Molecular Signatures of the Insulin-Like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers


 ,
, 


Abstract
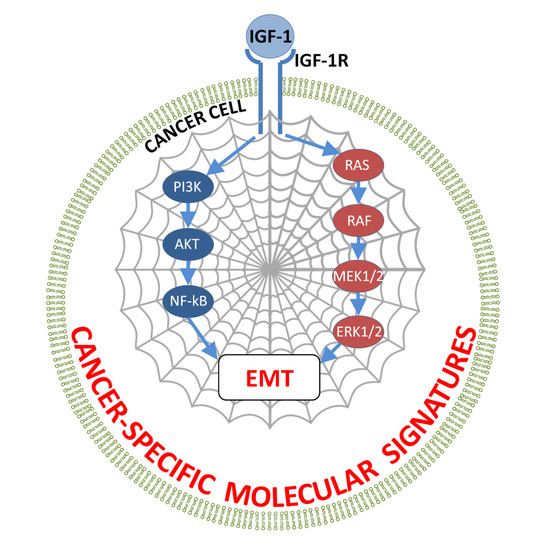
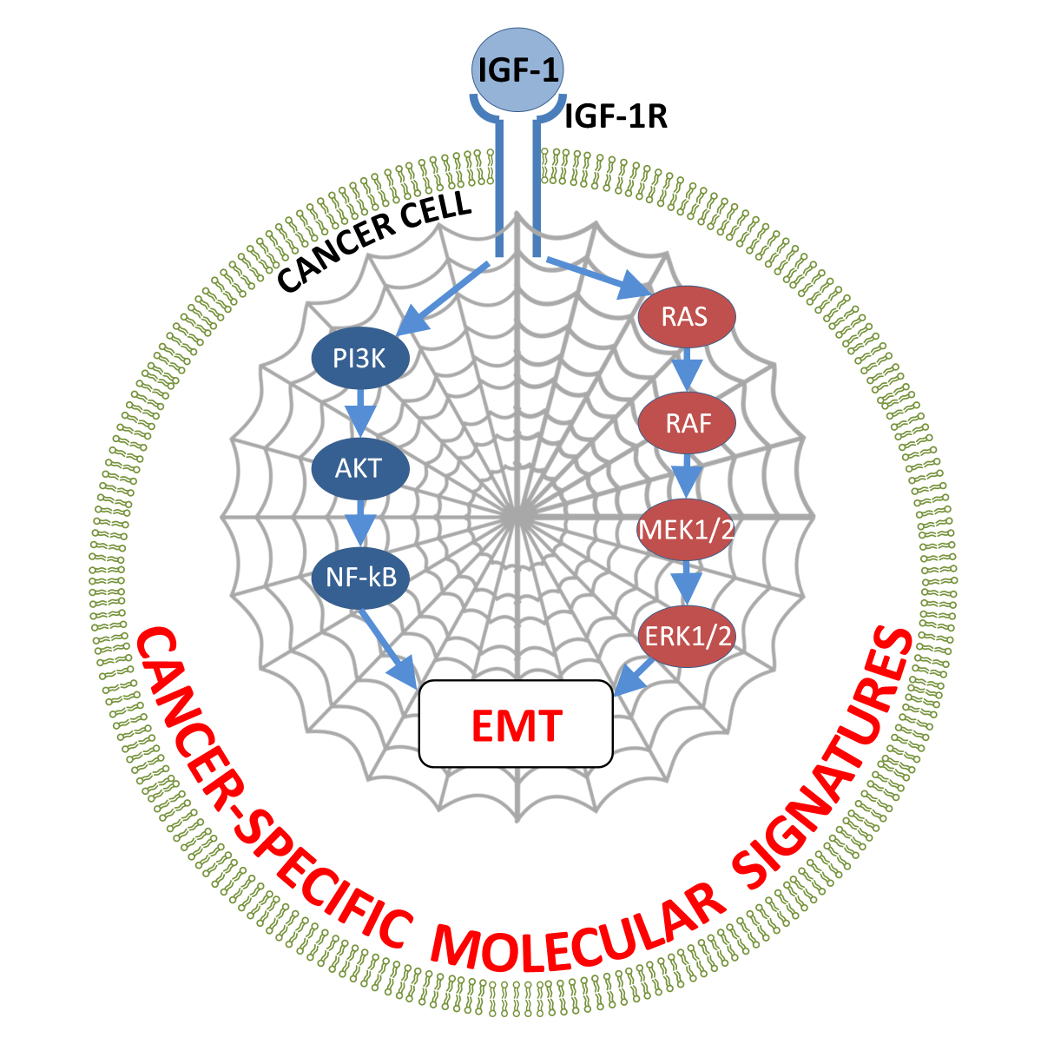{kind=link}
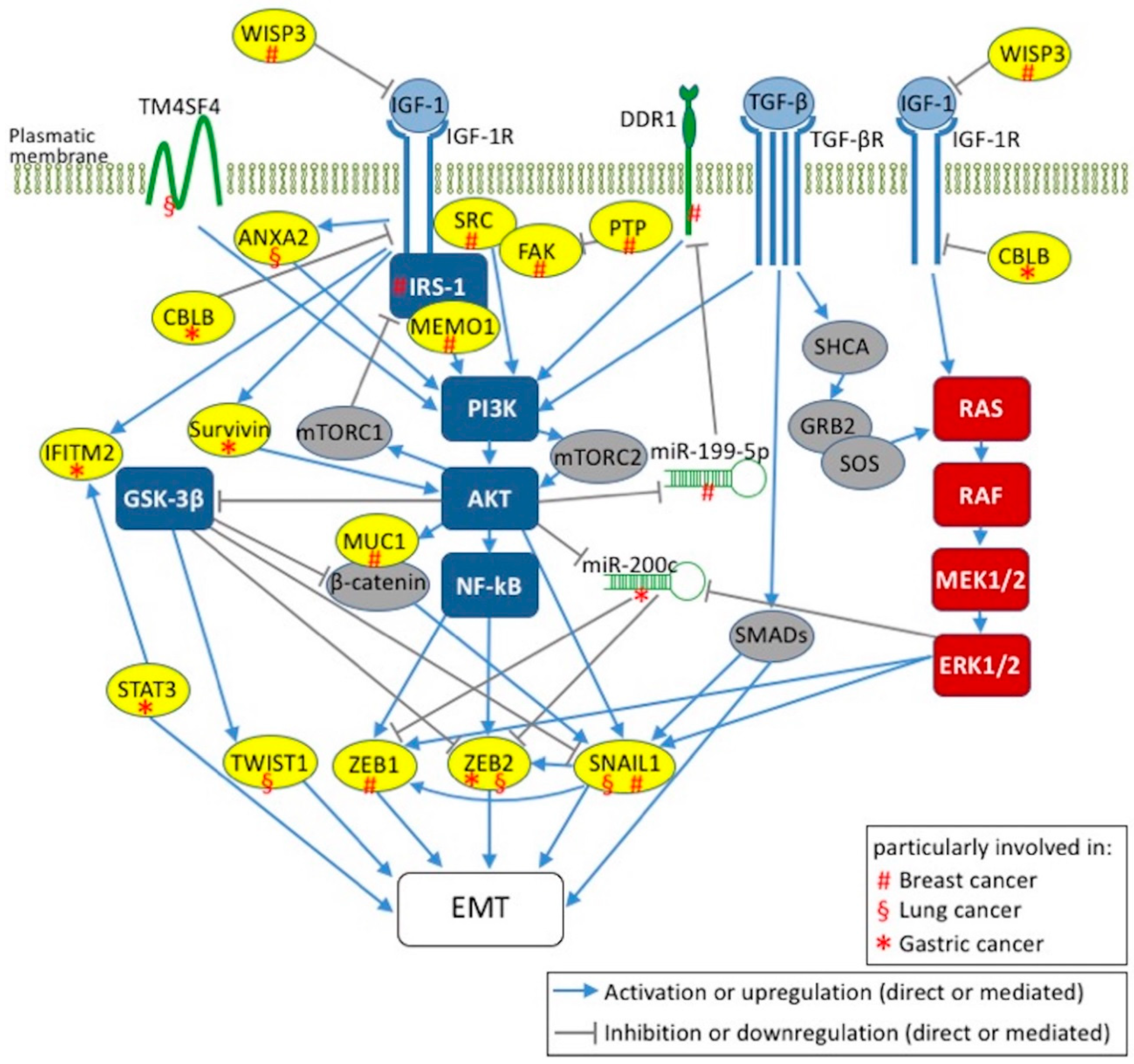{kind=link}
1. Introduction
2. The IGF System
3. The IGF System and Breast, Lung, and Gastric Cancers
4. Epithelial-Mesenchymal Transition (EMT)
4.1. Transcription Factors
4.2. Signaling Pathways
5. IGF-1 Signaling and EMT Activation in Breast, Lung, and Gastric Cancers
5.1. Breast Cancer
5.2. Lung Cancer
5.3. Gastric Cancer
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IGF-1 | Insulin-like growth factor 1 |
| IGF-1R | IGF-1 receptor |
| GH | Growth hormone |
| IGFBP | IGF binding protein 3 |
| EMT | Epithelial-mesenchymal transition |
| ECM | Extracellular matrix |
| CSC | Cancer stem cell |
| RTK | Receptor tyrosine kinase |
| NCAM | Neural cell adhesion molecule |
| MMP | Matrix metalloproteinase |
| bHLH | Helix-loop-helix |
| ZEB | Zinc finger E-box |
| TGF-β | Transforming growth factor-beta |
| GSK-3β | Glycogen synthase kinase 3-beta |
| NF-κB | Nuclear factor kappa B |
| PI3K | Phosphoinositide 3-kinase |
| AKT | AKT serine/threonine kinase |
| HIF1α | Hypoxia-inducible factor 1α |
| MAPK | Mitogen-activated protein kinase |
| ID1 | Inhibitor of DNA binding 1 |
| mTORC | Mammalian TOR complex |
| ERK | Extracellular signal-regulated kinases |
| SHCA | SRC homology 2 domain-containing-transforming A |
| SOS | Son of seven-less |
| TNF | Tumor necrosis factor |
| TRAF6 | TNF receptor-associated factor 6 |
| TAK1 | TGF-β-activated kinase 1 |
| BC | Breast cancer |
| DDR1 | Discoidin domain receptor 1 |
| MEMO1 | Mediator of ErbB2-driven cell motility 1 |
| IRS-1 | Insulin receptor substrate-1 |
| WISP3 | WNT1 inducible signaling pathway protein 3 |
| MUC1 | Mucin1 |
| EGFR | Epidermal growth factor receptor |
| FAK | Focal adhesion kinase |
| TNBC | Triple negative breast cancer |
| LC | Lung cancer |
| NSCLC | Non-small cell lung cancer |
| IKBα | NF-κB inhibitor alpha |
| SCLC | Small cell lung cancer |
| TKI | Tyrosine kinase inhibitors |
| CAF | Cancer-associated fibroblasts |
| ANXA2 | Annexin A2 |
| HGF | Hepatocyte growth factor |
| TM4SF4 | Transmembrane 4L six family member 4 |
| GC | Gastric cancer |
| CBLB | Cbl proto-oncogene B |
| CBLC | Cbl proto-oncogene C |
| IFITM2 | Interferon induced transmembrane protein 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
References
- Baserga, R.; Peruzzi, F.; Reiss, K. The igf-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor 1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Bruchim, I. The insulin-like growth factor-i receptor as an oncogene. Arch. Physiol. Biochem. 2009, 115, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G. Small is beautiful: Insulin-like growth factors and their role in growth, development, and cancer. J. Clin. Oncol. 2010, 28, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Batth, I.S.; Qu, X.; Xu, L.; Song, N.; Wang, R.; Liu, Y. Igf-ir signaling in epithelial to mesenchymal transition and targeting igf-ir therapy: Overview and new insights. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Overview of the igf-i system. Horm. Res. 2006, 65, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Inokuchi, M.; Takagi, Y.; Otsuki, S.; Fujimori, Y.; Yanaka, Y.; Kobayashi, K.; Higuchi, K.; Kojima, K.; Kawano, T. Relationship between expression of igfbp7 and clinicopathological variables in gastric cancer. J. Clin. Pathol. 2015, 68, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A.; Headey, S.J.; Norton, R.S. Igf-binding proteins—The pieces are falling into place. Trends Endocrinol. Metab. 2005, 16, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Brouwer-Visser, J.; Huang, G.S. Igf2 signaling and regulation in cancer. Cytokine Growth Factor Rev. 2015, 26, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Vishwamitra, D.; George, S.K.; Shi, P.; Kaseb, A.O.; Amin, H.M. Type i insulin-like growth factor receptor signaling in hematological malignancies. Oncotarget 2017, 8, 1814–1844. [Google Scholar] [CrossRef] [PubMed]
- Laviola, L.; Natalicchio, A.; Giorgino, F. The igf-i signaling pathway. Curr. Pharm. Des. 2007, 13, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Imperlini, E.; Mancini, A.; Alfieri, A.; Martone, D.; Caterino, M.; Orru, S.; Buono, P. Molecular effects of supraphysiological doses of doping agents on health. Mol. Biosyst. 2015, 11, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Harrela, M.; Koistinen, H.; Kaprio, J.; Lehtovirta, M.; Tuomilehto, J.; Eriksson, J.; Tolvanen, L.; Koskenvuo, M.; Leinonen, P.; Koistinen, R.; et al. Genetic and environmental components of interindividual variation in circulating levels of igf-i, igf-ii, igfbp-1, and igfbp-3. J. Clin. Investig. 1996, 98, 2612–2615. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth-factors. Endocr. Rev. 1994, 15, 80–101. [Google Scholar] [PubMed]
