PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity


{kind=link}
Abstract
1. Introduction
2. Adipose Tissue Physiology and Lipotoxicity
3. PPARs and Fat Mass Expansion and Function
4. PPARs and Pregnancy
5. PPARs and Aging
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| T2D | Type 2 diabetes |
| MetS | Metabolic Syndrome |
| AT | Adipose tissue |
| PPARs | Peroxisome proliferator-activated receptors |
| PPRE | Ppar response element |
| RXR | Retinoid X receptor |
| NF-κB | Nuclear factor kappa B |
| TZDs | Thiazolidenidiones |
| WAT | White adipose tissue |
| BAT | Brown adipose tissue |
| FFAs | Free fatty acids |
| TGs | Triglycerides |
| TNF-α | Tumor necrosis factor α |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| Ucp-1 | Uncoupling protein 1 |
| GDM | Gestational diabetes mellitus |
| CR | Caloric restriction |
References
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Curtis, R.K.; Jimenez-Linan, M.; Blount, M.; Yeo, G.S.H.; et al. PPAR gamma 2 Prevents Lipotoxicity by Controlling Adipose Tissue Expandability and Peripheral Lipid Metabolism. PLoS Genet. 2007, 3, e64. [Google Scholar] [CrossRef] [PubMed]
- Venteclef, N.; Jakobsson, T.; Steffensen, K.R.; Treuter, E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol. Metab. 2011, 22, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Wahli, W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J. Clin. Investig. 2006, 116, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Vitale, S.G.; Laganà, A.S.; Nigro, A.; La Rosa, V.L.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Corrado, F.; Buscema, M.; et al. Peroxisome Proliferator-Activated Receptor Modulation during Metabolic Diseases and Cancers: Master and Minions. PPAR Res. 2016, 2016, 6517313. [Google Scholar] [CrossRef] [PubMed]
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Saeland, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J. Immunol. 2007, 178, 4362–4372. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Schoonjans, K.; Ulven, S.M.; Weedon-Fekjaer, M.S.; Bentzen, T.G.; Kontnikova, H.; Auwerx, J.; Nebb, H.I. Adipose Tissue Expression of the Lipid Droplet-Associating Proteins31 S–S2 and Perilipin Is Controlled by Peroxisome Proliferator-Activated Receptor-γ. Diabetes 2004, 53, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wrahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Lalloyer, F.; Wouters, K.; Baron, M.; Caron, S.; Vallez, E.; Vanhoutte, J.; Baugé, E.; Shiri-Sverdlov, R.; Hofker, M.; Staels, B.; et al. Peroxisome proliferator-activated receptor-α gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Maes, M.; Zambon, A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome Proliferator-Activated Receptor δ Regulates Inflammation via NF-κB Signaling in Polymicrobial Sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Ferre, P. The Biology of Peroxisome Proliferator-Activated Receptors: Relationship With Lipid Metabolism and Insulin Sensitivity. Diabetes 2004, 53, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G.; Virtue, S.; Lelliott, C.; Boiani, R.; Campbell, M.; Christodoulides, C.; Perrin, C.; Jimenez-Linan, M.; Blount, M.; Dixon, J.; et al. The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-gamma2 isoform. Diabetes 2005, 54, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Kubota, T.; Kumagai, H.; Itoh, S.; Satoh, H.; Yano, W.; Ogata, H.; Tokuyama, K.; Takamoto, I.; et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J. Biol. Chem. 2006, 281, 8748–8755. