Abstract
Kabuki syndrome (KS) is a rare disorder characterized by multiple congenital anomalies and variable intellectual disability caused by mutations in KMT2D/MLL2 and KDM6A/UTX, two interacting chromatin modifier responsible respectively for 56–75% and 5–8% of the cases. To date, three KS patients with mosaic KMT2D deletions in blood lymphocytes have been described. We report on three additional subjects displaying KMT2D gene mosaics including one in which a single nucleotide change results in a new frameshift mutation (p.L1199HfsX7), and two with already-known nonsense mutations (p.R4484X and p.R5021X). Consistent with previously published cases, mosaic KMT2D mutations may result in mild KS facial dysmorphisms and clinical and neurobehavioral features, suggesting that these characteristics could represent the handles for genetic testing of individuals with slight KS-like traits.
1. Introduction
Kabuki syndrome (KS, MIM #147920) is a rare disorder characterized by a distinctive face, with long palpebral fissures and eversion of the lateral third of lower eyelids, short columella with broad and depressed nasal tip, prominent ears, and cleft or high-arched palate. Additional features include short stature, skeletal anomalies, congenital heart defects, renal malformations, anorectal anomalies, and abnormal dermatoglyphics [1,2].
Mutations in KMT2D have been identified as the main cause for KS [3], less frequent are mutations in KDM6A [4]. Since then, a number of studies have shown that mutations in KMT2D and KMD6A account for approximately 56–75% and 5–8% of KS cases, respectively [5].
To date, mosaic KMT2D mutations have been described in three KS patients [6]. Here we expand the number of mosaic KS patients by reporting three additional cases. Consistent with the published cases, mosaic KMT2D mutations are associated with classical KS facial dysmorphisms and clinical features, although some manifestations could be milder. KS is a multi-systemic disorder, not limited to a single organ or tissue of a common embryonic origin, and haploinsufficiency of KMT2D in just some cell types may be sufficient to produce deleterious phenotypic events.
2. Results
2.1. Clinical Description
Patient 1 (GM13-3816) is the third female child of healthy non-consanguineous parents (Table 1, and Figure 1A). At birth the mother was 34 years old, the father 36. A family history disclosed learning difficulties in an older brother, depression in the paternal grandmother, a psychotic disorder in a maternal uncle, and epilepsy in a maternal niece. The baby was born by vaginal delivery at term of an uneventful pregnancy. The birth weight was 3260 g (50th centile), length 50 cm (50–75th centile), and head circumference 34 cm (50th centile). Apgar scores were 9 and 10 at 1 and 5 min. We first evaluated the patient at the age of 6.1 years for learning difficulties and facial dysmorphisms. The weight was 26 kg (97th centile), height 119 cm (75–90th centile), and head circumference 49.5 cm (10th centile). Facial anomalies included sparse lateral eyebrows, long palpebral fissures with thick eyelids, mild palpebral ptosis, flat philtrum, thick everted lips, and large prominent ears with abnormal helix and a large pinna. The proximal fourth fingers’ phalanges were hypoplastic, with clinodactylous fifth fingers. She does not have fetal fingertip pads. Tapering fingers were present. Pigmentary anomalies were also present, including partial hypochromia of eyelashes, white cutaneous spots in the trunk midline and on the dorsum, a single café-au-lait spot on the abdomen. Body asymmetry was evident, the left side of the face being larger and the left lower limb 1 cm longer in comparison to the right one. Ophthalmogic examination, audiological brainstem audiometry, electroencephalography, 2-Dimensional color-Doppler echocardiography and renal ultrasonography were unremarkable.

Table 1.
Clinical features of patients with mosaic KMT2D mutations.
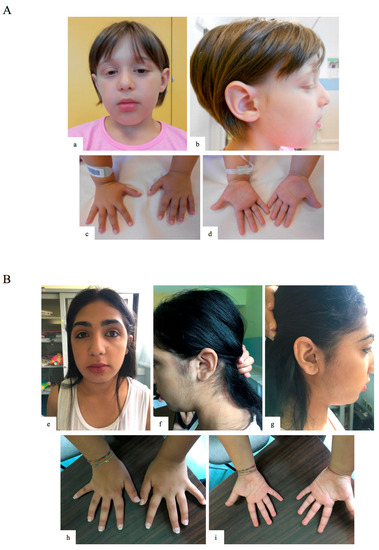
Figure 1.
Clinical features of patients 1 and 3. (a,b) Facial features of patient 1 (GM13-3816; c.15061C=/>T, p.R5021X heterozygous mosaic KTM2D mutation), including sparse lateral eyebrows, long palpebral fissures with thick eyelids, ptosis, flat philtrum, thick everted lips and large dysmorphic ears with abnormal helix and large pinna; (c,d) hypoplastic proximal phalanges of 4th fingers, clinodactylous 5th fingers, and tapering fingers; (e–g) facial features of patient 3 (KB 369; c.3596_3597=/del, p.L1199HfsX7 heterozygous mosaic KMT2D mutation), including sparse medial eyebrows, long palpebral fissures with thick eyelids, flat philtrum, and thick everted lips; (h,i) limb/skeletal anomalies with persistent fetal pads and clinodactylous 5th fingers, and tapering fingers.
