Using microRNA Networks to Understand Cancer

 ,
, 
Abstract

1. Introduction
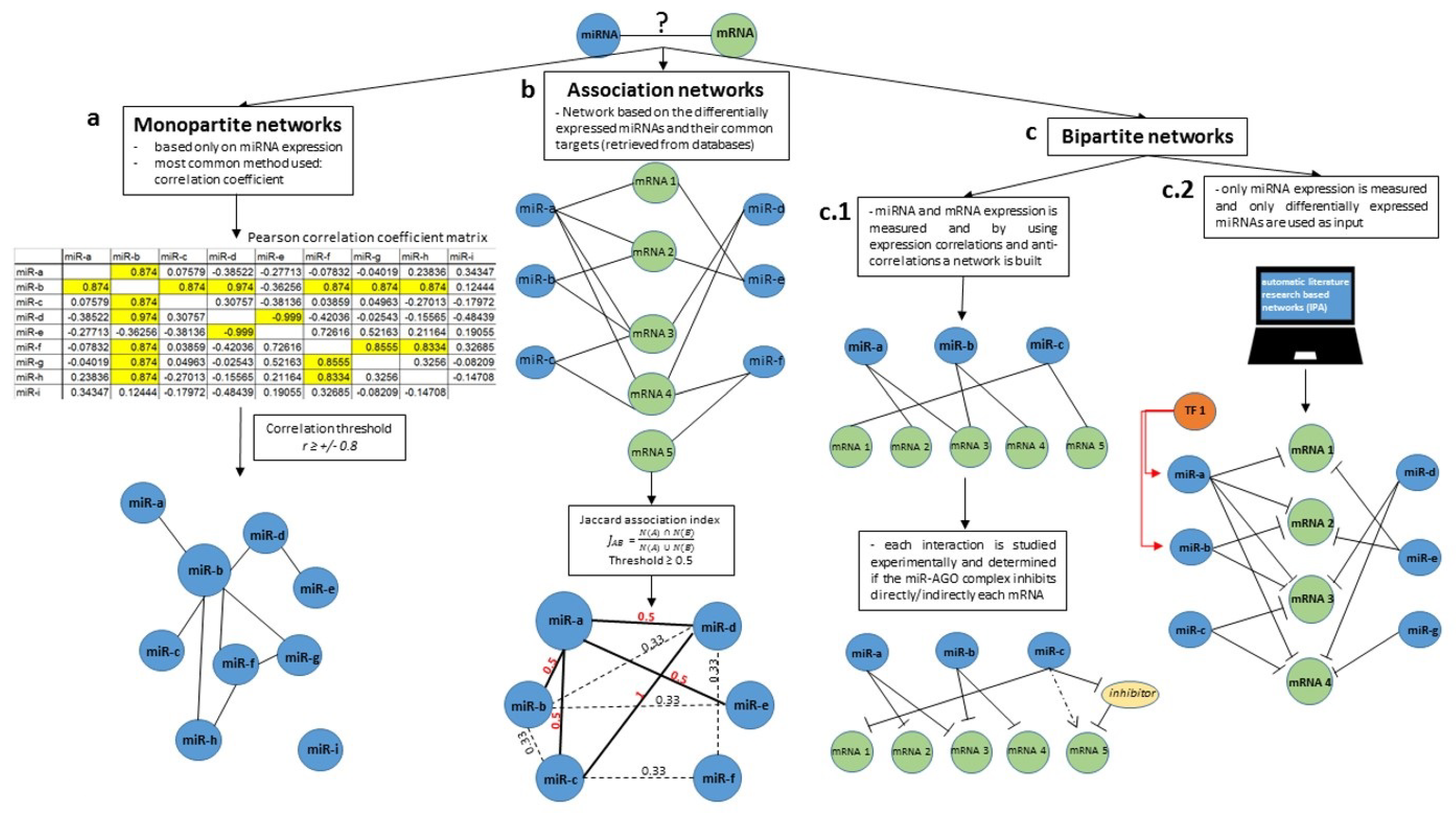
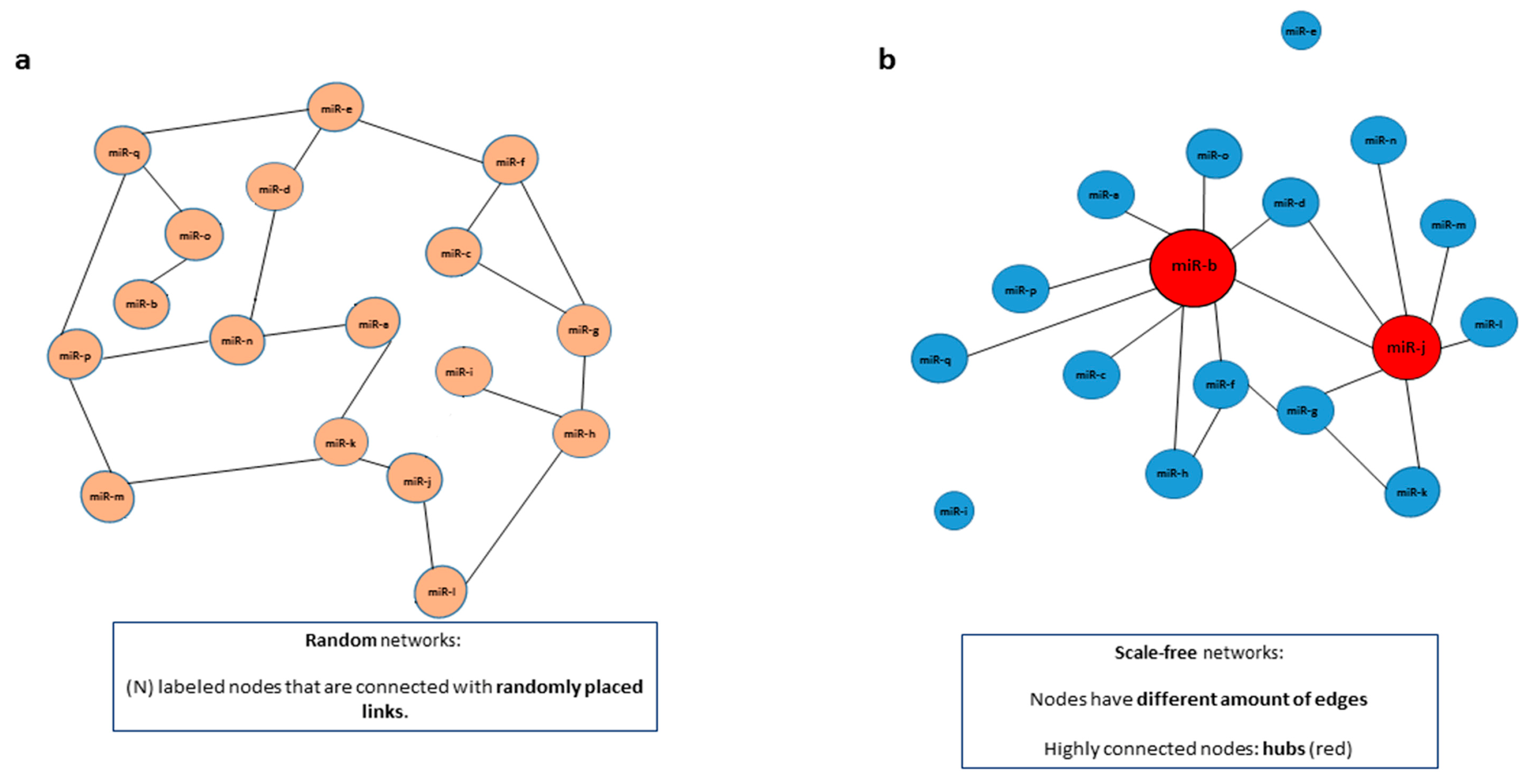
2. Understanding and Building miRNA Networks
3. Examples of miRNA Network Analyses
4. Therapeutic Perspectives of miRNA Networks in Cancer
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNAs | MicroRNAs |
| ncRNAs | Non-coding RNAs |
| Lin-4 | Lineage defective 4 |
| Let-7 | Lethal 7 |
| mRNAs | Messenger RNAs |
| circRNAs | Circular RNAs |
| lncRNAs | Long non-coding RNAs |
| OncomiRs | Oncogenic microRNAs |
| CLL | Chronic Lymphocytic Leukemia |
| AML | Acute myeloid leukemia |
| ceRNAs | Competitive endogenous RNAs |
| MREs | MiRNA response elements |
| EBV | Epstein-Barr virus |
| EMT | Epithelial-to-mesenchymal transition |
| PD-L1 | Programmed death-ligand 1 |
| MUC1 | Mucin 1 |
| Mimics | Mimic endogenous microRNAs |
| AntagomiRs | MicroRNA inhibitors |
| ceRNAs | Competitive endogenous microRNAs |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| Real-time qRT-PCR | Real-time reverse transcription polymerase chain reaction |
| IPA | Ingenuity Pathway Analysis |
| RBPs | RNA-binding proteins |
| CLIP-seq | Cross-linking Immunoprecipitation followed by deep sequencing |
| Ago | Argonaute |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Targeted Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Sun, T.; Hacisuleyman, E.; Fei, T.; Wang, X.; Brown, M.; Rinn, J.L.; Lee, M.G.; Chen, Y.; Kantoff, P.W.; et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016, 7, 10982. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Spizzo, R.; Nicoloso, M.S.; Croce, C.M.; Calin, G.A. SnapShot: MicroRNAs in Cancer. Cell 2009, 137, 586. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Anastasiadou, E.; Esteller, M.; He, L.; Slack, F.J. The Inescapable Influence of Noncoding RNAs in Cancer. Cancer Res. 2015, 75, 5206–5210. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bullrich, F.; Fujii, H.; Calin, G.; Mabuchi, H.; Negrini, M.; Pekarsky, Y.; Rassenti, L.; Alder, H.; Reed, J.C.; Keating, M.J.; et al. Characterization of the 13q14 tumor suppressor locus in CLL: Identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001, 61, 6640–6648. [Google Scholar] [PubMed]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, H.; Seto, M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 2005, 19, 2013–2016. [Google Scholar] [CrossRef] [PubMed]
- Gillies, J.K.; Lorimer, I.A. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2007, 6, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Garg, N.; Bigi, R.; Yadav, S.; Campese, A.F.; Lapenta, C.; Spada, M.; Cuomo, L.; Botta, A.; Belardelli, F.; et al. Epstein-Barr virus infection induces miR-21 in terminally differentiated malignant B cells. Int. J. Cancer 2015, 137, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Magdo, L.; Jurj, A.; Raduly, L.; Cojocneanu-Petric, R.; Moldovan, A.; Moldovan, C.; Florea, A.; Pasca, S.; Pop, L.A.; et al. The silent healer: MiR-205-5p up-regulation inhibits epithelial to mesenchymal transition in colon cancer cells by indirectly up-regulating E-cadherin expression. Cell Death Dis. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Stroopinsky, D.; Rosenblatt, J.; Anastasiadou, E.; Rajabi, H.; Washington, A.; Tagde, A.; Chu, J.H.; Coll, M.; Jiao, A.L.; et al. MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia 2017, 31, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.C.B.; Fu, X.; Calin, G.A. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol. Med. 2018, 15, 103–115. [Google Scholar] [CrossRef]
- Business Wire. Mirna Therapeutics Halts Phase 1 Clinical Study of MRX34. Available online: https://www.businesswire.com/news/home/20160920006814/en/Mirna-Therapeutics-Halts-Phase-1-Clinical-Study (accessed on 15 May 2018).