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Treatment of insulin resistance with peroxisome proliferator–activated receptor γ agonists. J. Clin. Investig. 2000, 106, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kayahara, T.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef] [PubMed]
- Berbée, J.F.P.; Boon, M.R.; Khedoe, P.P.S.J.; Bartelt, A.; Schlein, C.; Worthmann, A.; Kooijman, S.; Hoeke, G.; Mol, I.M.; John, C.; et al. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat. Commun. 2015, 6, 6356. [Google Scholar] [CrossRef] [PubMed]
- Pisani, D.F.; Barquissau, V.; Chambard, J.-C.; Beuzelin, D.; Ghandour, R.A.; Giroud, M.; Mairal, A.; Pagnotta, S.; Cinti, S.; Langin, D.; et al. Mitochondrial fission is associated with UCP1 activity in human brite/beige adipocytes. Mol. Metab. 2018, 7, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, A.; Yamauchi, T.; Takekawa, S.; Hada, Y.; Ito, Y.; Maki, T.; Kadowaki, T. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: Comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes 2005, 54, 3358–3370. [Google Scholar] [CrossRef] [PubMed]
- Veiga, F.M.S.; Graus-Nunes, F.; Rachid, T.L.; Barreto, A.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Anti-obesogenic effects of WY14643 (PPAR-alpha agonist): Hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie 2017, 140, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Gervois, P.P.; Verschuren, L.; Staels, B.; Princen, H.M.G.; Kooistra, T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood 2003, 101, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, Q.; Lin, R.; Zhang, J.; Ren, F.; Zhang, J.; Ji, M.; Li, Y. PPARα agonist fenofibrate attenuates TNF-α-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp. Cell Res. 2013, 319, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Rosell, M.; Díaz-Delfín, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef] [PubMed]
- Rachid, T.L.; Penna-de-Carvalho, A.; Bringhenti, I.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. PPAR-α agonist elicits metabolically active brown adipocytes and weight loss in diet-induced obese mice. Cell Biochem. Funct. 2015, 33, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Shearer, B.G.; Steger, D.J.; Way, J.M.; Stanley, T.B.; Lobe, D.C.; Grillot, D.A.; Iannone, M.A.; Lazar, M.A.; Willson, T.M.; Billin, A.N. Identification and characterization of a selective peroxisome proliferator-activated receptor beta/delta (NR1C2) antagonist. Mol. Endocrinol. 2008, 22, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.