Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment



 ,
,
Abstract
1. Introduction
2. Methods
3. Renal Cell Carcinoma
3.1. Definition
3.2. Current Treatment Options
3.3. MKIs in the Treatment of RCC
3.3.1. The Adverse Effect Hypertension
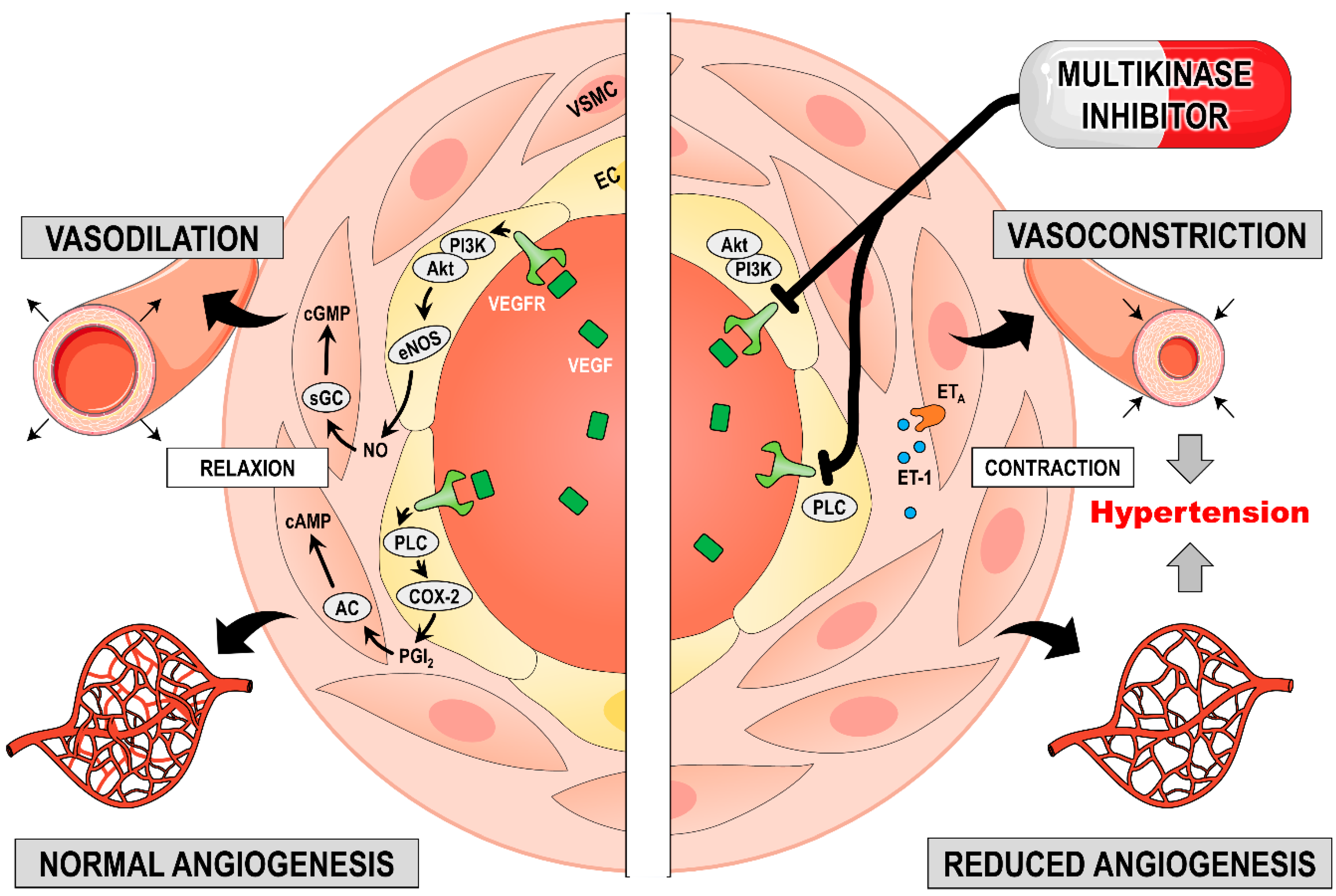
3.3.2. Induction of Hypertension
3.3.3. Sorafenib
3.3.4. Sunitinib
3.3.5. Lenvatinib
3.3.6. Axitinib
4. Results
4.1. Clinical Trials with Sorafenib, Sunitinib, Lenvatinib and Axitinib
4.2. Countermeasures against Drug-Induced Hypertension
4.3. Biomarkers in mRCC
5. Discussion
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | adenylate cyclase |
| AE(s) | adverse effect(s) |
| AKT | protein kinase B |
| ARCC | advanced renal cell carcinoma |
| ATP | adenosine triphosphate |
| cAMP | cyclic adenosine monophosphate |
| ccRCC | clear cell renal cell carcinoma |
| cGMP | cyclic guanosine monophosphate |
| CI | confidence interval |
| C-KIT | stem cell factor receptor |
| Cmax | peak concentration of a drug |
| COX-2 | cyclooxygenae-2) |
| CYP3A4 | cytochrome P450 3A4 |
| DBP | diastolic blood pressure |
| DSF | disease-free survival |
| EC | endothelial cell |
| EMA | European Medicines Agency |
| eNOS | endothelial nitric oxide synthase |
| ERK | extracellular signal-regulated kinase |
| ET-1 | endothelin-1 |
| ETA | endothelin receptor A |
| FDA | Food and Drug Administration |
| FGF | fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| FLT-3 | FMS-like tyrosine kinase 3 |
| GC | guanylyl cyclase |
| GTP | guanosine triphosphate |
| HIF | hypoxia-inducible factor |
| I.V. | intravenous |
| MEK | Mitogen-activated protein kinase |
| MET | hepatocyte growth factor receptor |
| MKI(s) | multikinase inhibitor(s) |
| mRCC | metastatic renal cell carcinoma/cancer |
| mTOR | mammalian target of rapamycin |
| NO | nitric oxide |
| OS | overall survival |
| PC | pericyte |
| PD-1 | programmed death-1 |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptor |
| PFS | progression-free survival |
| PGI2 | prostacyclin |
| PI3K | phosphoinositide 3-kinase |
| PLC | phospholipase C |
| pVHL | von Hippel-Lindau protein |
| RCC | renal cell carcinoma |
| RAF | rapidly accelerated fibrosarcoma kinase |
| RAS | rat sarcoma |
| RET | rearranged during transfection |
| RTK | tyrosine kinase receptor |
| SBP | systolic blood pressure |
| TC | tumour cell |
| TGFA | transforming growth factor alpha |
| TRAE | treatment-related adverse effect |
| TTP | time to progression |
| T1/2 | elimination half-life |
| UGT1A9 | uridine diphosphate glucuronosyltransferase 1A9 |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
| VHL | von Hippel-Lindau |
| VSMC | vascular smooth muscle cell |
References
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; De Simone, G.; Dominiczak, A. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef]
- Wermelt, J.A.; Schunkert, H. Management of arterial hypertension. Herz 2017, 42, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Mattei, J.; da Silva, R.D.; Sehrt, D.; Molina, W.R.; Kim, F.J. Targeted therapy in metastatic renal carcinoma. Cancer Lett. 2014, 343, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Randrup Hansen, C.; Grimm, D.; Bauer, J.; Wehland, M.; Magnusson, N.E. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 461. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, M.A.F.; Grimm, D.; Bauer, J.; Wehland, M.; Wise, P.; Magnusson, N.E.; Infanger, M.; Kruger, M. Hypertension caused by lenvatinib and everolimus in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 1736. [Google Scholar] [CrossRef] [PubMed]
- PubMed. National Center for Biotechnology Information, U.S. National Library of Medicine. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 26 November 2018).
- ClinicalTrials. U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ (accessed on 26 November 2018).
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Garfield, K.; LaGrange, C.A. Cancer, renal cell. In Statpearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2018. [Google Scholar]