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Giza, D.E.; Vasilescu, C.; Calin, G.A. MicroRNAs and ceRNAs: Therapeutic implications of RNA networks. Expert Opin. Biol. Ther. 2014, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.; Calin, G.A. Circular RNAs in Cancer—Lessons Learned From microRNAs. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Alon, U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet 2007, 8, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Fuxman Bass, J.I.; Diallo, A.; Nelson, J.; Soto, J.M.; Myers, C.L.; Walhout, A.J. Using networks to measure similarity between genes: Association index selection. Nat. Methods 2013, 10, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Batushansky, A.; Toubiana, D.; Fait, A. Correlation-Based Network Generation, Visualization, and Analysis as a Powerful Tool in Biological Studies: A Case Study in Cancer Cell Metabolism. Biomed. Res. Int. 2016, 2016, 8313272. [Google Scholar] [CrossRef] [PubMed]
- Vasilescu, C.; Dragomir, M.; Tanase, M.; Giza, D.; Purnichescu-Purtan, R.; Chen, M.; Yeung, S.J.; Calin, G.A. Circulating miRNAs in sepsis—A network under attack: An in-silico prediction of the potential existence of miRNA sponges in sepsis. PLoS ONE 2017, 12, e0183334. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.; Ciaramella, A.; Deniskina, N.; Del Mondo, C.; di Bernardo, D.; Donalek, C.; Longo, G.; Mangano, G.; Miele, G.; Raiconi, G.; et al. A multi-step approach to time series analysis and gene expression clustering. Bioinformatics 2006, 22, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Hartemink, A. Banjo: Bayesian Network Inference with Java Objects. Available online: https://users.cs.duke.edu/~amink/software/banjo/ (accessed on 17 June 2018).