-H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.B.; Zhang, H.; Rasmussen, T.H.; Petersen, R.K.; Flindt, E.N.; Kristiansen, K. Peroxisome proliferator-activated receptor delta (PPARdelta )-mediated regulation of preadipocyte proliferation and gene expression is dependent on cAMP signaling. J. Biol. Chem. 2001, 276, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Majithia, A.R.; Tsuda, B.; Agostini, M.; Gnanapradeepan, K.; Rice, R.; Peloso, G.; Patel, K.A.; Zhang, X.; Broekema, M.F.; Patterson, N.; et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016, 48, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zeve, D.; Suh, J.M.; Bosnakovski, D.; Kyba, M.; Hammer, R.E.; Tallquist, M.D.; Graff, J.M. White Fat Progenitor Cells Reside in the Adipose Vasculature. Science 2008, 322, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Semple, R.K.; Meirhaeghe, A.; Vidal-Puig, A.J.; Schwabe, J.W.R.; Wiggins, D.; Gibbons, G.F.; Gurnell, M.; Chatterjee, V.K.K.; O’Rahilly, S. A dominant negative human peroxisome proliferator-activated receptor (PPAR){alpha} is a constitutive transcriptional corepressor and inhibits signaling through all PPAR isoforms. Endocrinology 2005, 146, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Nora, E.D.; Grosse, J.; Manieri, M.; Stoeger, T.; Medina-Gomez, G.; Burling, K.; Wattler, S.; Russ, A.; Yeo, G.S.H.; et al. Leptin deficiency unmasks the deleterious effects of impaired peroxisome proliferator-activated receptor gamma function (P465L PPARgamma) in mice. Diabetes 2006, 55, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, G.; Yetukuri, L.; Velagapudi, V.; Campbell, M.; Blount, M.; Jimenez-Linan, M.; Ros, M.; Oresic, M.; Vidal-Puig, A. Adaptation and failure of pancreatic beta cells in murine models with different degrees of metabolic syndrome. Dis. Model. Mech. 2009, 2, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003, 52, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Murakami, K.; Motojima, K.; Komeda, K.; Ide, T.; Kubota, N.; Terauchi, Y.; Tobe, K.; et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (PPARgamma) deficiency and PPARgamma agonist improve insulin resistance. J. Biol. Chem. 2001, 276, 41245–41254. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Rhoades, B.; Shapiro, J.S.; Rich, A.S.; Kim, J.K.; Shulman, G.I.; Kaestner, K.H.; Lazar, M.A. Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev. Cell 2003, 5, 657–663. [Google Scholar] [CrossRef]
- Lefebvre, B.; Benomar, Y.; Guédin, A.; Langlois, A.; Hennuyer, N.; Dumont, J.; Bouchaert, E.; Dacquet, C.; Pénicaud, L.; Casteilla, L.; et al. Proteasomal degradation of retinoid X receptor alpha reprograms transcriptional activity of PPARgamma in obese mice and humans. J. Clin. Investig. 2010, 120, 1454–1468. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Xu, M.; Spiegelman, B.M. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J. Clin. Investig. 1997, 100, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.Q.; Phillips, C.T.; et al. A PPARγ-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast Growth Factor-21 Regulates PPARγ Activity and the Antidiabetic Actions of Thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional Control of Brown Fat Determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.E. Long-term Outcomes in Mothers Diagnosed With Gestational Diabetes Mellitus and Their Offspring. Clin. Obstet. Gynecol. 2007, 50, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Mericq, V.; Martinez-Aguayo, A.; Uauy, R.; Iñiguez, G.; Van der Steen, M.; Hokken-Koelega, A. Long-term metabolic risk among children born premature or small for gestational age. Nat. Rev. Endocrinol. 