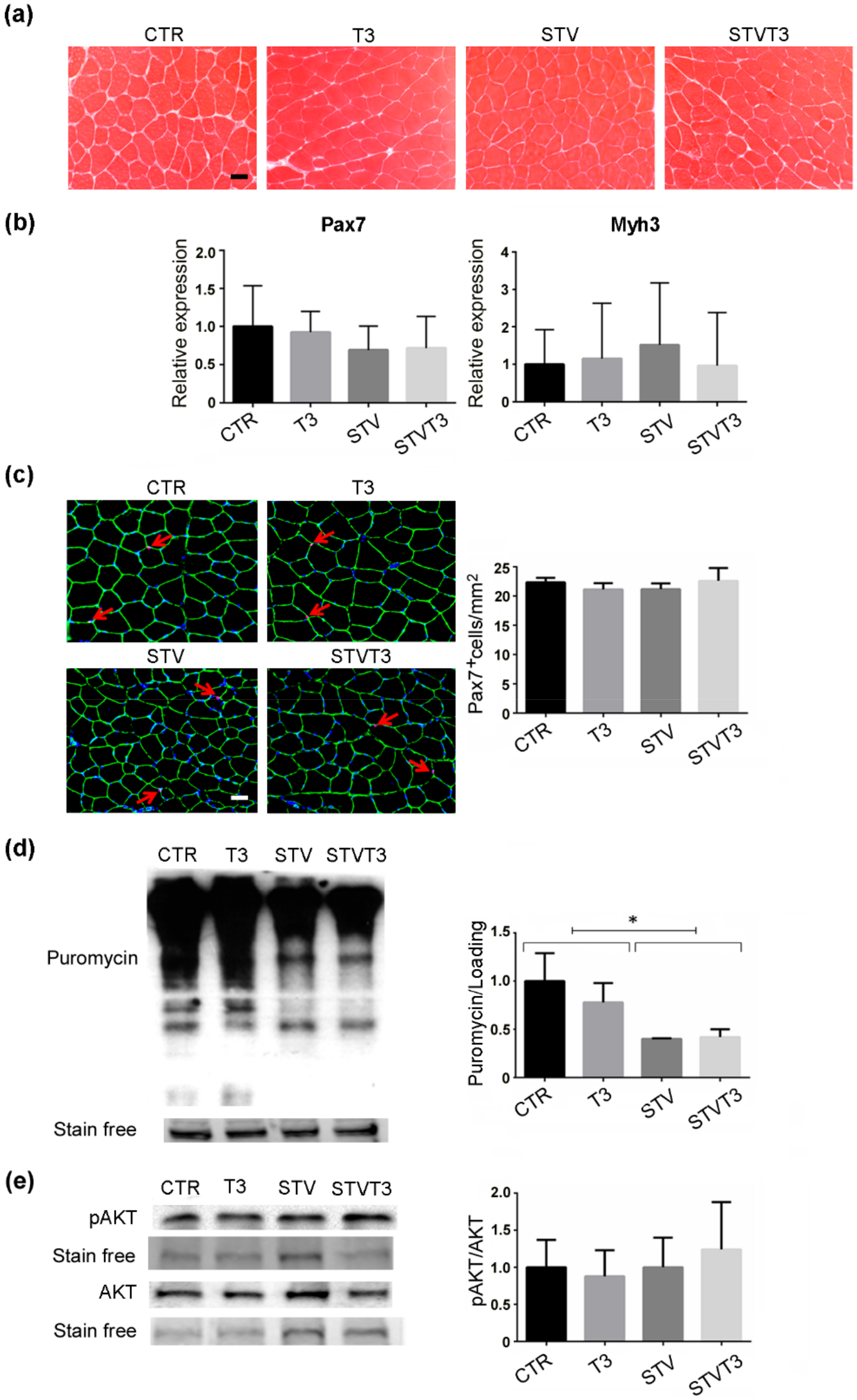
2.3. Thyroid Hormone Does Not Induce Skeletal Muscle Synthesis
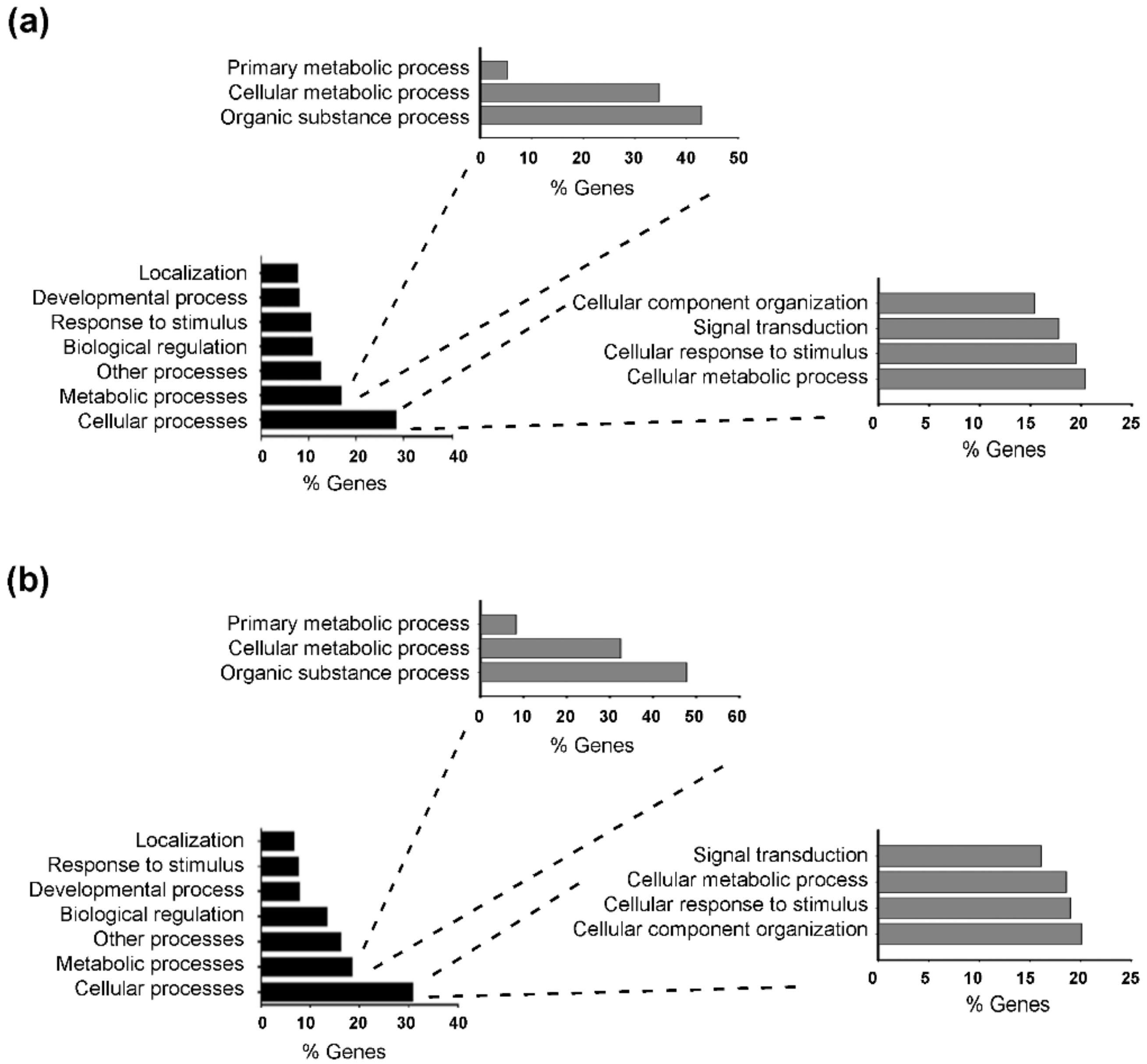
2.4. T3 Modulates Skeletal Muscle Gene Expression
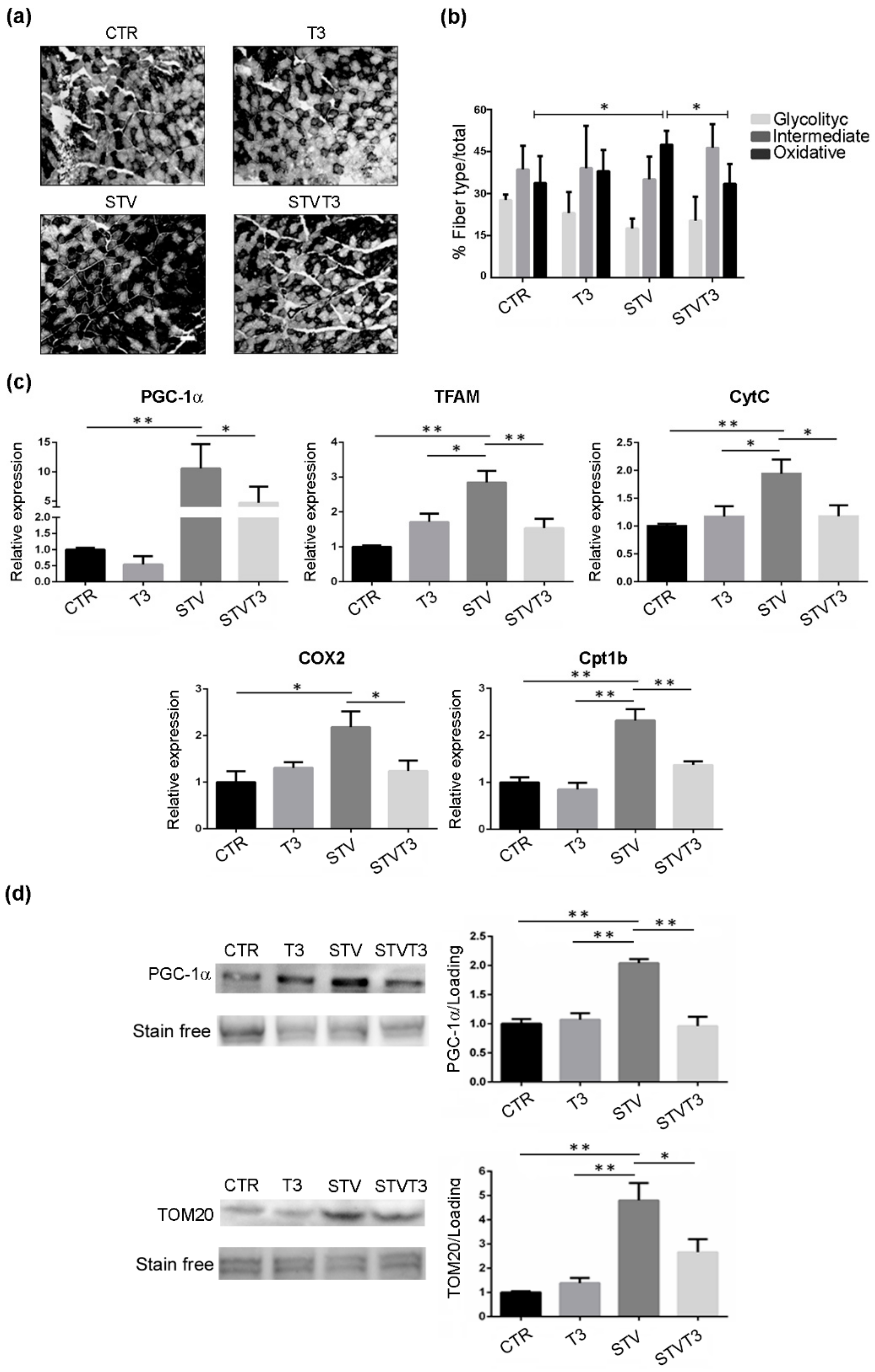
2.5. Thyroid Hormone Induces Metabolic Adaptation in Skeletal Muscle
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. Histology
4.2.1. Histological Sample Preparation
4.2.2. Immunofluorescence
4.3. Morphometric Analysis
4.4. RNA Isolation and Quantitative RT-PCR Analysis
4.5. RNA Sequencing
4.6. Bioinformatics Analysis
4.7. Proteasome Assay
4.8. Protein Extraction and Western Blot
4.9. SUnSET Assay
4.10. ELISA
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| THs | Thyroid hormones |
| TRs | Thyroid receptors |
| PGC-1α | Peroxisome proliferative activated receptor Gamma Coactivator 1 alpha |
| TA | Tibialis Anterior |
| ANOVA | Analysis of variance |
| STV | Starved |
| CTR | Control |
| Dio2 | Type-2 iodothyronine deiodinase |
| CSA | Cross-sectional area |
| Cpt1b | Carnitine palmitoyltransferase 1B |
References
- Visser, W.E.; Friesema, E.C.H.; Visser, T.J. Minireview: Thyroid Hormone Transporters: The Knowns and the Unknowns. Mol. Endocrinol. 2011, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Oetting, A.; Yen, P.M. New Insights into Thyroid Hormone Action. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Maruvada, P.; Baumann, C.T.; Hager, G.L.; Yen, P.M. Dynamic Shuttling and Intranuclear Mobility of Nuclear Hormone Receptors. J. Biol. Chem. 2003, 278, 12425–12432. [Google Scholar] [CrossRef] [PubMed]
- Osmak-Tizon, L.; Poussier, M.; Cottin, Y.; Rochette, L. Non-Genomic Actions of Thyroid Hormones: Molecular Aspects. Arch. Cardiovasc. Dis. 2014, 107, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kambe, F.; Moeller, L.C.; Refetoff, S.; Seo, H. Thyroid Hormone Induces Rapid Activation of Akt/Protein Kinase B-Mammalian Target of Rapamycin-P70 S6K Cascade through Phosphatidylinositol 3-Kinase in Human Fibroblasts. Mol. Endocrinol. 2005, 19, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kambe, F.; Yamauchi, M.; Seo, H. Thyroid-Hormone-Dependent Activation of the Phosphoinositide 3-Kinase/Akt Cascade Requires Src and Enhances Neuronal Survival. Biochem. J. 2009, 424, 201–209. [Google Scholar] [CrossRef]
- Verga Falzacappa, C.; Timperi, E.; Bucci, B.; Amendola, D.; Piergrossi, P.; D’Amico, D.; Santaguida, M.G.; Centanni, M.; Misiti, S. T(3) Preserves Ovarian Granulosa Cells from Chemotherapy-Induced Apoptosis. J. Endocrinol. 2012, 215, 281–289. [Google Scholar] [CrossRef]