- Randall, J.M.; Millard, F.; Kurzrock, R. Molecular aberrations, targeted therapy, and renal cell carcinoma: Current state-of-the-art. Cancer Metastasis Rev. 2014, 33, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of renal cell carcinoma. Eur. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Grignon, D.J.; Che, M. Clear cell renal cell carcinoma. Clin. Lab. Med. 2005, 25, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Beltran, A.; Carrasco, J.C.; Cheng, L.; Scarpelli, M.; Kirkali, Z.; Montironi, R. 2009 update on the classification of renal epithelial tumors in adults. Int. J. Urol. 2009, 16, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wu, Y.; Zhang, J.; Fang, Z.; Liu, Z.; Xu, Z.; Fan, Y. Nomograms for predicting long-term overall survival and disease-specific survival of patients with clear cell renal cell carcinoma. OncoTargets Ther. 2018, 11, 5535–5544. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of plin2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ren, Y.; Pang, L.; Qi, Y.; Jia, W.; Tao, L.; Hu, Z.; Zhao, J.; Zhang, H.; Li, L.; et al. Papillary renal cell carcinoma: A clinicopathological and whole-genome exon sequencing study. Int. J. Clin. Exp. Pathol. 2015, 8, 8311–8335. [Google Scholar] [PubMed]
- Motzer, R.J.; Basch, E. Targeted drugs for metastatic renal cell carcinoma. Lancet 2007, 370, 2071–2073. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Plantade, A.; Elson, P.; Negrier, S.; Ravaud, A.; Oudard, S.; Zhou, M.; Rini, B.I.; Bukowski, R.M.; Escudier, B. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J. Clin. Oncol. 2008, 26, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Nabi, S.; Kessler, E.R.; Bernard, B.; Flaig, T.W.; Lam, E.T. Renal cell carcinoma: A review of biology and pathophysiology. F1000Research 2018, 7, 307. [Google Scholar] [CrossRef]
- Gollob, J.A.; Wilhelm, S.; Carter, C.; Kelley, S.L. Role of Raf kinase in cancer: Therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin. Oncol. 2006, 33, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Erman, M.; Benekli, M.; Basaran, M.; Bavbek, S.; Buyukberber, S.; Coskun, U.; Demir, G.; Karabulut, B.; Oksuzoglu, B.; Ozkan, M.; et al. Renal cell cancer: Overview of the current therapeutic landscape. Expert Rev. Anticancer Ther. 2016, 16, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Gerendash, B.; Dizman, N.; Khan, A.; Pal, S.K. Advances in treatment of metastatic renal cell carcinoma. Curr. Opin. Urol. 2016, 26, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Glen, H. Lenvatinib therapy for the treatment of patients with advanced renal cell carcinoma. Future Oncol. 2016, 12, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: Results from a phase III trial. J. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Cicenas, E. Multi-kinase inhibitors, aurks and cancer. Med. Oncol. 2016, 33, 43. [Google Scholar] [CrossRef]
- Wehland, M.; Bauer, J.; Infanger, M.; Grimm, D. Target-based anti-angiogenic therapy in breast cancer. Curr. Pharm. Des. 2012, 18, 4244–4257. [Google Scholar] [CrossRef]
- Kristensen, T.B.; Knutsson, M.L.; Wehland, M.; Laursen, B.E.; Grimm, D.; Warnke, E.; Magnusson, N.E. Anti-vascular endothelial growth factor therapy in breast cancer. Int. J. Mol. Sci. 2014, 15, 23024–23041. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase iii treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Ravaud, A.; Motzer, R.J.; Pandha, H.S.; George, D.J.; Pantuck, A.J.; Patel, A.; Chang, Y.H.; Escudier, B.; Donskov, F.; Magheli, A.; et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N. Engl. J. Med. 2016, 375, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Rini, B.I.; Wilding, G.; Hudes, G.; Stadler, W.M.; Kim, S.; Tarazi, J.; Rosbrook, B.; Trask, P.C.; Wood, L.; Dutcher, J.P. Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J. Clin. Oncol. 2009, 27, 4462–4468. [Google Scholar] [CrossRef] [PubMed]