2017, 13, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Worda, C.; Leipold, H.; Gruber, C.; Kautzky-Willer, A.; Knöfler, M.; Bancher-Todesca, D. Decreased plasma adiponectin concentrations in women with gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2004, 191, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Resi, V.; Basu, S.; Haghiac, M.; Presley, L.; Minium, J.; Kaufman, B.; Bernard, S.; Catalano, P.; Hauguel-de Mouzon, S. Molecular inflammation and adipose tissue matrix remodeling precede physiological adaptations to pregnancy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E832–E840. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, J.; de Castro, J.; Bocos, C.; Herrera, E.; Ramos, M.P. Role of insulin receptor substrate-1 serine 307 phosphorylation and adiponectin in adipose tissue insulin resistance in late pregnancy. Endocrinology 2007, 148, 5933–5942. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Nizielski, S.E.; Shao, J.; Preston, L.; Qiao, L.; Friedman, J.E. Downregulated IRS-1 and PPARgamma in obese women with gestational diabetes: Relationship to FFA during pregnancy. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E522–E533. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; Martínez, N.; Fornes, D.; Higa, R.; Di Marco, I.; Basualdo, M.N.; Faingold, M.C.; Jawerbaum, A. PPAR activation as a regulator of lipid metabolism, nitric oxide production and lipid peroxidation in the placenta from type 2 diabetic patients. Mol. Cell. Endocrinol. 2013, 377, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M. Effect of pre-existing maternal obesity, gestational diabetes and adipokines on the expression of genes involved in lipid metabolism in adipose tissue. Metabolism 2014, 63, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cuenca, S.; Carobbio, S.; Velagapudi, V.R.; Barbarroja, N.; Moreno-Navarrete, J.M.; Tinahones, F.J.; Fernandez-Real, J.M.; Orešic, M.; Vidal-Puig, A. Peroxisome proliferator-activated receptor γ-dependent regulation of lipolytic nodes and metabolic flexibility. Mol. Cell. Biol. 2012, 32, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Vivas, Y.; Díez-Hochleitner, M.; Izquierdo-Lahuerta, A.; Corrales, P.; Horrillo, D.; Velasco, I.; Martínez-García, C.; Campbell, M.; Sevillano, J.; Ricote, M.; et al. Peroxisome Proliferator-Activated Receptor γ 2 Modulates Late-Pregnancy Homeostatic Metabolic Adaptations. Mol. Med. 2016, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Vivas, Y.; Martínez-García, C.; Izquierdo, A.; Garcia-Garcia, F.; Callejas, S.; Velasco, I.; Campbell, M.; Ros, M.; Dopazo, A.; Dopazo, J.; et al. Early peroxisome proliferator-activated receptor gamma regulated genes involved in expansion of pancreatic beta cell mass. BMC Med. Genom. 2011, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Yessoufou, A.; Hichami, A.; Besnard, P.; Moutairou, K.; Khan, N.A. Peroxisome Proliferator-Activated Receptor α Deficiency Increases the Risk of Maternal Abortion and Neonatal Mortality in Murine Pregnancy with or without Diabetes Mellitus: Modulation of T Cell Differentiation. Endocrinology 2006, 147, 4410–4418. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, M.; Capobianco, E.; Martínez, N.; Fernández, J.; Higa, R.; White, V.; Jawerbaum, A. Carbaprostacyclin, a PPARδ agonist, ameliorates excess lipid accumulation in diabetic rat placentas. Life Sci. 2010, 86, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Sadovsky, Y.; Shalom-Barak, T. PPAR Signaling in Placental Development and Function. PPAR Res. 2008, 2008, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; She, R.; Sha, W. Gestational diabetes mellitus is associated with decreased adipose and placenta peroxisome proliferator-activator receptor γ expression in a Chinese population. Oncotarget 2017, 8, 113928–113937. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhuang, X.; Jiang, M.; Guan, F.; Fu, Q.; Lin, J. ANGPTL4 mediates the protective role of PPARγ activators in the pathogenesis of preeclampsia. Cell Death Dis. 2017, 8, e3054. [Google Scholar] [CrossRef] [PubMed]
- Suwaki, N.; Masuyama, H.; Masumoto, A.; Takamoto, N.; Hiramatsu, Y. Expression and potential role of peroxisome proliferator-activated receptor gamma in the placenta of diabetic pregnancy. Placenta 2007, 28, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, J.M.; Andrés, A.; Ros, M.; Bogónez, E.; Arribas, C.; Fernández-Agulló, T.; De Solís, A.J.; Gallardo, N.; Martínez, C. Development of insulin resistance during aging: Involvement of central processes and role of adipokines. Curr. Protein Pept. Sci. 2011, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805. [Google Scholar] [CrossRef] [PubMed]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 161–196. [Google Scholar] [PubMed]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.R.; Zhang, X.; Zhang, Y.; Wu, J.; Su, D.; Han, J.Y.; Fang, X.; Yu, B.; Breyer, M.D.; Guan, Y. Peroxisome proliferator activated receptor alpha/gamma dual agonist tesaglitazar attenuates diabetic nephropathy in db/db mice. Diabetes 2007, 56, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic Dysregulation and Adipose Tissue Fibrosis: Role of Collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ma, X.; Verma, N.K.; Wang, D.; Gavrilova, O.; Proia, R.L.; Finkel, T.; Mueller, E. Ablation of PPARγ in subcutaneous fat exacerbates age-associated obesity and metabolic decline. Aging Cell 2018, 17, e12721. [Google Scholar] [CrossRef]
- Miard, S.; Dombrowski, L.; Carter, S.; Boivin, L.; Picard, F. Aging alters PPARgamma in rodent and human adipose tissue by modulating the balance in steroid receptor coactivator-1. Aging Cell 2009, 8, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choe, S.S.; Choi, A.H.; Kim, K.H.; Yoon, M.J.; Suganami, T.; Ogawa, Y.; Kim, J.B. Increase in Glucose-6-Phosphate Dehydrogenase in Adipocytes Stimulates Oxidative Stress and Inflammatory Signals. Diabetes 2006, 55, 2939–2949. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Mennes, E.; Dungan, C.M.; Frendo-Cumbo, S.; Williamson, D.L.; Wright, D.C. Aging-associated reductions in lipolytic and mitochondrial proteins in mouse adipose tissue are not rescued by metformin treatment. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, P.; Sjöström, L.; Hedlund, H.; Lundell, L.; Olbe, L. Influence of age, fat cell weight, and obesity on O2 consumption of human adipose tissue. Am. J. Physiol. 1989, 256, E467–E474. [Google Scholar] [CrossRef] [PubMed]