- Fontana, L.; Weiss, E.P.; Villareal, D.T.; Klein, S.; Holloszy, J.O. Long-term effects of calorie or protein restriction on serum igf-1 and igfbp-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Veldhuis, J.D.; Rood, J.; Smith, S.R.; Williamson, D.; Ravussin, E.; Team, P.C. The effect of caloric restriction interventions on growth hormone secretion in nonobese men and women. Aging Cell 2010, 9, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Matsubara, T.; Tobina, T.; Shindo, M.; Tokuyama, K.; Tanaka, K.; Tanaka, H. Effect of low-intensity aerobic exercise on insulin-like growth factor-i and insulin-like growth factor-binding proteins in healthy men. Int. J. Endocrinol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; King, B.; Ewert, E.; Su, X.; Mardiyati, N.; Zhao, Z.; Wang, W. Exercise activates p53 and negatively regulates igf-1 pathway in epidermis within a skin cancer model. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Hede, M.S.; Salimova, E.; Piszczek, A.; Perlas, E.; Winn, N.; Nastasi, T.; Rosenthal, N. E-peptides control bioavailability of igf-1. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- De Santi, M.; Annibalini, G.; Barbieri, E.; Villarini, A.; Vallorani, L.; Contarelli, S.; Berrino, F.; Stocchi, V.; Brandi, G. Human igf1 pro-forms induce breast cancer cell proliferation via the igf1 receptor. Cell. Oncol. 2016, 39, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Annibalini, G.; Contarelli, S.; De Santi, M.; Saltarelli, R.; Di Patria, L.; Guescini, M.; Villarini, A.; Brandi, G.; Stocchi, V.; Barbieri, E. The intrinsically disordered e-domains regulate the igf-1 prohormones stability, subcellular localisation and secretion. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Philippou, A.; Maridaki, M.; Pneumaticos, S.; Koutsilieris, M. The complexity of the igf1 gene splicing, posttranslational modification and bioactivity. Mol. Med. 2014, 20, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Kiess, W.; Paquette, J.; Koepf, G.; Wolf, E.; Deal, C. Proinsulin-like growth factor-ii overexpression does not alter monoallelic h19 gene expression in transfected human embryonic kidney fibroblasts. Biochem. Biophys. Res. Commun. 1999, 255, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor ii in oncogene-induced tumorigenesis. Nature 1994, 369, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor-i (igf-1) and type-1 igf receptor (igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Sell, C.; Rubini, M.; Rubin, R.; Liu, J.P.; Efstratiadis, A.; Baserga, R. Simian virus-40 large tumor-antigen is unable to transform mouse embryonic fibroblasts lacking type-1 insulin-like growth-factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11217–11221. [Google Scholar] [CrossRef] [PubMed]
- Bentov, I.; Werner, H. Igf, igf receptor and overgrowth syndromes. Pediatr. Endocrinol. Rev. 2004, 1, 352–360. [Google Scholar] [PubMed]
- Werner, H. For debate: The pathophysiological significance of igf-i receptor overexpression: New insights. Pediatr. Endocrinol. Rev. 2009, 7, 2–5. [Google Scholar] [PubMed]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Bruchim, I. Igf-1 and brca1 signalling pathways in familial cancer. Lancet Oncol. 2012, 13, E537–E544. [Google Scholar] [CrossRef]
- Plymate, S.R.; Bae, V.L.; Maddison, L.; Quinn, L.S.; Ware, J.L. Reexpression of the type 1 insulin-like growth factor receptor inhibits the malignant phenotype of simian virus 40 t antigen immortalized human prostate epithelial cells. Endocrinology 1997, 138, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Damon, S.E.; Plymate, S.R.; Carroll, J.M.; Sprenger, C.C.; Dechsukhum, C.; Ware, J.L.; Roberts, C.T. Transcriptional regulation of insulin-like growth factor-i receptor gene expression in prostate cancer cells. Endocrinology 2001, 142, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Lee, A.V. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J. Mammary Gland Biol. Neoplasia 2000, 5, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Schnarr, B.; Strunz, K.; Ohsam, J.; Benner, A.; Wacker, J.; Mayer, D. Down-regulation of insulin-like growth factor-i receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int. J. Cancer 2000, 89, 506–513. [Google Scholar] [CrossRef]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the igf system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Maor, S.; Mayer, D.; Yarden, R.I.; Lee, A.V.; Sarfstein, R.; Werner, H.; Papa, M.Z. Estrogen receptor regulates insulin-like growth factor-i receptor gene expression in breast tumor cells: Involvement of transcription factor sp1. J. Endocrinol. 2006, 191, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Schayek, H.; Seti, H.; Greenberg, N.M.; Sun, S.H.; Werner, H.; Plymate, S.R. Differential regulation of insulin-like growth factor-i receptor gene expression by wild type and mutant androgen receptor in prostate cancer cells. Mol. Cell. Endocrinol. 2010, 323, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Hellawell, G.O.; Turner, G.D.; Davies, D.R.; Poulsom, R.; Brewster, S.F.; Macaulay, V.M. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002, 62, 2942–2950. [Google Scholar] [PubMed]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. Igf signaling defects as causes of growth failure and iugr. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Kruis, T.; Klammt, J.; Galli-Tsinopoulou, A.; Wallborn, T.; Schlicke, M.; Muller, E.; Kratzsch, J.; Korner, A.; Odeh, R.; Kiess, W.; et al. Heterozygous mutation within a kinase-conserved motif of the insulin-like growth factor i receptor causes intrauterine and postnatal growth retardation. J. Clin. Endocrinol. Metab. 2010, 95, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Wallborn, T.; Wuller, S.; Klammt, J.; Kruis, T.; Kratzsch, J.; Schmidt, G.; Schlicke, M.; Muller, E.; van de Leur, H.S.; Kiess, W.; et al. A heterozygous mutation of the insulin-like growth factor-i receptor causes retention of the nascent protein in the endoplasmic reticulum and results in intrauterine and postnatal growth retardation. J. Clin. Endocrinol. Metab. 2010, 95, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Feng, Z.H.; Mak, T.W.; You, H.; Jin, S.K. Coordination and communication between the p53 and igf-1-akt-tor signal transduction pathways. Genes Dev. 2006, 20, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Maor, S.B.; Abramovitch, S.; Erdos, M.R.; Brody, L.C.; Werner, H. Brca1 suppresses insulin-like growth factor-i receptor promoter activity: Potential interaction between brca1 and sp1. Mol. Genet. Metab. 