- Oliva, F.; Berardi, A.C.; Misiti, S.; Verga Falzacappa, C.; Falzacappa, C.V.; Iacone, A.; Maffulli, N. Thyroid Hormones Enhance Growth and Counteract Apoptosis in Human Tenocytes Isolated from Rotator Cuff Tendons. Cell Death Dis. 2013, 4, e705. [Google Scholar] [CrossRef]
- Verga Falzacappa, C.; Patriarca, V.; Bucci, B.; Mangialardo, C.; Michienzi, S.; Moriggi, G.; Stigliano, A.; Brunetti, E.; Toscano, V.; Misiti, S. The TRbeta1 Is Essential in Mediating T3 Action on Akt Pathway in Human Pancreatic Insulinoma Cells. J. Cell. Biochem. 2009, 106, 835–848. [Google Scholar] [CrossRef]
- Verga Falzacappa, C.; Mangialardo, C.; Raffa, S.; Mancuso, A.; Piergrossi, P.; Moriggi, G.; Piro, S.; Stigliano, A.; Torrisi, M.R.; Brunetti, E.; et al. The Thyroid Hormone T3 Improves Function and Survival of Rat Pancreatic Islets during in Vitro Culture. Islets 2010, 2, 96–103. [Google Scholar] [CrossRef]
- Oliva, F.; Berardi, A.C.; Misiti, S.; Maffulli, N.; Oliva, F. Thyroid Hormones and Tendon: Current Views and Future Perspectives. Concise Review. Muscles Ligaments Tendons J. 2013, 3, 201–203. [Google Scholar] [PubMed]
- Canipari, R.; Mangialardo, C.; Di Paolo, V.; Alfei, F.; Ucci, S.; Russi, V.; Santaguida, M.G.; Virili, C.; Segni, M.; Misiti, S.; et al. Thyroid Hormones Act as Mitogenic and pro Survival Factors in Rat Ovarian Follicles. J. Endocrinol. Investig. 2019, 42, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Simonides, W.S.; van Hardeveld, C. Thyroid Hormone as a Determinant of Metabolic and Contractile Phenotype of Skeletal Muscle. Thyroid 2008, 18, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Bloise, F.F.; Cordeiro, A.; Ortiga-Carvalho, T.M. Role of Thyroid Hormone in Skeletal Muscle Physiology. J. Endocrinol. 2018, R57–R68. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Kim, N.-H.; Milanesi, A. Thyroid Hormone Signaling in Muscle Development, Repair and Metabolism. J. Endocrinol. Diabetes Obes. 2014, 2, 1046. [Google Scholar] [PubMed]
- Thompson, L.V. Skeletal Muscle Adaptations with Age, Inactivity, and Therapeutic Exercise. J. Orthop. Sport. Phys. Ther. 2002, 32, 44–57. [Google Scholar] [CrossRef]
- Baldwin, K.M.; Haddad, F.; Pandorf, C.E.; Roy, R.R.; Edgerton, V.R. Alterations in Muscle Mass and Contractile Phenotype in Response to Unloading Models: Role of Transcriptional/Pretranslational Mechanisms. Front. Physiol. 2013, 4, 284. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and Molecular Mechanisms of Muscle Atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef]