- Milling, R.V.; Grimm, D.; Krüger, M.; Grosse, J.; Kopp, S.; Bauer, J.; Infanger, M.; Wehland, M. Pazopanib, cabozantinib, and vandetanib in the treatment of progressive medullary thyroid cancer with a special focus on the adverse effects on hypertension. Int. J. Mol. Sci. 2018, 19, 3258. [Google Scholar] [CrossRef]
- Laursen, R.; Wehland, M.; Kopp, S.; Pietsch, J.; Infanger, M.; Grosse, J.; Grimm, D. Effects and role of multikinase inhibitors in thyroid cancer. Curr. Pharm. Des. 2016, 22, 5915–5926. [Google Scholar] [CrossRef]
- Ancker, O.V.; Wehland, M.; Bauer, J.; Infanger, M.; Grimm, D. The adverse effect of hypertension in the treatment of thyroid cancer with multi-kinase inhibitors. Int. J. Mol. Sci. 2017, 18, 625. [Google Scholar] [CrossRef]
- Larochelle, P.; Kollmannsberger, C.; Feldman, R.D.; Schiffrin, E.L.; Poirier, L.; Patenaude, F.; Ruether, D.; Myers, M.; Bjarnason, G. Hypertension management in patients with renal cell cancer treated with anti-angiogenic agents. Curr. Oncol. 2012, 19, 202–208. [Google Scholar] [CrossRef]
- Robinson, E.S.; Khankin, E.V.; Karumanchi, S.A.; Humphreys, B.D. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin. Nephrol. 2010, 30, 591–601. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef]
- Kappers, M.H.; van Esch, J.H.; Sluiter, W.; Sleijfer, S.; Danser, A.H.; van den Meiracker, A.H. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 2010, 56, 675–681. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol. Res. 2017, 120, 116–132. [Google Scholar] [CrossRef]
- European Medicines Agency. Nexavar (Sorafenib). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/nexavar (accessed on 4 November 2018).
- Samadi, A.K.; Mukerji, R.; Shah, A.; Timmermann, B.N.; Cohen, M.S. A novel ret inhibitor with potent efficacy against medullary thyroid cancer in vivo. Surgery 2010, 148, 1228–1236. [Google Scholar] [CrossRef]
- Edginton, A.N.; Zimmerman, E.I.; Vasilyeva, A.; Baker, S.D.; Panetta, J.C. Sorafenib metabolism, transport, and enterohepatic recycling: Physiologically based modeling and simulation in mice. Cancer Chemother. Pharmacol. 2016, 77, 1039–1052. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Huang, X.; Li, Y.; Wu, M.; Liu, J. The drug-drug interaction of sorafenib mediated by p-glycoprotein and cyp3a4. Xenobiotica 2016, 46, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Pratz, K.W.; Xu, L.; Wright, J.J.; Smith, B.D.; Rudek, M.A. Plasma protein binding of sorafenib, a multi kinase inhibitor: In vitro and in cancer patients. Invest. New Drugs 2012, 30, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Lathia, C.; Lettieri, J.; Cihon, F.; Gallentine, M.; Radtke, M.; Sundaresan, P. Lack of effect of ketoconazole-mediated cyp3a inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother. Pharmacol. 2006, 57, 685–692. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Sutent (Sunitinib). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/sutent (accessed on 4 November 2018).
- Yu, H.; Steeghs, N.; Kloth, J.S.; de Wit, D.; van Hasselt, J.G.; van Erp, N.P.; Beijnen, J.H.; Schellens, J.H.; Mathijssen, R.H.; Huitema, A.D. Integrated semi-physiological pharmacokinetic model for both sunitinib and its active metabolite su12662. Br. J. Clin. Pharmacol. 2015, 79, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Haznedar, J.O.; Patyna, S.; Bello, C.L.; Peng, G.W.; Speed, W.; Yu, X.; Zhang, Q.; Sukbuntherng, J.; Sweeny, D.J.; Antonian, L.; et al. Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: Comparative plasma kinetics in non-clinical species. Cancer Chemother. Pharmacol. 2009, 64, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Toyama, Y.; Ueyama, J.; Nomura, H.; Tsukiyama, I.; Saito, H.; Hisada, T.; Matsuura, K.; Hasegawa, T. Contribution of plasma proteins, albumin and alpha 1-acid glycoprotein, to pharmacokinetics of a multi-targeted receptor tyrosine kinase inhibitor, sunitinib, in analbuminemic rats. Anticancer Res. 2014, 34, 2283–2289. [Google Scholar] [PubMed]
- European Medicines Agency. Lenvima (Lenvatinib). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lenvima (accessed on 4 November 2018).