2000, 69, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.S.P.; Cockman, M.E.; Sullivan, M.; Protheroe, A.; Turner, G.D.H.; Roberts, I.S.; Pugh, C.W.; Werner, H.; Macaulay, V.M. The vhl tumor suppressor inhibits expression of the igf1r and its loss induces igf1r upregulation in human clear cell renal carcinoma. Oncogene 2007, 26, 6499–6508. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Meisel-Sharon, S.; Bruchim, I. Oncogenic fusion proteins adopt the insulin-like growth factor signaling pathway. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Karnieli, E.; Werner, H.; Rauscher, F.J.; Benjamin, L.E.; LeRoith, D. The igf-i receptor gene promoter is a molecular target for the ewing’s sarcoma wilms’ tumor 1 fusion protein. J. Biol. Chem. 1996, 271, 19304–19309. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Idelman, G.; Rubinstein, M.; Pattee, P.; Nagalla, S.R.; Roberts, C.T. A novel ews-wt1 gene fusion product in desmoplastic small round cell tumor is a potent transactivator of the insulin-like growth factor-i receptor (igf-ir) gene. Cancer Lett. 2007, 247, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Ayalon, D.; Glaser, T.; Werner, H. Transcriptional regulation of igf-i receptor gene expression by the pax3-fkhr oncoprotein. Growth Horm. IGF Res. 2001, 11, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sharon, S.M.; Pozniak, Y.; Geiger, T.; Werner, H. Tmprss2-erg fusion protein regulates insulin-like growth factor-1 receptor (igf1r) gene expression in prostate cancer: Involvement of transcription factor sp1. Oncotarget 2016, 7, 51375–51392. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.E.; Jelinek, T.; Kaleko, M.; Siddle, K.; Weber, M.J. C-phosphorylation and activation of the igf-i receptor in src-transformed cells. J. Biol. Chem. 1994, 269, 27315–27321. [Google Scholar] [PubMed]
- Reiss, K.; Ferber, A.; Travali, S.; Porcu, P.; Phillips, P.D.; Baserga, R. The protooncogene c-myb increases the expression of insulin-like growth factor-i and insulin-like growth factor-i receptor messenger-rnas by a transcriptional mechanism. Cancer Res. 1991, 51, 5997–6000. [Google Scholar] [PubMed]
- Travali, S.; Reiss, K.; Ferber, A.; Petralia, S.; Mercer, W.E.; Calabretta, B.; Baserga, R. Constitutively expressed c-myb abrogates the requirement for insulinlike growth factor 1 in 3t3 fibroblasts. Mol. Cell. Biol. 1991, 11, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.O.; Park, J.G.; Lee, Y.I. Increased expression of the insulin-like growth factor i (igf-i) receptor gene in hepatocellular carcinoma cell lines: Implications of igf-i receptor gene activation by hepatitis b virus x gene product. Cancer Res. 1996, 56, 3831–3836. [Google Scholar] [PubMed]
- Jones, R.A.; Campbell, C.I.; Petrik, J.J.; Moorehead, R.A. Characterization of a novel primary mammary tumor cell line reveals that cyclin d1 is regulated by the type i insulin-like growth factor receptor. Mol. Cancer Res. 2008, 6, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Guha, N.; Sonksen, P.H.; Holt, R.I.G. Igf-i abuse in sport: Current knowledge and future prospects for detection. Growth Horm. Igf Res. 2009, 19, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Orrù, S.; Nigro, E.; Mandola, A.; Alfieri, A.; Buono, P.; Daniele, A.; Mancini, A.; Imperlini, E. A functional interplay between igf-1 and adiponectin. Int. J. Mol. Sci. 2017, 18, 2145. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.P. Growth hormone doping in sports: A critical review of use and detection strategies. Endocr. Rev. 2012, 33, 155–186. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.K.; Gravholt, C.H.; Orskov, H.; Rasmussen, M.H.; Christiansen, J.S.; Jorgensen, J.O.L. Dose dependency of the pharmacokinetics and acute lipolytic actions of growth hormone. J. Clin. Endocrinol. Metab. 2002, 87, 4691–4698. [Google Scholar] [CrossRef] [PubMed]
- Chikani, V.; Ho, K.K.Y. Action of gh on skeletal muscle function: Molecular and metabolic mechanisms. J. Mol. Endocrinol. 2014, 52, R107–R123. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Imperlini, E.; Alfieri, A.; Spaziani, S.; Martone, D.; Parisi, A.; Orru, S.; Buono, P. Dht and igf-1 in peripheral blood lymphocytes: New markers for the biological passport of athletes. J. Biol. Regul. Homeost. Agents 2013, 27, 757–770. [Google Scholar] [PubMed]
- Spaziani, S.; Imperlini, E.; Mancini, A.; Caterino, M.; Buono, P.; Orru, S. Insulin-like growth factor 1 receptor signaling induced by supraphysiological doses of igf-1 in human peripheral blood lymphocytes. Proteomics 2014, 14, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Imperlini, E.; Spaziani, S.; Mancini, A.; Caterino, M.; Buono, P.; Orru, S. Synergistic effect of dht and igf-1 hyperstimulation in human peripheral blood lymphocytes. Proteomics 2015, 15, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, A.; Alam, N.; Trerotola, M.; Languino, L.R. Insulin-like growth factor 1 stimulation of androgen receptor activity requires beta(1a) integrins. J. Cell. Physiol. 2012, 227, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.; Plymate, S.R. Interaction of igf signaling and the androgen receptor in prostate cancer progression. J. Cell. Biochem. 2006, 99, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Freier, S.; Weiss, O.; Eran, M.; Flyvbjerg, A.; Dahan, R.; Nephesh, I.; Safra, T.; Shiloni, E.; Raz, I. Expression of the insulin-like growth factors and their receptors in adenocarcinoma of the colon. Gut 1999, 44, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Meyerhardt, J.A.; Chan, A.T.; Ng, K.; Chan, J.A.; Wu, K.; Pollak, M.N.; Giovannucci, E.L.; Fuchs, C.S. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, E.; Shomron, N. Canalization of development by micrornas. Nat. Genet. 2006, 38. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.Q.; Chen, Z.J.; Jin, Y.; Heidbreder, C.E.; Kolokythas, A.; Wang, A.X.; Dai, Y.; Zhou, X.F. Microrna-7 targets igf1r (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem. J. 2010, 432, 199–205. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, E.L.; Parrish, J.K.; Irwin, A.E.; Niemeyer, B.F.; Kern, H.B.; Birks, D.K.; Jedlicka, P. A novel oncogenic mechanism in ewing sarcoma involving igf pathway targeting by ews/fli1-regulated micrornas. Oncogene 2011, 30, 4910–4920. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Plas, D.R.; Thompson, C.B. Akt-dependent transformation: There is more to growth than just surviving. Oncogene 2005, 24, 7435–7442. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mtor in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of igf1r in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Youlden, D.R.; Cramb, S.M.; Dunn, N.A.M.; Muller, J.M.; Pyke, C.M.; Baade, P.D. The descriptive epidemiology of female breast cancer: An international comparison of screening, incidence, survival and mortality. Cancer Epidemiol. 2012, 36, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer in women: Burden and trend. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Niksic, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Esteve, J.; et al. Global surveillance of trends in cancer survival 2000-14 (concord-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, Y.; Sun, X.J.; Wang, B.Y.; Wang, Z.H.; Luo, J.F.; Wang, L.P.; Zhang, S.; Cao, J.; Tao, Z.H.; et al. Biomarker assessment of the cbcsg006 trial: A randomized phase iii trial of cisplatin plus gemcitabine compared with paclitaxel plus gemcitabine as first-line therapy for patients with metastatic triple-negative breast cancer. Ann. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatrilova, R.; Kruzliak, P.; Stefanicka, P.; Busselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharm. 2018, 101, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Ouban, A.; Muraca, P.; Yeatman, T.; Coppola, D. Expression and distribution of insulin-like growth factor-1 receptor in human carcinomas. Hum. Pathol. 2003, 34, 803–808. [Google Scholar] [CrossRef]
- Shimizu, C.; Hasegawa, T.; Tani, Y.; Takahashi, F.; Takeuchi, M.; Watanabe, T.; Ando, M.; Katsumata, N.; Fujiwara, Y. Expression of insulin-like growth factor 1 receptor in primary breast cancer: Immunohistochemical analysis. Hum. Pathol. 2004, 35, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-i and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W. Insulin-like growth factor 1 (igf1), igf binding protein 3 (igfbp3), and breast cancer risk: Pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010, 11, 530–542. [Google Scholar] [PubMed]
- Duggan, C.; Wang, C.Y.; Neuhouser, M.L.; Xiao, L.; Smith, A.W.; Reding, K.W.; Baumgartner, R.N.; Baumgartner, K.B.; Bernstein, L.; Ballard-Barbash, R.; et al. Associations of insulin-like growth factor and insulin-like growth factor binding protein-3 with mortality in women with breast cancer. Int. J. Cancer 2013, 132, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Campbell, C.I.; Gunther, E.J.; Chodosh, L.A.; Petrik, J.J.; Khokha, R.; Moorehead, R.A. Transgenic overexpression of igf-ir disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene 2007, 26, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The role of the insulin-like growth factor-1 system in breast cancer. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Motallebnezhad, M.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Nickho, H.; Samadi-Kafil, H.; Shamsasenjan, K.; Yousefi, M. The insulin-like growth factor-i receptor (igf-ir) in breast cancer: Biology and treatment strategies. Tumour Biol. 2016, 37, 11711–11721. [Google Scholar] [CrossRef] [PubMed]
- Hartog, H.; Boezen, H.M.; de Jong, M.M.; Schaapveld, M.; Wesseling, J.; van der Graaf, W.T. Prognostic value of insulin-like growth factor 1 and insulin-like growth factor binding protein 3 blood levels in breast cancer. Breast 2013, 22, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Yardley, D.A.; Layman, R.; Sparano, J.A.; Chuang, E.; Northfelt, D.W.; Schwartz, G.N.; Youssoufian, H.; Tang, S.; Novosiadly, R.; et al. Clinical and translational results of a phase ii, randomized trial of an anti-igf-1r (cixutumumab) in women with breast cancer that progressed on endocrine therapy. Clin. Cancer Res. 2015, 22, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Kurzrock, R.; Patnaik, A.; Aisner, J.; Warren, T.; Leong, S.; Benjamin, R.; Eckhardt, S.G.; Eid, J.E.; Greig, G.; Habben, K.; et al. A phase i study of weekly r1507, a human monoclonal antibody insulin-like growth factor-i receptor antagonist, in patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 2458–2465. [Google Scholar] [CrossRef] [PubMed]