- Glass, D.J. Molecular Mechanisms Modulating Muscle Mass. Trends Mol. Med. 2003, 9, 344–350. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal Muscle Hypertrophy and Atrophy Signaling Pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple Types of Skeletal Muscle Atrophy Involve a Common Program of Changes in Gene Expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Heszele, M.F.C.; Price, S.R. Insulin-Like Growth Factor I: The Yin and Yang of Muscle Atrophy. Endocrinology 2004, 145, 4803–4805. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear Factor-Kappa B Signaling in Skeletal Muscle Atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle in Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms Regulating Skeletal Muscle Growth and Atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy Is Defective in Collagen VI Muscular Dystrophies, and Its Reactivation Rescues Myofiber Degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal Muscle Autophagy and Apoptosis during Aging: Effects of Calorie Restriction and Life-Long Exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. PGC-1 Coactivators: Inducible Regulators of Energy Metabolism in Health and Disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha Protects Skeletal Muscle from Atrophy by Suppressing FoxO3 Action and Atrophy-Specific Gene Transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S.; Lecker, S.H.; Jagoe, R.T. Proteolysis in Illness-Associated Skeletal Muscle Atrophy: From Pathways to Networks. Crit. Rev. Clin. Lab. Sci. 2011, 48, 49–70. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Mammalian Skeletal Muscle Fiber Type Transitions. Int. Rev. Cytol. 1997, 170, 143–223. [Google Scholar]
- Pette, D.; Staron, R.S. Transitions of Muscle Fiber Phenotypic Profiles. Histochem. Cell Biol. 2001, 115, 359–372. [Google Scholar]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef]
- Salvatore, D.; Simonides, W.S.; Dentice, M.; Zavacki, A.M.; Larsen, P.R. Thyroid Hormones and Skeletal Muscle–New Insights and Potential Implications. Nat. Rev. Endocrinol. 2014, 10, 206–214. [Google Scholar] [CrossRef]
- Verga Falzacappa, C.; Mangialardo, C.; Madaro, L.; Ranieri, D.; Lupoi, L.; Stigliano, A.; Torrisi, M.R.; Bouchè, M.; Toscano, V.; Misiti, S. Thyroid Hormone T3 Counteracts STZ Induced Diabetes in Mouse. PLoS ONE 2011, 6, e19839. [Google Scholar] [CrossRef]
- Muscat, G.E.O.; Mynett-johnson, L.; Dowhan, D.; Downes, M. Activation of MyoD Gene Transcription by 3,5,3′-Triiodo-L-Thyronine: A Direct Role for the Thyroid Hormone and Retinoid X Receptors. Nucleic Acids Res. 1994, 22, 583–591. [Google Scholar] [CrossRef]