- Gupta, A.; Jarzab, B.; Capdevila, J.; Shumaker, R.; Hussein, Z. Population pharmacokinetic analysis of lenvatinib in healthy subjects and patients with cancer. Br. J. Clin. Pharmacol. 2016, 81, 1124–1133. [Google Scholar] [CrossRef]
- Dubbelman, A.C.; Rosing, H.; Nijenhuis, C.; Huitema, A.D.; Mergui-Roelvink, M.; Gupta, A.; Verbel, D.; Thompson, G.; Shumaker, R.; Schellens, J.H.; et al. Pharmacokinetics and excretion of (14)c-lenvatinib in patients with advanced solid tumors or lymphomas. Invest. New Drugs 2015, 33, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, R.C.; Aluri, J.; Fan, J.; Martinez, G.; Thompson, G.A.; Ren, M. Effect of rifampicin on the pharmacokinetics of lenvatinib in healthy adults. Clin. Drug Investig. 2014, 34, 651–659. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Albiges, L.; Gizzi, M.; Carton, E.; Escudier, B. Axitinib in metastatic renal cell carcinoma. Expert Rev. Anticancer Ther. 2015, 15, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Gross-Goupil, M.; Francois, L.; Quivy, A.; Ravaud, A. Axitinib: A review of its safety and efficacy in the treatment of adults with advanced renal cell carcinoma. Clin. Med. Insights Oncol. 2013, 7, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Herbst, R.S.; Liu, G.; Park, J.W.; Kies, M.S.; Steinfeldt, H.M.; Pithavala, Y.K.; Reich, S.D.; Freddo, J.L.; Wilding, G. Phase I trial of the oral antiangiogenesis agent ag-013736 in patients with advanced solid tumors: Pharmacokinetic and clinical results. J. Clin. Oncol. 2005, 23, 5474–5483. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Uemura, H.; Fujimoto, H.; Kanayama, H.O.; Shinohara, N.; Nakazawa, H.; Imai, K.; Umeyama, Y.; Ozono, S.; Naito, S.; et al. Key predictive factors of axitinib (ag-013736)-induced proteinuria and efficacy: A phase II study in japanese patients with cytokine-refractory metastatic renal cell carcinoma. Eur. J. Cancer 2011, 47, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Rixe, O.; Bukowski, R.M.; Michaelson, M.D.; Wilding, G.; Hudes, G.R.; Bolte, O.; Motzer, R.J.; Bycott, P.; Liau, K.F.; Freddo, J.; et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: A phase II study. Lancet Oncol. 2007, 8, 975–984. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (axis): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Rini, B.I.; Quinn, D.I.; Baum, M.; Wood, L.S.; Tarazi, J.; Rosbrook, B.; Arruda, L.S.; Cisar, L.; Roberts, W.G.; Kim, S.; et al. Hypertension among patients with renal cell carcinoma receiving axitinib or sorafenib: Analysis from the randomized phase iii axis trial. Target. Oncol 2015, 10, 45–53. [Google Scholar] [CrossRef]
- De Jesus-Gonzalez, N.; Robinson, E.; Moslehi, J.; Humphreys, B.D. Management of antiangiogenic therapy-induced hypertension. Hypertension 2012, 60, 607–615. [Google Scholar] [CrossRef]
- Martignoni, G.; Brunelli, M.; Segala, D.; Munari, E.; Gobbo, S.; Cima, L.; Borze, I.; Wirtanen, T.; Sarhadi, V.K.; Atanesyan, L.; et al. Validation of 34betae12 immunoexpression in clear cell papillary renal cell carcinoma as a sensitive biomarker. Pathology 2017, 49, 10–18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lou, N.; Ruan, A.M.; Qiu, B.; Bao, L.; Xu, Y.C.; Zhao, Y.; Sun, R.L.; Zhang, S.T.; Xu, G.H.; Ruan, H.L.; et al. Mir-144–3p as a novel plasma diagnostic biomarker for clear cell renal cell carcinoma. Urol. Oncol. 2017, 35, 36.e7–36.e14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhu, J.; George, D.J.; Nixon, A.B. Metastatic clear cell renal cell carcinoma: Circulating biomarkers to guide antiangiogenic and immune therapies. Urol. Oncol. 2016, 34, 510–518. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology. Available online: https://www.nccn.org/professionals/physician_gls/f_guidelines.asp (accessed on 26 November 2018).
- Haddad, A.Q.; Luo, J.H.; Krabbe, L.M.; Darwish, O.; Gayed, B.; Youssef, R.; Kapur, P.; Rakheja, D.; Lotan, Y.; Sagalowsky, A.; et al. Prognostic value of tissue-based biomarker signature in clear cell renal cell carcinoma. BJU Int. 2017, 119, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Pires-Luis, A.S.; Costa-Pinheiro, P.; Ferreira, M.J.; Antunes, L.; Lobo, F.; Oliveira, J.; Henrique, R.; Jeronimo, C. Identification of clear cell renal cell carcinoma and oncocytoma using a three-gene promoter methylation panel. J. Transl. Med. 2017, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Dudani, S.; Savard, M.F.; Heng, D.Y.C. An update on predictive biomarkers in metastatic renal cell carcinoma. Eur. Urol. Focus 2019. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Haas, N.B. Axitinib in the treatment of renal cell carcinoma: Patient selection and perspectives. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kollmannsberger, C. Sunitinib side effects as surrogate biomarkers of efficacy. Can. Urol. Assoc. J. 2016, 10, S245–S247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heng, D.Y.; Xie, W.; Regan, M.M.; Harshman, L.C.; Bjarnason, G.A.; Vaishampayan, U.N.; Mackenzie, M.; Wood, L.; Donskov, F.; Tan, M.H.; et al. External validation and comparison with other models of the international metastatic renal-cell carcinoma database consortium prognostic model: A population-based study. Lancet Oncol. 2013, 14, 141–148. [Google Scholar] [CrossRef]