- Hartemink, A.J. Reverse engineering gene regulatory networks. Nat. Biotechnol. 2005, 23, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.A.; Yu, J.; Smulders, T.V.; Hartemink, A.J.; Jarvis, E.D. Computational inference of neural information flow networks. PLoS Comput. Biol. 2006, 2, e161. [Google Scholar] [CrossRef] [PubMed]
- Vila-Casadesus, M.; Gironella, M.; Lozano, J.J. MiRComb: An R Package to Analyse miRNA-mRNA Interactions. Examples across Five Digestive Cancers. PLoS ONE 2016, 11, e0151127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Edwards, A.; Fan, W.; Flemington, E.K.; Zhang, K. miRNA-mRNA correlation-network modules in human prostate cancer and the differences between primary and metastatic tumor subtypes. PLoS ONE 2012, 7, e40130. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Piepoli, A.; Tavano, F.; Copetti, M.; Mazza, T.; Palumbo, O.; Panza, A.; di Mola, F.F.; Pazienza, V.; Mazzoccoli, G.; Biscaglia, G.; et al. Mirna expression profiles identify drivers in colorectal and pancreatic cancers. PLoS ONE 2012, 7, e33663. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Galasso, M.; Costinean, S.; Tagliavini, L.; Gamberoni, G.; Drusco, A.; Marchesini, J.; Mascellani, N.; Sana, M.E.; Abu Jarour, R.; et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010, 20, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Kose, F.; Weckwerth, W.; Linke, T.; Fiehn, O. Visualizing plant metabolomic correlation networks using clique-metabolite matrices. Bioinformatics 2001, 17, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.X.; Li, Y.S.; Lv, J.Y.; Ma, Y.; Shao, T.T.; Xu, L.D.; Wang, Y.Y.; Du, L.; Zhang, Y.P.; et al. MiRNA-miRNA synergistic network: Construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res. 2011, 39, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Kulyte, A.; Belarbi, Y.; Lorente-Cebrian, S.; Bambace, C.; Arner, E.; Daub, C.O.; Heden, P.; Ryden, M.; Mejhert, N.; Arner, P. Additive effects of microRNAs and transcription factors on CCL2 production in human white adipose tissue. Diabetes 2014, 63, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Slabakova, E.; Culig, Z.; Remsik, J.; Soucek, K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017, 8, e3100. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res 2009, 37, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Chindelevitch, L.; Ziemek, D.; Enayetallah, A.; Randhawa, R.; Sidders, B.; Brockel, C.; Huang, E.S. Causal reasoning on biological networks: Interpreting transcriptional changes. Bioinformatics 2012, 28, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Felciano, R.M.; Bavari, S.; Richards, D.R.; Billaud, J.N.; Warren, T.; Panchal, R.; Kramer, A. Predictive systems biology approach to broad-spectrum, host-directed drug target discovery in infectious diseases. Pac. Symp. Biocomput. 2013, 17–28. [Google Scholar] [CrossRef]
- Kumar, R.; Blakemore, S.J.; Ellis, C.E.; Petricoin, E.F., 3rd; Pratt, D.; Macoritto, M.; Matthews, A.L.; Loureiro, J.J.; Elliston, K. Causal reasoning identifies mechanisms of sensitivity for a novel AKT kinase inhibitor, GSK690693. BMC Genom. 2010, 11, 419. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Thomson, T.M.; Sewer, A.; Drubin, D.A.; Mathis, C.; Weisensee, D.; Pratt, D.; Hoeng, J.; Peitsch, M.C. Assessment of network perturbation amplitudes by applying high-throughput data to causal biological networks. BMC Syst. Biol. 2012, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J., Jr.; Butte, A.J.; Hoberman, S.; Joshi, M.; Levy, J.; Pappo, J. A computational model to define the molecular causes of type 2 diabetes mellitus. Diabetes Technol. Ther. 2005, 7, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.S.; Pósfai, M.R. Network Science; Cambridge University Press: Cambridge, UK, 2016; 456p. [Google Scholar]
- Aguda, B.D.; Kim, Y.; Piper-Hunter, M.G.; Friedman, A.; Marsh, C.B. MicroRNA regulation of a cancer network: Consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc. Natl. Acad. Sci. USA 2008, 105, 19678–19683. [Google Scholar] [CrossRef] [PubMed]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Lin, K.Y.; Chen, Y.Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Richards, E.J.; Challa, S.; Xu, C.X.; Permuth-Wey, J.; Lancaster, J.M.; Coppola, D.; Sellers, T.A.; Djeu, J.Y.; et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2014, 33, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Nie, H.; Wang, M.; Su, L.P.; Li, J.F.; Yu, Y.Y.; Yan, M.; Qu, Q.L.; Zhu, Z.G.; Liu, B.Y. microRNA-155 is downregulated in gastric cancer cells and involved in cell metastasis. Oncol. Rep. 2012, 27, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Palma, C.A.; Al Sheikha, D.; Lim, T.K.; Bryant, A.; Vu, T.T.; Jayaswal, V.; Ma, D.D. MicroRNA-155 as an inducer of apoptosis and cell differentiation in Acute Myeloid Leukaemia. Mol. Cancer 2014, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Ren, Q.; Liu, T.; Huang, Y.; Wang, J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013, 587, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Shapiro, A.; Kosik, K.S. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008, 68, 8164–8172. [Google Scholar] [CrossRef] [PubMed]
- Bhajun, R.; Guyon, L.; Pitaval, A.; Sulpice, E.; Combe, S.; Obeid, P.; Haguet, V.; Ghorbel, I.; Lajaunie, C.; Gidrol, X. A statistically inferred microRNA network identifies breast cancer target miR-940 as an actin cytoskeleton regulator. Sci. Rep. 2015, 5, 8336. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Kim, W.; Lee, J.; Youn, B. Network-based approaches for anticancer therapy (Review). Int. J. Oncol. 2013, 43, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, N.; Carthew, R.W. Biological robustness and the role of microRNAs: A network perspective. Curr. Top. Dev. Biol. 2012, 99, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, W.F.; Lim, T.L.; Hart, T. PICKLES: The database of pooled in-vitro CRISPR knockout library essentiality screens. Nucleic Acids Res. 2018, 46, D776–D780. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Basic Nomenclature | Definitions |
|---|---|
| Nodes | Elements of a network. |
| Edges | Interactions/connections between elements of a network. |
| Hubs | Nodes with a high degree of interactions. |
| Association index | Method used for the quantification of similarity between nodes of the same species. The most common used association indices are: Jaccard, Simpson, Geometric and Cosin. |
| Monopartite network | Graph that contains only one species of nodes, such as a miRNA network. |
| Bipartite network | Graph that contains two species, with edges representing their interaction pattern, such as miRNA-mRNA networks. |
| Multipartite network | Graph that contains more than two species of nodes. |
| Association networks | Networks in which two nodes of the same type are connected only if their similarity calculated using an association index is above a selected threshold. |
| Scale free network | A network that follows a power-law distribution, with some nodes that have a higher number of connections (hubs). |
| Random network | A network with (N) labeled nodes that are connected with randomly distributed links (edges). |
| Undirected network | A graph in which the edges between nodes have no orientation. |
| Directed network | A graph in which edges between nodes have orientation. An edge is depicted as an arrow, is directed from node a to node b and is not equivalent to an edge from node b to node a. |
| Network Type | Method | Examples of Software/Webtools | Reference |
|---|---|---|---|
| Monopartite network | Correlation coefficient | Any statistics analysis software (GraphPad Prism, IBM SPSS, R) | [49,50] |
| Hierarchical clustering | IBM SPSS, R | [50,51,52] * | |
| Bayesian inference | Banjo (Bayesian network inference with Java objects) https://users.cs.duke.edu/~amink/software/banjo/ | [53,54,55] | |
| Association networks | Association indexes | GAIN http://csbio.cs.umn.edu/similarity_index/login.php | [48] |
| Bipartite networks | Experimental approach (correlation coefficient) | Any statistics analysis software (GraphPad Prism, IBM SPSS, R) | [56,57] |
| Automatic literature search | IPA Qiagen | [58] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. https://doi.org/10.3390/ijms19071871
Dragomir M, Mafra ACP, Dias SMG, Vasilescu C, Calin GA. Using microRNA Networks to Understand Cancer. International Journal of Molecular Sciences. 2018; 19(7):1871. https://doi.org/10.3390/ijms19071871
Chicago/Turabian StyleDragomir, Mihnea, Ana Carolina P. Mafra, Sandra M. G. Dias, Catalin Vasilescu, and George A. Calin. 2018. "Using microRNA Networks to Understand Cancer" International Journal of Molecular Sciences 19, no. 7: 1871. https://doi.org/10.3390/ijms19071871
APA StyleDragomir, M., Mafra, A. C. P., Dias, S. M. G., Vasilescu, C., & Calin, G. A. (2018). Using microRNA Networks to Understand Cancer. International Journal of Molecular Sciences, 19(7), 1871. https://doi.org/10.3390/ijms19071871