- Iwaki, H.; Sasaki, S.; Matsushita, A.; Ohba, K.; Matsunaga, H. Essential Role of TEA Domain Transcription Factors in the Negative Regulation of the MYH 7 Gene by Thyroid Hormone and Its Receptors. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Jakubiec-Puka, A.; Ciechomska, I.; Mackiewicz, U.; Langford, J.; Chomontowska, H. Effect of Tyroid Hormone Oh the Myosin Heavy Chain Isoforms in Slaw and Fast Muscles of the Rat. Acta Biochim. Pol. 1999, 46, 823–835. [Google Scholar] [PubMed]
- Lesmana, R.; Sinha, R.A.; Singh, B.K.; Zhou, J.; Ohba, K.; Wu, Y.; Yau, W.W.; Bay, B.-H.; Yen, P.M. Thyroid Hormone Stimulation of Autophagy Is Essential for Mitochondrial Biogenesis and Activity in Skeletal Muscle. Endocrinology 2016, 157, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, A.; Lee, J.-W.; Yang, A.; Liu, Y.-Y.; Sedrakyan, S.; Cheng, S.; Perin, L.; Brent, G.A. Thyroid Hormone Receptor Alpha Is Essential to Maintain the Satellite Cell Niche During Skeletal Muscle Injury and Sarcopenia of Aging. Thyroid 2017, 27, 1316–1322. [Google Scholar] [CrossRef]
- Milanesi, A.; Lee, J.-W.; Kim, N.-H.; Liu, Y.-Y.; Yang, A.; Sedrakyan, S.; Kahng, A.; Cervantes, V.; Tripuraneni, N.; Cheng, S.; et al. Thyroid Hormone Receptor α Plays an Essential Role in Male Skeletal Muscle Myoblast Proliferation, Differentiation, and Response to Injury. Endocrinology 2016, 157, 4–15. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a Nonradioactive Method to Monitor Protein Synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Lai, K.-M.V.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional Activation of Akt in Adult Skeletal Muscle Induces Rapid Hypertrophy. Mol. Cell. Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic Control through the PGC-1 Family of Transcription Coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Virmani, A.; Pinto, L.; Bauermann, O.; Zerelli, S.; Diedenhofen, A.; Binienda, Z.K.; Ali, S.F.; van der Leij, F.R. The Carnitine Palmitoyl Transferase (CPT) System and Possible Relevance for Neuropsychiatric and Neurological Conditions. Mol. Neurobiol. 2015, 52, 826–836. [Google Scholar] [CrossRef]
- Kandarian, S.C.; Jackman, R.W. Intracellular Signaling during Skeletal Muscle Atrophy. Muscle Nerve 2006, 33, 155–165. [Google Scholar] [CrossRef]
- Ezeoke, C.C.; Morley, J.E. Pathophysiology of Anorexia in the Cancer Cachexia Syndrome. J. Cachexia Sarcopenia Muscle 2015, 6, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Beal, J.; Flynn, N. AIDS-Associated Anorexia. J. Physicians Assoc. AIDS Care 1995, 2, 19–22. [Google Scholar] [PubMed]