- Graham, J.; Dudani, S.; Heng, D.Y.C. Prognostication in kidney cancer: Recent advances and future directions. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Suzuki, K.; Terakawa, T.; Furukawa, J.; Harada, K.; Hinata, N.; Nakano, Y.; Fujisawa, M. C-reactive protein and the neutrophil-to-lymphocyte ratio are prognostic biomarkers in metastatic renal cell carcinoma patients treated with nivolumab. Int. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kovacova, J.; Juracek, J.; Poprach, A.; Kopecky, J.; Fiala, O.; Svoboda, M.; Fabian, P.; Radova, L.; Brabec, P.; Buchler, T.; et al. Mir-376b-3p is associated with long-term response to sunitinib in metastatic renal cell carcinoma patients. Cancer Genom. Proteom. 2019, 16, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Kong, W.; Dong, B.; Zhang, J.; Chen, Y.; Xue, W.; Huang, Y.; Zhou, L.; Huang, J. Comparison of efficacy, safety, and quality of life between sorafenib and sunitinib as first-line therapy for chinese patients with metastatic renal cell carcinoma. Chin. J. Cancer 2017, 36, 64. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Hutson, T.E.; Escudier, B.; Esteban, E.; Bjarnason, G.A.; Lim, H.Y.; Pittman, K.B.; Senico, P.; Niethammer, A.; Lu, D.R.; Hariharan, S.; et al. Randomized phase III trial of temsirolimus versus sorafenib as second-line therapy after sunitinib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2014, 32, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Eisen, T.; Broderick, S.; Stadler, W.M.; Jones, R.J.; Garcia, J.A.; Vaishampayan, U.N.; Picus, J.; Hawkins, R.E.; et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (aspen): A multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016, 17, 378–388. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The alliance a031203 cabosun trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef]
- Budolfsen, C.; Faber, J.; Grimm, D.; Krüger, M.; Bauer, J.; Wehland, M.; Infanger, M.; Magnusson, N.E. Tyrosine kinase inhibitor-induced hypertension: Role of hypertension as a biomarker in cancer treatment. Curr. Vasc. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Donskov, F.; Michaelson, M.D.; Puzanov, I.; Davis, M.P.; Bjarnason, G.A.; Motzer, R.J.; Goldstein, D.; Lin, X.; Cohen, D.P.; Wiltshire, R.; et al. Sunitinib-associated hypertension and neutropenia as efficacy biomarkers in metastatic renal cell carcinoma patients. Br. J. Cancer 2015, 113, 1571–1580. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| RCC Subtype | Percent of RCC Cases |
|---|---|
| Clear cell | >80% |
| Papillary | 10–15% |
| Chromophobe | 5% |
| Collecting duct | <1% |
| Medullary | Rare |
| Mucinous | Rare |
| Xp11 | Rare |
| Tumour Stage | Surgery | |
|---|---|---|
| Partial nephrectomy | T1 | Complete removal of the primary tumour, leaving the largest amount of healthy renal tissue |
| Radical nephrectomy | T1 and T2 Tumour ≤ 5 cm in the inferior pole | Removal of the renal, perirenal fat tissue, adrenal gland and regional lymph nodes |
| Drug | Pathway Interaction | Treatment Target | Mechanism | Administration |
|---|---|---|---|---|
| Targeted therapies | ||||
| Sorafenib | Multikinase inhibition | VEGFR (1, 2, 3), PDGFR (α + β), Raf, C-KIT, RET | Anti-angiogenetic, anti-lymphangiogenic, inhibition of tumour growth | p. o. |
| Sunitinib | Multikinase inhibition | VEGFR (1, 2, 3), PDGFR (α + β), c-KIT, FLT-3, RET | Anti-angiogenetic, anti-lymphangiogenic, inhibition of tumour growth | p. o. |
| Pazopanib | Multikinase inhibition | VEGFR (1, 2, 3), PDGFR (α + β), RET, c-KIT | Anti-angiogenetic, anti-lymphangiogenic, inhibition of tumour growth | p. o. |
| Axitinib | Multikinase inhibition | VEGFR (1, 2, 3), c-KIT, PDGFR-β | Anti-angiogenetic, anti-lymphangiogenic, inhibition of tumour growth | p. o. |
| Lenvatinib | Multikinase inhibition | VEGFR-2, FGFR (1, 2, 3, 4), PDGFR-α, c-KIT, RET | Anti-angiogenetic, inhibition of tumour growth | p. o. |
| Tivozanib | Multikinase inhibitor | VEGFR (1, 2, 3), PDGFR-β, c-KIT | Anti-angiogenetic, anti-lymphangiogenic, inhibition of tumour growth | p. o. |
| Cabozantinib | Multikinase inhibition | MET, VEGFR-2, RET | Anti-angiogenetic, inhibition of cell migration and invasion | p. o. |
| Everolimus | mTOR inhibition | mTOR, HIF(1–2), VEGF | Cellular metabolism, cell growth, apoptosis and angiogenesis regulation | p. o. |
| Temsirolimus | mTOR inhibition | mTOR, HIF(1–2), VEGF | Cellular metabolism, cell growth, apoptosis and angiogenesis regulation | I.V. |
| Bevacizumab | Anti-VEGF monoclonal antibody | VEGF | Anti-angiogenetic | I.V. |
| Immunotherapy | ||||
| Interferon-α | Immune system activation | Leucocytes | Immunologic, antiproliferation, antiviral and antiangiogenic | I.V. |
| High-dose IL-2 | Immune system activation | Leucocytes | Activation of the immune system leading to tumour regression | I.V. |
| Nivolumab | Programmed death 1 (PD-1)-antibody | PD-1 at T-lymphocytes | Blocking the PD-1/PD-L1 resulting in cellular immune response inhibition and restoration of antitumour immunity | I.V. |
| Drug | Adverse Effects (10 Most Common) |
|---|---|
| Sorafenib | Diarrhoea, rash, hand–foot syndrome, alopecia, fatigue, nausea, hypertension, pruritus, dry skin, vomiting |
| Sunitinib | Diarrhoea, hand–foot syndrome, hypertension, fatigue, dysgeusia, mucositis, dyspepsia, stomatitis, neutropenia |
| Lenvatinib | Diarrhoea, nausea, decreased appetite, hypertension, weight loss, fatigue, vomiting, hypothyroidism, abdominal pain, stomatitis |
| Axitinib | Diarrhoea, hypertension, fatigue, decreased appetite, nausea, dysphoria, palmar–plantar erythrodysesthesia, weight loss, vomiting, asthenia |
| Title and ClinicalTrials.Gov NCT Number | Design and Study Size | Objective | Status and Conclusion |
|---|---|---|---|
| “A multicenter uncontrolled study of sorafenib in patients with unresectable and/or metastatic renal cell carcinoma” NCT00586105 | Interventional, non-randomised, open-label, multicentre, phase III study; 39 participants | This trial investigated the efficacy, safety, tolerability and pharmacokinetic profile of sorafenib in patients with an unresectable and/or mRCC. | Completed The trial showed a PFS of 5.5 months (95% CI [4.1–7.4]) and an OS of 7.8 months (95% CI [0.9–13.4]). Hypertension was reported in 17.95% of the patients. |