- Graier, W.F.; Malli, R.; Kostner, G.M. Mitochondrial protein phosphorylation: Instigator or target of lipotoxicity? Trends Endocrinol. Metab. 2009, 20, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Wolfrum, C.; Scheideler, M. Age-Induced Changes in White, Brite, and Brown Adipose Depots: A Mini-Review. Gerontology 2018, 64, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Okamatsu-Ogura, Y.; Machida, K.; Tsubota, A.; Nio-Kobayashi, J.; Kimura, K. Impaired adrenergic agonist-dependent beige adipocyte induction in aged mice. Obesity 2017, 25, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Park, S.; Yu, B.P.; Chung, H.Y. Modulation of PPAR in aging, inflammation, and calorie restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Hoener, P.A.; Jow, L.; Bilakovics, J.; Klausing, K.; Mais, D.E.; Faulkner, A.; Croston, G.E.; Paterniti, J.R. A selective peroxisome proliferator-activated receptor-gamma (PPARgamma) modulator blocks adipocyte differentiation but stimulates glucose uptake in 3T3-L1 adipocytes. Mol. Endocrinol. 2000, 14, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.V. Diabetes, TZDs, and Bone: A Review of the Clinical Evidence. PPAR Res. 2006, 2006, 24502. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Fabbiano, S.; Suá Rez-Zamorano, N.; Rigo, D.E.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; González-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, F.; Gavete, M.L.; Fermín, Y.; Pérez, C.; Gallardo, N.; Alvarez, C.; Andrés, A.; Ros, M.; Carrascosa, J.M. Effect of age and moderate food restriction on insulin sensitivity in Wistar rats: Role of adiposity. J. Endocrinol. 2007, 194, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Atzmon, G.; Rajala, M.W.; Berg, A.H.; Scherer, P.; Rossetti, L.; Barzilai, N. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: An adipokine-mediated process? Diabetes 2002, 51, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Masternak, M.M.; Bartke, A. PPARs in Calorie Restricted and Genetically Long-Lived Mice. PPAR Res. 2007, 2007, 28436. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Investig. 2004, 114, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
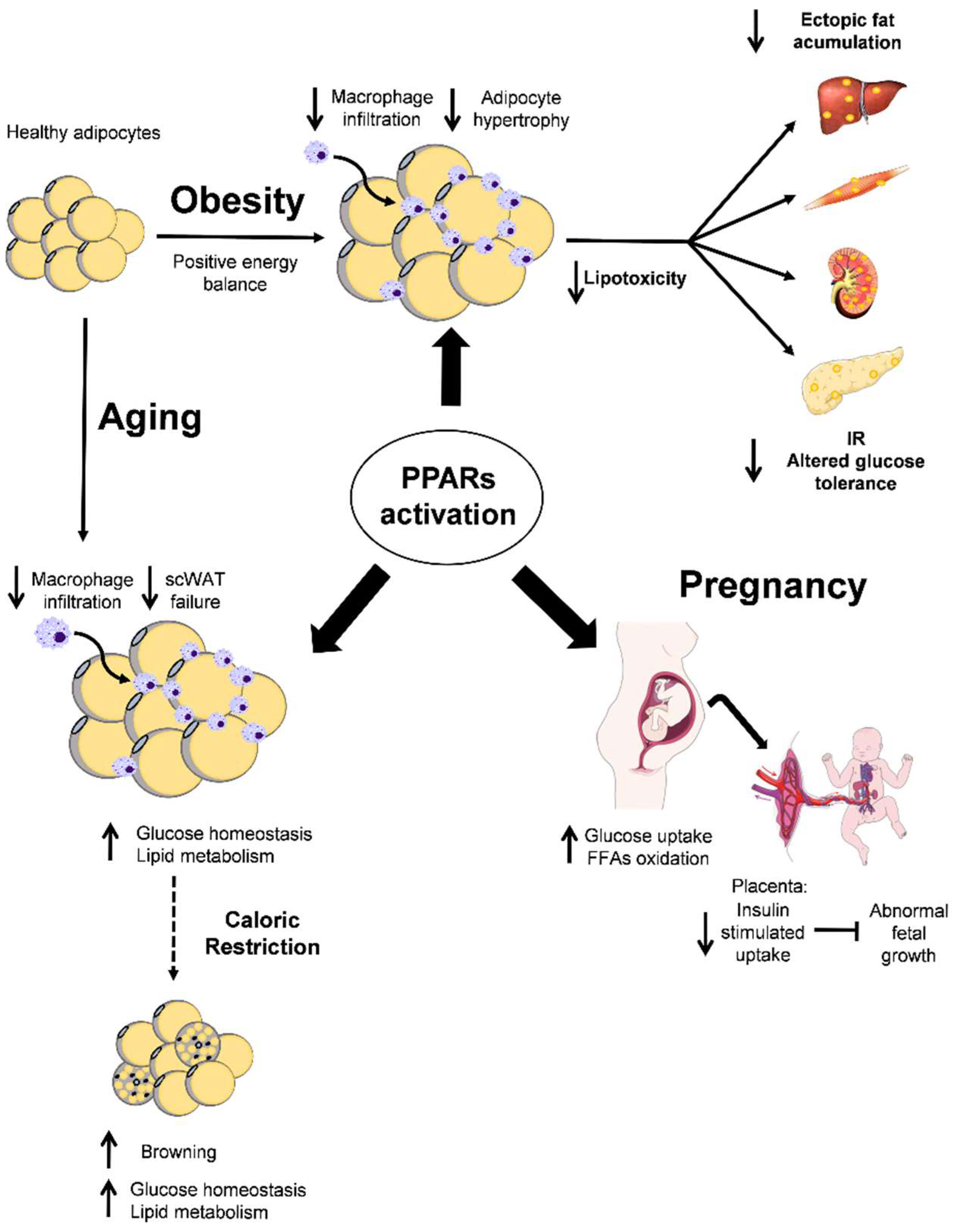
 PPARs activation;
PPARs activation;  . effects or PPARs activation;
. effects or PPARs activation;  CR effect;
CR effect;  inhibition;
inhibition;  increase or
increase or  decrease effect.
PPARs activation; . effects or PPARs activation; CR effect; inhibition; increase or decrease effect.
decrease effect.
PPARs activation; . effects or PPARs activation; CR effect; inhibition; increase or decrease effect.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. https://doi.org/10.3390/ijms19072124
Corrales P, Vidal-Puig A, Medina-Gómez G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. International Journal of Molecular Sciences. 2018; 19(7):2124. https://doi.org/10.3390/ijms19072124
Chicago/Turabian StyleCorrales, Patricia, Antonio Vidal-Puig, and Gema Medina-Gómez. 2018. "PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity" International Journal of Molecular Sciences 19, no. 7: 2124. https://doi.org/10.3390/ijms19072124
APA StyleCorrales, P., Vidal-Puig, A., & Medina-Gómez, G. (2018). PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. International Journal of Molecular Sciences, 19(7), 2124. https://doi.org/10.3390/ijms19072124