- Li, J.; Goldberg, A. Effects of Food Deprivation on Protein Synthesis and Degradation in Rat Skeletal Muscles. Am. J. Physiol. Content 1976, 231, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Tata, B.J.R.; Ernster, L.; Hedman, R. The Action of Thyroid Hormones at the Cell Level. Biochem. J. 1963, 408–428. [Google Scholar] [CrossRef]
- Bach-Huynh, T.-G.; Nayak, B.; Loh, J.; Soldin, S.; Jonklaas, J. Timing of Levothyroxine Administration Affects Serum Thyrotropin Concentration. J. Clin. Endocrinol. Metab. 2009, 94, 3905–3912. [Google Scholar] [CrossRef]
- Abdalla, S.M.; Bianco, A.C. Defending Plasma T3 Is a Biological Priority. Clin. Endocrinol. (Oxf.) 2014, 81, 633–641. [Google Scholar] [CrossRef]
- Jonklaas, J.; Burman, K.D.; Wang, H.; Latham, K.R. Single Dose T3 Administration: Kinetics and Effects on Biochemical and Physiologic Parameters. Ther. Drug Monit. 2015, 37, 110. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin-Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Sandri, M. Autophagy in Skeletal Muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef]
- Sandri, M. Protein Breakdown in Muscle Wasting: Role of Autophagy-Lysosome and Ubiquitin-Proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of Autophagy and the Ubiquitin-Proteasome System by the FoxO Transcriptional Network during Muscle Atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; White, L.J.; Larsen, A.E.; Léger, B.; Russell, A.P. The Role and Regulation of MAFbx/Atrogin-1 and MuRF1 in Skeletal Muscle Atrophy. Pflügers Arch. Eur. J. Physiol. 2011, 461, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Toschi, A.; Murfuni, I.; Pelosi, L.; Sica, G.; Adamo, S.; Scicchitano, B.M. Local Overexpression of V1a-Vasopressin Receptor Enhances Regeneration in Tumor Necrosis Factor-Induced Muscle Atrophy. Biomed Res. Int. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Rizzuto, E.; Musarò, A. Counteracting Muscle Wasting in Aging and Neuromuscular Diseases: The Critical Role of IGF-1. Aging (Albany NY) 2009, 1, 451–457. [Google Scholar] [CrossRef]
- Muscat, G.E.O.; Downes, M.; Dowhan, D.H. Regulation of Vertebrate Muscle Differentiation by Thyroid Hormone: The Role of ThemyoD Gene Family. BioEssays 1995, 17, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Marsili, A.; Ambrosio, R.; Guardiola, O.; Sibilio, A.; Paik, J.-H.; Minchiotti, G.; DePinho, R.A.; Fenzi, G.; Larsen, P.R.; et al. The FoxO3/Type 2 Deiodinase Pathway Is Required for Normal Mouse Myogenesis and Muscle Regeneration. J. Clin. Investig. 2010, 120, 4021–4030. [Google Scholar] [CrossRef] [PubMed]