| “A phase III randomized study of BAY43-9006 in patients with unresectable and/or metastatic renal cell cancer” NCT00073307 | Interventional, randomised, parallel assignment, phase III; 903 participants | This study investigated the efficacy, safety and pharmacokinetics of patients with unresectable and/or mRCC treated with sorafenib. | Completed Sorafenib treatment in patients with mRCC showed significant improvement compared to the placebo group. Hypertension developed in 16.41% of the patients who received sorafenib. |
| “Sorafenib dose escalation in treatment-naïve patients with metastatic renal cell carcinoma: a non-randomized, open-label, Phase 2b study” NCT00618982 | International, non-randomised, open label, uncontrolled, multicentre phase IIb study; 83 participants | This study investigated the efficacy and safety of sorafenib in patients with mRCC who had no prior systemic treatment. | Completed Patients treated with sorafenib showed clinical benefits. Hypertension was found in 48.2% of the patients. |
| Title and ClinicalTrials.Gov NCT Number | Design and Study Size | Objective | Status |
|---|---|---|---|
| “A study of abemaciclib in combination with sunitinib in metastatic renal cell carcinoma” NCT03905889 | Interventional, non-randomised, open label phase Ib study; 22 participants | To investigate the combination of abemaciclib with sunitinib. Primary outcome measures: Maximal tolerated dose and toxicity and pharmacokinetic assessment. | Recruiting |
| “A study of NKTR-214 in combination with nivolumab compared with the investigator’s choice of a tyrosine kinase inhibitor (TKI) therapy (either sunitinib or cabozantinib monotherapy) for advanced metastatic renal cell carcinoma” (RCC) NCT03729245 | Interventional, randomised, open label phase III trial; 600 participants | To investigate NKTR-214 in combination with nivolumab compared to the investigator’s choice of TKI (including sunitinib). Primary outcome measures: ORR and OS. | Recruiting |
| “A study of nivolumab combined with cabozantinib compared to sunitinib in previously untreated advanced or metastatic renal cell carcinoma (CheckMate 9ER)” NCT03141177 | Interventional, randomised, open label phase III trial; 638 participants | This study investigates the efficacy and safety of nivolumab combined with cabozantinib compared to sunitinib. Primary outcome measure: PFS. Secondary outcome measures: OS, ORR and AEs. | Active, not recruiting. |
| “Real-world clinical patterns of care and outcomes among mRCC patients receiving sunitinib as first line therapy. (OPTIMISE)” NCT03140176 | Observational, prospective study; 140 participants | This study aims to investigate efficacy, adverse events and health-related quality of life in patients receiving sunitinib. Primary outcome measures: PFS and time to failure (TTF). | Recruiting |
| “Biomarker study of patients with metastatic ccRCC undergoing sequential therapy with 1st line sunitinib and 2nd line axitinib (SuAx)” NCT03592199 | Interventional, open label phase II study; 30 participants | To investigate potential prognostic and/or predictive biomarkers. Primary outcome measure: RR. | Recruiting |
| “Study to evaluate efficacy and safety of sunitinib in renal cell carcinoma progressed to 1L immunotherapy treatment. (INMUNOSUN)” NCT03066427 | Interventional, open label phase II study; 23 participants | To investigate the activity of sunitinib after treatment with new immunotherapy regimens that are currently developed in phase III trials. Primary outcome measure: ORR. Secondary outcome measures: time to progression (TTP), duration of response, OR, etc. | Recruiting |
| “Cabozantinib or sunitinib malate in treating participants with metastatic variant histology renal cell carcinoma” NCT03541902 | Interventional, randomised, open label phase II trial; 84 participants | To compare the safety and efficacy of cabozantinib and sunitinib. Primary outcome measure: PFS. Secondary outcome measures: ORR, OS, AE rate. | Recruiting |
| “Alternative schedule sunitinib in metastatic renal cell carcinoma: cardiopulmonary exercise testing (ASSET)” NCT03109015 | Interventional, randomised, open label phase II trial; 30 participants | To compare sunitinib administration of a 2/1 schedule (2 weeks of treatment followed by 1 week without) to a 4/2 schedule (4 weeks of treatment followed by 2 weeks without) on cardiopulmonary function. Primary outcome measure: Change in VO2 peak from baseline to week 12. | Recruiting |
| “Role of PRoactivE Coaching on PAtient REported outcome in advanced or metastatic RCC treated with sunitinib (PREPARE)” NCT03013946 | Interventional, randomised, open label phase III study; 430 participants | To evaluate the effect of a 24-week concomitant coaching program. Primary outcome measure: Quality of life assessment. | Recruiting |
| “Study of patients with metastatic and/or advanced renal cell carcinoma, treated with sunitinib/axitinib.” NCT04033991 | Observational, retrospective cohort study; 841 participants | To investigate patients treated with first-line sunitinib and second-line axitinib. Primary outcome measures: PFS (first-line treatment with sunitinib) and PFS (second-line treatment with axitinib). | Not yet recruiting |
| “Impact of sunitinib bioavailability on toxicity and treatment efficacy in patients treated for metastatic renal cancer (BIOSUNTOX)” NCT03846128 | Observational, prospective cohort study; 64 participants | To measure the plasma concentration of sunitinib and its active metabolite desethyl-sunitinib (DES) and evaluate the safety and efficacy at the end of each cycle. Primary outcome measures: Hazard ratio for severe toxicity (grade 3–4 clinical and/or biological) according to the plasma sunitinib concentration. | Not yet recruiting |