The neurobehavioral profile was evaluated through standardized instruments at the age of eight years and seven months. The cognitive profile was assessed using WISC IV [7]. In the verbal comprehension index (VCI) and perceptual reasoning index (PRI), the patient achieved average scores (VCI 108; PRI 87). The working memory index (WMI) and processing speed index (PSI) disclosed scores one standard deviation (SD) below the mean (WMI 82; PSI: 76). The general intellectual ability was in the average range (Total Intelligence Quozient (TIQ) 87). Adaptive behaviour was evaluated using the ABAS-II (Adaptive Behavior Assessment System—Second Edition) questionnaire parent form [8]. In the conceptual (CON) and practical domain (PR), the patient’s scores were below two SD from the mean (respectively CON: 65 and PR: 68), while in the social domain (SO) the score was one SD below the mean (SO: 81). The general adaptive composite (GAC) score was 65, two SD below the mean. The patient presented learning disabilities, with a score below one SD from the mean in the MT-2 reading task [9] in the speed and accuracy indexes, and a score two SD below the mean in the writing accuracy index of the BVSCO-2 task (Batteria per la Valutazione della Scrittura e della Competenza Ortografica—2) [10]. The psychopathological profile was investigated by psychiatric evaluation and psychodiagnostic evaluation through K-SADS-PL [11]. The patient met the criteria for generalized anxiety disorder and multiple phobias (bugs, fireworks, loud noises). She also had bizarre behaviour (talking to herself, soliloquy) and emotional dysregulation (Table 2).
Table 2.
Summary assessment of neurobehavioral tests.
Patient 2 (KB450) is the female, first child of healthy non-consanguineous parents, with the mother 33 years old and the father 37 years old (Table 1). The family history was uneventful for disabilities. The baby was born by caesarean section due to breech presentation, at term of an uncomplicated pregnancy. Birth weight was 3345 g (75th centile), length 51 cm (90th centile), and head circumference 35 cm (75th centile). Apgar scores were 9 and 10 at 1 and 5 min. A grade 3/6 L ejection systolic murmur was noted on clinical evaluation. A cardiac ultrasonography, on the first day of the patient’s life, showed a non-restrictive intraventricular subaortic defect. Kabuki syndrome’s evocative facial dysmorphisms were evident in the perinatal period, including elongated palpebral fissures extending laterally, hypertelorism, axial hypotonia and joint hypermobility, fetal finger pads, micrognathia, long and flat philtrum, low set and cup-shaped ears. Hip’s ultrasonography disclosed a right hip dysplasia. Tapering fingers were not present. Renal and brain ultrasonography, ophtalmologic examination, and audiological brainstem audiometry were all unremarkable. An X-ray at birth showed a paracardiac opacity in the right lung, 2.6 cm in diameter, interpreted by chest tomography as a small diaphragmatic hernia. The infant underwent a closure procedure of ventricular septal defects and correction of diaphragmatic hernia at one month of life with an uneventful postoperative course. Discharged at two months of life with a suspicion of KS, the patient underwent a cardiological and neuropsychiatric follow-up.
The neurobehavioral profile was assessed between six and eight years of life. The cognitive profile was evaluated using WISC IV [7]. In the WMI and PSI, the patient achieved average scores (respectively 91 and 87). In PSI and VCI, the disclosed scores were one SD below the mean (respectively 74 and 76). The general intellectual ability was one SD below the mean (Total IQ) TIQ of 84. Adaptive behaviour was evaluated using ABAS-II. In the conceptual and social domains, the patient’s scores were average (CON: 112, SO: 108), while in the practical domain the score was one SD above the mean (PR: 120). The patient presented learning disabilities with a score below one SD in the MT-2 in the reading/comprehension task [9] and below two SD in the writing accuracy index of the BVSCO-2 task [10]. Computation abilities were evaluated with the AC-MT 6–11 tests [13], reaching a total score between the fifth and tenth centile. The psychopathological profile was investigated through psychiatric evaluation, which showed mild emotional dysregulation.
Patient 3 (KB 369) is the first female child of non-consanguineous parents (Table 1 and Figure 1B). At birth, the HCV-positive mother was 36 years old and the father 27. A younger brother has a mild intellectual disability. The baby was born by vaginal delivery at term of an uneventful pregnancy. The birth weight was 3230 g (50th centile), length 50 cm (50–75th centile). Apgar scores was 9 at 1 min. At three and a half years she was diagnosed having premature puberty and was treated with Enantone up to 12 years of age. We first evaluated the patient at five years of age, because of her tendency to isolation and poor social participation. The neurobehavioral profile was assessed at 11 years (Table 2). The cognitive profile was evaluated using WISC III [7], showing an IQ below two SD of the mean (TIQ 46, verbal IQ 58, performance IQ 46). Adaptive behaviour was assessed using ABAS-II. In the practical domain, the patient’s scores were two SD below the mean (PR: 58), while in conceptual domain the score was one SD below the mean (CON: 72). In the social domain, the score was average (SO: 88). The psychopathological profile was investigated through both psychiatric and psychodiagnostic evaluation through K-SADS-PL [11]. The patient met the criteria for generalized anxiety disorder and multiple phobias (stuffed animals, dolls and some real animals). She also had bizarre behaviour (soliloquy). At age of 12, ADOS (Autism Diagnostic Observation Schedule) Module 3 disclosed autistic-like behaviour. The weak academic competence level (reading, writing, comprehension skills) achieved by the patient did not permit a structured assessment.