- Pernitsky, A.N.; McIntosh, L.M.; Anderson, J.E. Hyperthyroidism Impairs Early Repair in Normal but Not Dystrophic Mdx Mouse Tibialis Anterior Muscle. An in Vivo Study. Biochem. Cell Biol. 1996, 74, 315–324. [Google Scholar] [CrossRef]
- Weitzel, J.M.; Iwen, K.A.H.; Seitz, H.J. Regulation of Mitochondrial Biogenesis by Thyroid Hormone. Exp. Physiol. 2003, 88, 121–128. [Google Scholar] [CrossRef]
- Hildebrandt, A.L.; Neufer, P.D. Exercise Attenuates the Fasting-Induced Transcriptional Activation of Metabolic Genes in Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1078–E1086. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle Type and Fiber Type Specificity in Muscle Wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- De Lange, P.; Ragni, M.; Silvestri, E.; Moreno, M.; Schiavo, L.; Lombardi, A.; Farina, P.; Feola, A.; Goglia, F.; Lanni, A. Combined CDNA Array/RT-PCR Analysis of Gene Expression Profile in Rat Gastrocnemius Muscle: Relation to Its Adaptive Function in Energy Metabolism during Fasting. FASEB J. 2004, 18, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional Co-Activator PGC-1 Alpha Drives the Formation of Slow-Twitch Muscle Fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Moreno, M.; Silvestri, E.; Lombardi, A.; Goglia, F.; Lanni, A. Fuel Economy in Food-Deprived Skeletal Muscle: Signaling Pathways and Regulatory Mechanisms. FASEB J. 2007, 21, 3431–3441. [Google Scholar] [CrossRef] [PubMed]
- Kraus, B.; Pette, D. Quantification of MyoD, Myogenin, MRF4 and Id-1 by Reverse-Transcriptase Polymerase Chain Reaction in Rat Muscles–Effects of Hypothyroidism and Chronic Low-Frequency Stimulation. Eur. J. Biochem. 1997, 247, 98–106. [Google Scholar] [CrossRef]
- Hughes, S.M.; Chi, M.M.; Lowry, O.H.; Gundersen, K. Myogenin Induces a Shift of Enzyme Activity from Glycolytic to Oxidative Metabolism in Muscles of Transgenic Mice. J. Cell Biol. 1999, 145, 633–642. [Google Scholar] [CrossRef]
- Jamart, C.; Raymackers, J.-M.; Li An, G.; Deldicque, L.; Francaux, M. Prevention of Muscle Disuse Atrophy by MG132 Proteasome Inhibitor. Muscle Nerve 2011, 43, 708–716. [Google Scholar] [CrossRef]
- Supinski, G.S.; Vanags, J.; Callahan, L.A. Effect of Proteasome Inhibitors on Endotoxin-Induced Diaphragm Dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L994–L1001. [Google Scholar] [CrossRef]