| “Registry of complete responses to sunitinib in Spanish patients with metastatic renal cell carcinoma (ATILA)” NCT03916458 | Observational, retrospective study; 90 participants | To investigate patients treated with first-line sunitinib who obtained complete response. Primary outcome measures: Percentage of patients with a good, intermediate and poor prognosis who achieved complete response in the investigator′s opinion or at least two consecutive CT scans. | Not yet recruiting |
| “A biomarker driven trial with nivolumab and ipilimumab or VEGFR tKi in naïve metastatic kidney cancer (BIONIKK)” NCT02960906 | Interventional, randomised, open label phase II study; 150 participants | To compare nivolumab monotherapy, nivolumab combined with ipilimumab, or TKI: sunitinib or pazopanib. Primary outcome measures: ORR evaluation according to molecular groups (ccRCC1 to 4) and assigned treatment. | Recruiting |
| “Quality of life assessment in daily clinical oncology practice for patients with advanced renal cell carcinoma (QUANARIE)” NCT03062410 | Interventional, open label study; 56 participants | To investigate and evaluate the use of HRQoL assessment in daily clinical practice. Primary outcome measure: Rate of completed questionnaires at 12 months. Secondary outcome measures: Exhaustiveness, acceptability, effectiveness and physician satisfaction. | Recruiting |
| “Savolitinib vs. sunitinib in MET-driven PRCC” NCT03091192 | Interventional, randomised, open label phase III study; 60 participants | To compare savolitinib to sunitinib in Mesenchymal Epithelial Transition (MET)-driven papillary renal cell carcinoma. Primary outcome measure: PFS. Secondary outcome measures: OS, ORR, DoR, etc. | Active, not recruiting |
| “Evaluation of a promising new combination of protein kinase inhibitors on organotypic cultures of human renal tumours (COMBOREIN)” NCT03571438 | Interventional, non-randomised, open label study; 100 participants | To compare the treatment of a cell culture with a combination of CK2 and ATM inhibitors serine/threonine kinase combination with sunitinib, pazopanib and temsirolimus. Primary outcome measures: Death cell rate on organotypic cultures of human renal tumours. | Recruiting |
| Title and ClinicalTrials.Gov Number | Design and Study Size | Objective | Status |
|---|---|---|---|
| “Trial to assess safety and efficacy of lenvatinib in combination with everolimus in participants with renal cell carcinoma” NCT03173560 | Interventional, randomised, open label phase II study; 338 participants | To compare and evaluate the efficacy and safety of two different treatment regimens with lenvatinib in combination with everolimus. Primary outcome measures: ORR and percentage of participants with intolerable Grade 2 or any ≥Grade 3 TEAEs. | Recruiting |
| “Lenvatinib and everolimus in renal cell carcinoma (RCC)” NCT03324373 | Interventional, open label phase I study; 15 participants | To investigate the use of lenvatinib in combination with everolimus in orally advanced and metastatic renal cell carcinoma, prior to cytoreductive nephrectomy. | Recruiting |
| “A phase 2 trial to evaluate efficacy and safety of lenvatinib in combination with everolimus in subjects with unresectable advanced or metastatic non clear cell renal cell carcinoma (nccRCC) who have not received any chemotherapy for advanced disease” NCT02915783 | Interventional, open label phase II study; 31 participants | To investigate lenvatinib in combination with everolimus in patients with unresectable advanced or metastatic nccRCC and who have not received prior chemotherapy for advanced disease. Primary outcome measure: ORR. Secondary outcome measures: PFS and OS. | Active, not recruiting |
| “Phase 1b trial of lenvatinib plus pembrolizumab in subjects with selected solid tumours” NCT03006887 | Interventional, open label phase I study; 6 participants | To investigate the safety of lenvatinib in combination with pembrolizumab in patients with selected solid tumours (primarily clear cell renal cell carcinoma). Primary outcome measures: AE and DLT. | Active, not recruiting |
| Title and ClinicalTrials.Gov NCT Number | Design and Study Size | Objective | Status |
|---|---|---|---|
| “Study of nivolumab and axitinib in patients with advanced renal cell carcinoma” NCT03172754 | Interventional, non-randomised, open label phase I/II study; 98 participants | To investigate axitinib in combination with nivolumab. Primary outcome measures: Incidence of treatment-related adverse events, ORR. | Recruiting |
| “Study of axitinib for reducing extent of venous tumour thrombus in renal cancer with venous invasion (NAXIVA)” NCT03494816 | Interventional, open label phase II study; 20 participants | To investigate axitinib in patients with metastatic and non-metastatic renal cell carcinoma and venous invasion. Primary outcome measure: Improvement in Mayo classification. | Recruiting |
| “Biomarker study of pts with metastatic ccRCC undergoing sequential therapy with 1st line sunitinib and 2nd line axitinib (SuAx)” NCT03592199 | Interventional, open label phase II study; 30 participants | To investigate potential prognostic and/or predictive biomarkers. Primary outcome measure: RR. | Recruiting |
| “Axitinib with or without anti-ox40 antibody pf-04518600 in treating patients with metastatic kidney cancer” NCT03092856 | Interventional, randomised, double-blind phase II study; 104 participants | To investigate axitinib in combination with anti-OX40 antibody PF-04518600 compared to axitinib and placebo. Primary outcome measure: PFS. Secondary outcome measures: Incidence of unacceptable toxicity and ORR. | Recruiting |