- Atzori, F.; Tabernero, J.; Cervantes, A.; Prudkin, L.; Andreu, J.; Rodriguez-Braun, E.; Domingo, A.; Guijarro, J.; Gamez, C.; Rodon, J.; et al. A phase i pharmacokinetic and pharmacodynamic study of dalotuzumab (mk-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6304–6312. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.H.; Lenz, H.J.; Saleh, M.N.; Mackenzie, M.J.; Knost, J.A.; Pathiraja, K.; Langdon, R.B.; Yao, S.L.; Lu, B.D. A randomized, phase ii study of the anti-insulin-like growth factor receptor type 1 (igf-1r) monoclonal antibody robatumumab (sch 717454) in patients with advanced colorectal cancer. Cancer Med. 2014, 3, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.D.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The international epidemiology of lung cancer: Latest trends, disparities, and tumor characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Agullò-Ortuño, M.T.; Diaz-Garcia, C.V.; Agudo-Lopez, A.; Perez, C.; Cortijo, A.; Paz-Ares, L.; Lopez-Rios, F.; Pozo, F.; de Castro, J.; Cortes-Funes, H.; et al. Relevance of insulin-like growth factor 1 receptor gene expression as a prognostic factor in non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.G.; Lu, P.F.; Liang, Z.; Zhang, Z.F.; Shi, W.C.; Cai, X.B.; Chen, C.Y. Increased insulin-like growth factor 1 receptor (igf1r) expression in small cell lung cancer and the effect of inhibition of igf1r expression by rnai on growth of human small cell lung cancer nci-h446 cell. Growth Factors 2015, 33, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Bilgin, E.; Tastekin, D.; Erturk, K.; Duranyildiz, D. Serum igf-1 and igfbp-3 levels as clinical markers for patients with lung cancer. Biomed. Rep. 2016, 4, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Izycki, T.; Chyczewska, E.; Naumnik, W.; Ossolinska, M. Serum levels of igf-i and igfbp-3 in patients with lung cancer during chemotherapy. Oncol. Res. 2006, 16, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.A.; Sun, Y.; Palmer, J.; Solomides, C.; Huang, L.C.; Shyr, Y.; Dicker, A.P.; Lu, B. Igfbp3 modulates lung tumorigenesis and cell growth through igf1 signaling. Mol. Cancer Res. 2017, 15, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Xu, L.; Li, C.; Zhao, L.; Ma, Y.J.; Zheng, H.C.; Li, Z.; Zhang, Y.; Wang, R.Y.; Liu, Y.P.; et al. Ubiquitin ligase cbl-b represses igf-i-induced epithelial mesenchymal transition via zeb2 and microrna-200c regulation in gastric cancer cells. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, L.; Manchi, M.; De Re, V.; Dolcetti, R.; Canzonieri, V. Proposed molecular and mirna classification of gastric cancer. Int. J. Mol. Sci. 2018, 19, 1683. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Maciejewski, R.; Sitarz, R. The pattern of signatures in gastric cancer prognosis. Int. J. Mol. Sci. 2018, 19, 1658. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, D.; Polom, K.; Neri, A.; Roviello, F. Clinical impact of molecular classifications in gastric cancer. Updates Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Liu, H.; Yu, J.; Shi, G.Y.; Zhao, L.Y.; Li, G.X. Prognostic and predictive blood biomarkers in gastric cancer and the potential application of circulating tumor cells. World J. Gastroenterol. 2018, 24, 2236–2246. [Google Scholar] [CrossRef] [PubMed]
- Orditura, M.; Galizia, G.; Sforza, V.; Gambardella, V.; Fabozzi, A.; Laterza, M.M.; Andreozzi, F.; Ventriglia, J.; Savastano, B.; Mabilia, A.; et al. Treatment of gastric cancer. World J. Gastroenterol. 2014, 20, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rao, H.; Liu, J.; Geng, Q.; Guo, J.; Kong, P.; Li, S.; Liu, X.; Sun, X.; Zhan, Y.; et al. Lymph nodes ratio based nomogram predicts survival of resectable gastric cancer regardless of the number of examined lymph nodes. Oncotarget 2017, 8, 45585–45596. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Shin, A.; Kim, S.G.; Hwang, J.A.; Hong, S.H.; Lee, Y.S.; Kwon, H.C. Relationship between insulin-like growth factor axis gene polymorphisms and clinical outcome in advanced gastric cancer patients treated with folfox. Oncotarget 2016, 7, 31204–31214. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Chen, Z.K.; Wu, S.B.; Chen, J.X.; Li, X.L.; Li, J.; Yin, J.Y.; Chen, Z.H. Expression levels of insulin-like growth factor-1 and multidrug resistance-associated protein-1 indicate poor prognosis in patients with gastric cancer. Digestion 2009, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion—Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.X.; Zhang, X.Y.; Xu, C.J. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget 2017, 8, 81558–81571. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.A.; Danen, E.H.J.; Beltman, J.B. Deciphering epithelial-mesenchymal transition regulatory networks in cancer through computational approaches. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metast. Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of wnt receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. P120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Mol. Biol. Cadherins 2013, 116, 409–432. [Google Scholar]
- Theveneau, E.; Mayor, R. Cadherins in collective cell migration of mesenchymal cells. Curr. Opin. Cell Biol. 2012, 24, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.J.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Pursell, B.; Lu, S.L.; Chang, T.K.; Mercurio, A.M. Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J. Cell Sci. 2009, 122, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.P.; Brumwell, A.N.; Chapman, H.A. Integrin alpha 3 beta 1-dependent beta-catenin phosphorylation links epithelial smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Maschler, S.; Wirl, G.; Spring, H.; Bredow, D.V.; Sordat, I.; Beug, H.; Reichmann, E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene 2005, 24, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Mise, N.; Savai, R.; Yu, H.Y.