- Pigna, E.; Renzini, A.; Greco, E.; Simonazzi, E.; Fulle, S.; Mancinelli, R.; Moresi, V.; Adamo, S. HDAC4 Preserves Skeletal Muscle Structure Following Long-Term Denervation by Mediating Distinct Cellular Responses. Skelet. Muscle 2018, 8. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of Autophagy in Skeletal Muscle Uncovers the Accumulation of Ubiquitinated Proteins and Their Potential Role in Muscle Damage in Pompe Disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The Mitochondrial Metabolic Reprogramming Agent Trimetazidine as an “exercise Mimetic” in Cachectic C26-Bearing Mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Pin, F.; Gorini, S.; Pontecorvo, L.; Ferri, A.; Mollace, V.; Costelli, P.; Rosano, G. Improvement of Skeletal Muscle Performance in Ageing by the Metabolic Modulator Trimetazidine. J. Cachexia Sarcopenia Muscle 2016, 7, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Cerveró, C.; Montull, N.; Tarabal, O.; Piedrafita, L.; Esquerda, J.E.; Calderó, J. Chronic Treatment with the AMPK Agonist AICAR Prevents Skeletal Muscle Pathology but Fails to Improve Clinical Outcome in a Mouse Model of Severe Spinal Muscular Atrophy. Neurotherapeutics 2016, 13, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and Cellular Mechanisms of Skeletal Muscle Atrophy: An Update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Vezzi, F.; Del Fabbro, C.; Tomescu, A.I.; Policriti, A. RNA: A Fast and Accurate Short Reads Numerical Aligner. Bioinformatics 2012, 28, 123–124. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 5–7. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering Splice Junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Articles Differential Analysis of Gene Regulation at Transcript Resolution with RNA-Seq. Nat. Biotechnol. 2012, 31, 46–53. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | CTR | T3 |
|---|---|---|
| GLUT 4 | 1 ± 0.15 | 1.83 ± 0.45 * |
| UCP3 | 1 ± 0.33 | 2.14 ± 0.23 * |
| MyoD | 1 ± 0.32 | 1.5 ± 0.22 * |
| Myh7 | 1 ± 0.20 | 0.60 ± 0.15 * |
| Myh1 | 1 ± 0.29 | 3.40 ± 0.61 ** |
| Gene | Sequence | |
|---|---|---|
| Atrogin-1 | F: GCA AAC ACT GCC ACA TTC TCT C | |
| R: CCT GAG GGG AAG TGA GAC G | ||
| MuRF-1 | F: ACC TGC TGG TGG AAA ACA TC | |
| R: CTT CGT GTT CCT TGC ACA TC | ||
| LC3b | F: CAC TGC TCT GTC TTG TGT AGG TTG | |
| R: TCG TTG TGC CTT TAT TAG TGC ATC | ||
| P62 | F: CCC AGT GTC TTG GCA TTC TT | |
| R: AGG GAA AGC AGA GGA AGC TC | ||
| Pax7 | F: TCC CCC TGG AAG TGT CCA | |
| R: TGG GAA ACA CGG AGC TGA | ||
| Myh3 | F: AGC AGC TCA ATC AGC TCA | |
| R: TGG TCG TAA TCA GCA GCA | ||
| PGC-1α | F: TGA GTA ACC GGA GGC ATT CTC T | |