| “Axitinib and nivolumab in treating patients with unresectable or metastatic TFE/translocation renal cell carcinoma” NCT03595124 | Interventional, randomised, open label phase II study; 87 participants | To investigate the efficacy of axitinib in combination with nivolumab in treating unresectable or metastatic TFE/translocation renal cell carcinoma. Primary outcome measure: PFS. | Recruiting |
| “Prior axitinib as a determinant of outcome of renal surgery (PADRES)” NCT03438708 | Interventional, open label phase II study; 50 participants | To evaluate axitinib in patients with strong PN indication (but it cannot be done due to anatomic considerations or concerns of residual renal function). Primary outcome measures: Reduction in tumour diameter, ORR, effect on tumour morphometry and feasibility of PN. | Recruiting |
| “Neoadjuvant axitinib and avelumab for patients with localized clear-cell RCC” NCT03341845 | Interventional, open label phase II study; 40 participants | To investigate axitinib in combination with avelumab in patients with intermediate to high-risk non-metastatic RCC. Primary outcome measure: Number of patients with partial remission. | Recruiting |
| “Study of patients with metastatic and/or advanced renal cell carcinoma, treated with sunitinib/axitinib” NCT04033991 | Observational, retrospective cohort study; 841 participants | To investigate patients treated with first-line sunitinib and second-line axitinib. Primary outcome measures: PFS (first-line treatment with sunitinib) and PFS (second-line treatment with axitinib). | Not yet recruiting |
| “Tolerability and pharmacokinetics of toripalimab in combination with axitinib in patients with kidney cancer and melanoma” NCT03086174 | Interventional, open label phase Ib study; 24 participants | To investigate dose-escalation, tolerability and pharmacokinetics study evaluating anti-PD-1 mAb for injection in combination with axitinib in patients with advanced kidney cancer and melanoma who have failed in routine systemic treatment. | Not yet recruiting |
| “Post marketing surveillance study to observe safety and efficacy of Inlyta in South Korea” NCT02156895 | Observational, prospective case-only study; 100 participants | To evaluate the efficacy and safety of axitinib in advanced renal cell carcinoma. Primary outcome measure: Adverse event incidence. | Recruiting |
| “A study of Anti-PD-1 combinations of D-CIK immunotherapy and axitinib in advanced renal carcinoma” NCT03736330 | Interventional, open label phase II study; 24 participants | To investigate the safety and efficacy of immunotherapy (anti-PD-1 (pembrolizumab) activated D-CIK) in combination with axitinib. Primary outcome measure: ORR. Secondary outcome measures: PFS, OS, DoR, etc. | Recruiting |
| Study | References |
|---|---|
| International mRCC Database Consortium (IMDC) risk model | Heng et al. 2013 [77], Dudani et al. 2019 [73], Graham et al. 2018 [78], |
| C-reactive protein and the neutrophil-to-lymphocyte ratio are prognostic biomarkers in mRCC | Suzuki et al. 2019 [79] |
| MiR-376b-3p Is Associated with Long-term Response to Sunitinib in Metastatic Renal Cell Carcinoma Patients | Kovacova et al. 2019 [80] |
| 34betaE12 Immuno-Expression in Clear Cell Papillary RCC | Martignoni et al. 2017 [67] |
| MiR-144-3p Plasma Diagnostic Biomarker for ccRCC. | Lou et al. 2017 [68] |
| Circulating Biomarkers to Guide Antiangiogenic and Immune Therapies | Zhang et al. 2016 [69] |
| Tissue-Based Biomarker Signature in ccRCC | Haddad et al. 2017 [71] |
| Identification of ccRCC Using a Three-Gene Promoter Methylation Panel | Pires-Luis et al. 2017 [72] |
| Trial | Drugs | PFS (Months (95% CI)) | OS (Months (95% CI)) | Hypertension (%) |
|---|---|---|---|---|
| Motzer et al. [83] | sorafenib vs. axitinib | 5.7 (4.7–6.5) vs. 8.3 (6.7–9.2) p < 0.0001 | 19.2 (17.5–22.3) vs. 20.1 (16.7–23.4) p = 0.3744 | 30 vs. 42 |
| Hutson et al. [84] | sorafenib vs. temsirolimus | 3.9 (2.8–4.2 vs. 4.3 (4–5.4) p = 0.19 | 16.6 (13.6–18.7) vs. 12.3 (10.1–14.8) p = 0.01 | Not reported |
| Motzer et al. [28] | sorafenib vs. tivozanib | 9.1 (7.3–9.5) vs. 11.9 (9.3–14.7) p = 0.042 | 29.3 vs. 28.8 p = 0.105 | 34 vs. 44 |
| Motzer et al. [85] | sunitinib vs. pazopanib | 9.5 (9.3–11.1) vs. 8.4 (8.3–10.9) | 29.3 (25.3–32.5) vs. 28.4 (26.2–35.6) p = 0.28 | 40.5 vs. 46.21 |
| Choueiri et al. [87] | sunitinib vs. cabozantinib | 5.6 (3.4–8.1) vs. 8.2 (6.2–8.8) p = 0.012 | 21.8 (16.3–27.0) vs. 30.3 (14.6–35) | 68.1 vs. 80.8 |
| Armstrong et al. [86] | sunitinib vs. everolimus | 8.3 (5.8–11.4) vs. 5.6 (5.5–6) p = 0.16 | 31.5 (14.8–NR) vs. 13.2 (9.7–37.9) p = 0.60 | 46 vs. 2 |
| Motzer et al. [35] | lenvatinib vs. lenvatinib + everolimus | 7.4 (5.6–10.2) vs. 14.6 (5.9–20.1) p = 0.12 | 18.4 (13.3–NR) vs. 25.5 (20.8–25.5) p = 0.32 | 48 vs. 41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bæk Møller, N.; Budolfsen, C.; Grimm, D.; Krüger, M.; Infanger, M.; Wehland, M.; E. Magnusson, N. Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment. Int. J. Mol. Sci. 2019, 20, 4712. https://doi.org/10.3390/ijms20194712
Bæk Møller N, Budolfsen C, Grimm D, Krüger M, Infanger M, Wehland M, E. Magnusson N. Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment. International Journal of Molecular Sciences. 2019; 20(19):4712. https://doi.org/10.3390/ijms20194712
Chicago/Turabian StyleBæk Møller, Nanna, Cecilie Budolfsen, Daniela Grimm, Marcus Krüger, Manfred Infanger, Markus Wehland, and Nils E. Magnusson. 2019. "Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment" International Journal of Molecular Sciences 20, no. 19: 4712. https://doi.org/10.3390/ijms20194712
APA StyleBæk Møller, N., Budolfsen, C., Grimm, D., Krüger, M., Infanger, M., Wehland, M., & E. Magnusson, N. (2019). Drug-Induced Hypertension Caused by Multikinase Inhibitors (Sorafenib, Sunitinib, Lenvatinib and Axitinib) in Renal Cell Carcinoma Treatment. International Journal of Molecular Sciences, 20(19), 4712. https://doi.org/10.3390/ijms20194712