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin is a transforming growth factor-beta (tgf-beta)/smad3 target gene that regulates lung cancer cell motility via integrin alpha 5 beta 1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type i induces disruption of e-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; de Herreros, A.G. The transcription factor snail is a repressor of e-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing e-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase lsd1 in snail-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates e-cadherin repression by the recruitment of the sin3a/histone deacetylase 1 (hdac1)/hdac2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. Ezh2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with hdac1/hdac2 and snail to inhibit e-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez-Munoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for e-cadherin repression by the snail1 transcription factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with suv39h1 is critical for snail-mediated e-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.F.; Wu, Y.D.; Yao, J.; Wang, Y.F.; Yu, Y.H.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with snail and is critical for snail-mediated e-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Hsu, D.S.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhou, J.; Fu, J.J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J.M. Phosphorylation of serine 68 of twist1 by mapks stabilizes twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of smad signaling through a differential recruitment of coactivators and corepressors by zeb proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between snail1 and twist in the regulation of zeb1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. Tgf-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Piek, E.; Zavadil, J.; Liang, D.; Xie, D.L.; Heyer, J.; Pavlidis, P.; Kucherlapati, R.; Roberts, A.B.; Bottinger, E.P. Hierarchical model of gene regulation by transforming growth factor beta. Proc. Natl. Acad. Sci. USA 2003, 100, 10269–10274. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Bottinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [PubMed]
- Jorda, M.; Olmeda, D.; Vinyals, A.; Valero, E.; Cubillo, E.; Llorens, A.; Canoe, A.; Fabra, A. Upregulation of mmp-9 in mdck epithelial cell line in response to expression of the snail transcription factor. J. Cell Sci. 2005, 118, 3371–3385. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by delta ef1 proteins in epithelial-mesenchymal transition induced by tgf-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.; Chen, C.R.; Massague, J. A self-enabling tgf beta response coupled to stress signaling: Smad engages stress response factor atf3 for id1 repression in epithelial cells. Mol. Cell 2003, 11, 915–926. [Google Scholar] [CrossRef]
- Nawshad, A.; Medici, D.; Liu, C.C.; Hay, E.D. Tgf beta 3 inhibits e-cadherin gene expression in palate medial-edge epithelial cells through a smad2-smad4-lef1 transcription complex. J. Cell Sci. 2007, 120, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-beta 1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in tgf-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-smad tgf-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. Tgf-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Emergence of the phosphoinositide 3-kinase-akt-mammalian target of rapamycin axis in transforming growth factor-beta-induced epithelial-mesenchymal transition. Cells Tissues Organs 2011, 193, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. Tgf-beta-induced activation of mtor complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in tgf-beta-induced epithelial to mesenchymal transition is regulated by activation of the mtor pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; Garcia de Herreros, A.; Bellacosa, A.; Larue, L. Activation of nf-kappa b by akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. Tgf-beta activates erk map kinase signalling through direct phosphorylation of shca. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Bowen, K.A.; Liu, J.Y.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.Y.; et al. Mtorc1 and mtorc2 regulate emt, motility, and metastasis of colorectal cancer via rhoa and rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. Braf and ras oncogenes regulate rho gtpase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Litzenburger, B.C.; Cui, X.; Delgado, D.A.; Grabiner, B.C.; Lin, X.; Lewis, M.T.; Gottardis, M.M.; Wong, T.W.; Attar, R.M.; et al. Constitutively active type i insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by nf-kappab and snail. Mol. Cell. Biol. 2007, 27, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Canonici, A.; Steelant, W.; Rigot, V.; Khomitch-Baud, A.; Boutaghou-Cherid, H.; Bruyneel, E.; Van Roy, F.; Garrouste, F.; Pommier, G.; Andre, F. Insulin-like growth factor-i receptor, e-cadherin and alpha v integrin form a dynamic complex under the control of alpha-catenin. Int. J. Cancer 2008, 122, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like growth factor-l-dependent up-regulation of zeb1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of akt1 and akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Guvakova, M.A.; Surmacz, E. The activated insulin-like growth factor i receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of fak, cas, and paxillin. Exp. Cell. Res. 1999, 251, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Matà, R.; Palladino, C.; Nicolosi, M.L.; Lo Presti, A.R.; Malaguarnera, R.; Ragusa, M.; Sciortino, D.; Morrione, A.; Maggiolini, M.; Vella, V.; et al. Igf-i induces upregulation of ddr1 collagen receptor in breast cancer cells by suppressing mir-199a-5p through the pi3k/akt pathway. Oncotarget 2016, 7, 7683–7700. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Chen, J. Memo1, a new irs1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene 2013, 32, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Lorenzatti, G.; Huang, W.; Pal, A.; Cabanillas, A.M.; Kleer, C.G. Ccn6 (wisp3) decreases zeb1-mediated emt and invasion by attenuation of igf-1 receptor signaling in breast cancer. J. Cell Sci. 2011, 124, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Wang, M.; Ou, Y.; Zhao, Y. Igf-1-induced epithelial-mesenchymal transition in mcf-7 cells is mediated by muc1. Cell Signal 2014, 26, 2131–2137. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.A.; Damjanovski, S. Igf-1 increases invasive potential of mcf 7 breast cancer cells and induces activation of latent tgf-beta1 resulting in epithelial to mesenchymal transition. Cell Commun. Signal 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Schiemann, W.P. Tgf-beta stimulation of emt programs elicits non-genomic er-alpha activity and anti-estrogen resistance in breast cancer cells. J. Cancer Metast. Treat 2017, 3, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Parvani, J.G.; Schiemann, W.P. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J. Mammary Gland Biol. Neoplasia 2010, 15, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. Fak activation is required for igf1r-mediated regulation of emt, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757–4772. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Imperlini, E.; Scudiero, O.; Monaco, M.L.; Polito, R.; Mazzarella, G.; Orru, S.; Bianco, A.; Daniele, A. Differentially expressed and activated proteins associated with non small cell lung cancer tissues. Respir. Res. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.H.; Fu, L.W. Mechanisms of resistance to egfr tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, J.J.; Zeng, Y.Y.; Zhang, X.; Hu, Q.T.; Zheng, J.H.; Chen, B.; Xie, B.; Zhang, W.M. Implication of epithelial-mesenchymal transition in igf1r-induced resistance to egfr-tkis in advanced non-small cell lung cancer. Oncotarget 2015, 6, 44332–44345. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.Y.; Gow, C.H.; Yang, P.C. Egfr mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. Egfr mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without t790m mutations in egfr mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.C.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.J.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to egfr inhibitors. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.M.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to egfr inhibitors occasionally harbor braf gene mutations but lack mutations in kras, nras, or mek1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to egfr tyrosine kinase inhibitors in cancer cells is mediated by loss of igf-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly acquired resistance to egfr tyrosine kinase inhibitors in nsclc cell lines through de-repression of fgfr2 and fgfr3 expression. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Shien, K.; Yamamoto, H.; Soh, J.; Miyoshi, S.; Toyooka, S. Drug resistance to egfr tyrosine kinase inhibitors for non-small cell lung cancer. Acta Med. Okayama 2014, 68, 191–200. [Google Scholar] [PubMed]
- Byers, L.A.; Diao, L.X.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.H.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to egfr and pi3k inhibitors and identifies axl as a therapeutic target for overcoming egfr inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Cufi, S.; Oliveras-Ferraros, C.; Torres-Garcia, V.Z.; Corominas-Faja, B.; Cuyas, E.; Bonavia, R.; Visa, J.; Martin-Castillo, B.; Barrajon-Catalan, E.; et al. Igf-1r/epithelial-to-mesenchymal transition (emt) crosstalk suppresses the erlotinib-sensitizing effect of egfr exon 19 deletion mutations. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Repellin, C.E.; Shimamura, T.; Capelletti, M.; Zejnullahu, K.; Ercan, D.; Christensen, J.G.; Wong, K.K.; Gray, N.S.; Janne, P.A. Resistance to irreversible egf receptor tyrosine kinase inhibitors through a multistep mechanism involving the igf1r pathway. Cancer Res. 2013, 73, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.M.; Zeng, S.S.; Wang, Z.T.; Wu, M.H.; Ma, Y.H.; Ye, X.X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and egfr-tki resistance of non-small cell lung cancers via hgf/igf-1/anxa2 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Selzer, E.; Kornek, G. Targeted drugs in combination with radiotherapy for the treatment of solid tumors: Current state and future developments. Expert Rev. Clin. Pharmacol. 2013, 6, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Kim, S.Y.; Lee, J.; Cho, E.W.; Kim, I.G. Tm4sf4 overexpression in radiation-resistant lung carcinoma cells activates igf1r via elevation of igf1. Oncotarget 2014, 5, 9823–9837. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.; Nagaraju, G.P.; Rajitha, B.; Lammata, S.; Jella, K.K.; Buchwald, Z.S.; Lakka, S.S.; Ali, A.N. Matrix metalloproteinases: Their functional role in lung cancer. Carcinogenesis 2017, 38, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.L.; Pantel, K.; Kang, Y.B. Tumor metastasis: Moving new biological insights into the clinic. Nat. Med. 2013, 19, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kobayashi, I.; Murakami, A.; Kato, M.; Minakata, K.; Nara, T.; Hashimoto, M.; Yagishita, S.; Baskoro, H.; et al. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial-mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2014, 455, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Li, J.B.; Wu, D.W.; Han, G. The involvement of survivin in insulin-like growth factor 1-induced epithelial-mesenchymal transition in gastric cancer. Tumor Biol. 2016, 37, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Xu, L.; Zhao, L.; Ma, Y.J.; Zhu, Z.T.; Liu, Y.P.; Qu, X.J. Insulin-like growth factor-i induces epithelial to mesenchymal transition via gsk-3 beta and zeb2 in the bgc-823 gastric cancer cell line. Oncol. Lett. 2015, 9, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, R.; Yuan, L.Z.; Wang, S.Q.; Li, X.Y.; Ma, H.R.; Zhou, M.Y.; Pan, C.Q.; Zhang, J.W.; Huang, N.; et al. Igf1/igf1r/stat3 signaling-inducible ifitm2 promotes gastric cancer growth and metastasis. Cancer Lett. 2017, 393, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ohuchida, K.; Mizumoto, K.; Sato, N.; Kayashima, T.; Fujita, H.; Nakata, K.; Tanaka, M. Microrna, hsa-mir-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Romero-Pérez, L.; Lopez-Garcia, M.A.; Diaz-Martin, J.; Biscuola, M.; Castilla, M.A.; Tafe, L.J.; Garg, K.; Oliva, E.; Matias-Guiu, X.; Soslow, R.A.; et al. Zeb1 overexpression associated with e-cadherin and microrna-200 downregulation is characteristic of undifferentiated endometrial carcinoma. Mod. Pathol. 2013, 26, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between zeb1-sip1 and the microrna-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.H.; Kang, Y.B. The mir-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of e-cadherin transcriptional repressors zeb1 and zeb2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Chen, X.Y.; Wang, R.; Xiao, P.; Xu, Z.H.; Chen, L.; Hang, W.W.; Ruan, A.M.; Yang, H.M.; Zhang, X.P. Microrna-200c modulates the epithelial-to-mesenchymal transition in human renal cell carcinoma metastasis. Oncol. Rep. 2013, 30, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Raja, S.M.; Chen, G.S.; Virmani, S.; Williams, S.H.; Clubb, R.J.; Mukhopadhyay, C.; Rainey, M.A.; Ying, G.G.; Dimri, M.; et al. Negative regulation of egfr-vav2 signaling axis by cbl ubiquitin ligase controls egf receptor-mediated epithelial cell adherens junction dynamics and cell migration. J. Biol. Chem. 2011, 286, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Liu, Y.P.; Ma, Y.J.; Zhang, Y.; Li, Y.C.; Hou, K.Z. Up-regulation of the cbl family of ubiquitin ligases is involved in atra and bufalin-induced cell adhesion but not cell differentiation. Biochem. Biophys. Res. Commun. 2008, 367, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Fournier, T.M.; Lamorte, L.; Maroun, C.R.; Lupher, M.; Band, H.; Langdon, W.; Park, M. Cbl-transforming variants trigger a cascade of molecular alterations that lead to epithelial mesenchymal conversion. Mol. Biol. Cell 2000, 11, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Sehat, B.; Andersson, S.; Girnita, L.; Larsson, O. Identification of c-cbl as a new ligase for insulin-like growth factor-i receptor with distinct roles from mdm2 in receptor ubiquitination and endocytosis. Cancer Res. 2008, 68, 5669–5677. [Google Scholar] [CrossRef] [PubMed]
- Numata, K.; Oshima, T.; Sakamaki, K.; Yoshihara, K.; Aoyama, T.; Hayashi, T.; Yamada, T.; Sato, T.; Cho, H.; Shiozawa, M.; et al. Clinical significance of igf1r gene expression in patients with stage ii/iii gastric cancer who receive curative surgery and adjuvant chemotherapy with s-1. J. Cancer Res. Clin. Oncol. 2016, 142, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Han, X.D.; Qiu, Y.; Xiong, J.; Yu, Y.; Wang, B.; Zhu, Z.Z.; Qian, B.P.; Chen, Y.X.; Wang, S.F.; et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2012, 105, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Denduluri, S.K.; Idowu, O.; Wang, Z.L.; Liao, Z.; Yan, Z.J.; Mohammed, M.K.; Ye, J.X.; Wei, Q.; Wang, J.; Zhao, L.G.; et al. Insulin-like growth factor (igf) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; DeSantos, V.; Ferry, R.J., Jr.; Kurzrock, R. Early drug development of inhibitors of the insulin-like growth factor-i receptor pathway: Lessons from the first clinical trials. Mol. Cancer Ther. 2008, 7, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Belfiore, A. The insulin receptor: A new target for cancer therapy. Front. Endocrinol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Olmos, D.; Molife, L.R.; de Bono, J.S. Targeting the insulin-like growth factor signaling pathway: Figitumumab and other novel anticancer strategies. Expert Opin. Investig. Drugs 2011, 20, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. Anticancer igf1r classes take more knocks. Nat. Rev. Drug. Dis. 2013, 12, 250. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cevenini, A.; Orrù, S.; Mancini, A.; Alfieri, A.; Buono, P.; Imperlini, E. Molecular Signatures of the Insulin-Like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers. Int. J. Mol. Sci. 2018, 19, 2411. https://doi.org/10.3390/ijms19082411
Cevenini A, Orrù S, Mancini A, Alfieri A, Buono P, Imperlini E. Molecular Signatures of the Insulin-Like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers. International Journal of Molecular Sciences. 2018; 19(8):2411. https://doi.org/10.3390/ijms19082411
Chicago/Turabian StyleCevenini, Armando, Stefania Orrù, Annamaria Mancini, Andreina Alfieri, Pasqualina Buono, and Esther Imperlini. 2018. "Molecular Signatures of the Insulin-Like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers" International Journal of Molecular Sciences 19, no. 8: 2411. https://doi.org/10.3390/ijms19082411
APA StyleCevenini, A., Orrù, S., Mancini, A., Alfieri, A., Buono, P., & Imperlini, E. (2018). Molecular Signatures of the Insulin-Like Growth Factor 1-Mediated Epithelial-Mesenchymal Transition in Breast, Lung and Gastric Cancers. International Journal of Molecular Sciences, 19(8), 2411. https://doi.org/10.3390/ijms19082411