| R: TGA GGA CCG CTA GCA AGT TTG | ||
| Dio2 | F: CCT CCT AGA TGC CTA CAA ACAG | |
| R: TGA TTC AGG ATT GGA GAC GTG | ||
| Myh7 | F: AGT CCC AGG TCA ACA AGC TG | |
| R: TTC CAC CTA AAG GGC TGT TG | ||
| Myh2 | F: AGT CCC AGG TCA ACA AGC TG | |
| R: GCA TGA CCA AAG GTT TCA CA | ||
| Myh1 | F: AGT CCC AGG TCA ACA AGC TG | |
| R: CAC ATT TTG CTC ATC TCT TTG G | ||
| Myh4 | F: AGT CCC AGG TCA ACA AGC TG | |
| R: TTT CTC CTG TCA CCT CTC AAC A | ||
| TFAM | F: AAA GCT TCC AGG AGG CAA | |
| R: GCC ATC TGC TCT TCC CAA | ||
| CytC | F: CCA GTG CCA CAC TGT GGA | |
| R: GTC TTC CGC CCG AAC AGA | ||
| COX2 | F: AAG ACG CCA CAT CCC CTA | |
| R: CGT AGA GAG GGG AGA GCA | ||
| Cpt1b | F: TCT CCA TGC GAC TGG TCG AT | |
| R: GAG ACG GAC ACA GAT AGC CC | ||
| MyoD | F: ACC CAG GAA CTG GGA ATG GA | |
| R: AAG TCG TCT GCT GTC TCA AA | ||
| Glut4 | F: ATG GCT GTC GCT GGT TTC TC | |
| R: ACC CAT ACG ATC CGC AAC AT | ||
| UCP3 | F: TGA CCT GCG CCC AGC | |
| R: CCC AGG CGT ATC ATG GCT | ||
| 18s | F: GCA ATT ATT CCC CAT GAA CG | |
| R: GGG ACT TAA TCA ACG CAA GC | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ucci, S.; Renzini, A.; Russi, V.; Mangialardo, C.; Cammarata, I.; Cavioli, G.; Santaguida, M.G.; Virili, C.; Centanni, M.; Adamo, S.; et al. Thyroid Hormone Protects from Fasting-Induced Skeletal Muscle Atrophy by Promoting Metabolic Adaptation. Int. J. Mol. Sci. 2019, 20, 5754. https://doi.org/10.3390/ijms20225754
Ucci S, Renzini A, Russi V, Mangialardo C, Cammarata I, Cavioli G, Santaguida MG, Virili C, Centanni M, Adamo S, et al. Thyroid Hormone Protects from Fasting-Induced Skeletal Muscle Atrophy by Promoting Metabolic Adaptation. International Journal of Molecular Sciences. 2019; 20(22):5754. https://doi.org/10.3390/ijms20225754
Chicago/Turabian StyleUcci, Sarassunta, Alessandra Renzini, Valentina Russi, Claudia Mangialardo, Ilenia Cammarata, Giorgia Cavioli, Maria Giulia Santaguida, Camilla Virili, Marco Centanni, Sergio Adamo, and et al. 2019. "Thyroid Hormone Protects from Fasting-Induced Skeletal Muscle Atrophy by Promoting Metabolic Adaptation" International Journal of Molecular Sciences 20, no. 22: 5754. https://doi.org/10.3390/ijms20225754
APA StyleUcci, S., Renzini, A., Russi, V., Mangialardo, C., Cammarata, I., Cavioli, G., Santaguida, M. G., Virili, C., Centanni, M., Adamo, S., Moresi, V., & Verga-Falzacappa, C. (2019). Thyroid Hormone Protects from Fasting-Induced Skeletal Muscle Atrophy by Promoting Metabolic Adaptation. International Journal of Molecular Sciences, 20(22), 5754. https://doi.org/10.3390/ijms20225754