At the age of 13, the family pediatrician required new medical advice for the presence of dysmorphisms. Her weight was 51 kg (>50th centile), height 153 cm (<25 centile), and head circumference 53.5 cm (<50th centile). Facial anomalies included long palpebral fissures with thick eyelids, flat philtrum, and thick everted lips (Figure 1B). Limb/skeletal anomalies were also present, including persistent fetal pads, clinodactylous of 5th fingers, and tapering fingers. Early breast development was present. Neurologic brain MRI, ophthalmologic, brainstem audiometry, electroencephalography, colour Doppler echocardiography, and renal ultrasonography evaluations were all unremarkable.
2.2. Mutational Analysis
Patient GM13-3816 disclosed a c.15061C=/>T, p.R5021X heterozygous variant in KTM2D gene [14], in a small fraction of alleles from the blood cells. Pyrosequencing analysis estimated a 16% of mosaic in peripheral blood, corresponding to a ~32% mosaic in leukocytes (Figure 2A). By testing other tissue, we found 15% mutated alleles in urine cells, 2% in buccal swab, and no mosaic in hairs (data not shown).
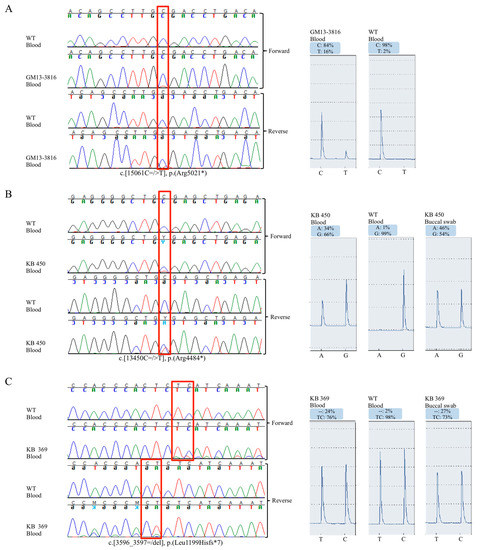
Figure 2.
Pyrosequencing analysis confirmed the mosaic mutations. (A–C) On the left, blood DNA Sanger sequencing electropherogram of indicated patients and healthy controls. Red frame indicates the mosaic nucleotide variants. On the right, pyrosequencing peak profile of patients and control with percentage quantification of mosaic nucleotide variants. Note that for KB 450 the reverse sequence is reported for Pyrosequencing.
In patient KB450 and KB369 we identified the nonsense c.13450C=/>T (p.R4484X) mutation [15], and the novel frameshift c.3596_3597=/del (p.L1199HfsX7) mutation, respectively, both in mosaic.
Pyrosequencing was used to confirm and quantify the mosaicism in different tissues. KMT2D c.13450C=/>T (p.R4484X) mutation was found in ~34% of peripheral blood alleles (in ~68% of leucocytes) and in ~46% of saliva cells (~92% in epithelial cells) (Figure 2B). The c.3596_3597=/del (p.L1199HfsX7) mutation was found in 20% of peripheral blood alleles (~40% in leucocytes), and in ~27% of saliva cells (~54% in epithelial cells) (Figure 2C).
Due to the unavailability of DNA we were not able to sequence any relatives from patients GM13-3816 and KB450, while both KB369 parents were negative for c.3596_3597=/del (p.L1199HfsX7).
3. Discussion
To date, only three KS patients with mosaic KMT2D deletions have been reported, two with intragenic deletions and one with a whole gene deletion, while no case has been described with mosaic KDM6A point mutation [6]. Here, we describe three additional KS subjects with mosaic KTM2D mutations. The patients with mosaic mutations described by Banka disclosed no significant difference in clinical features and cognitive profiles, in respect to non-mosaic patients [6]. Consistently, two of the three mosaic cases reported here showed distinct KS clinical characteristics, including facial features, as opposed to the GM13-3816 patient, who showed the lowest percentage of mutated cells in leukocytes (Figure 1 and Table 1). Although genotype–phenotype correlations based on few known subjects are premature, it seems plausible that both the type of mutation and the proportion of mutated cells have a direct impact on clinical outcome. Notably, five of the six known patients displayed a stature in the high–normal range (>50th centile), suggesting that mosaic mutations could have a mild effect their stature.
The neurobehavioral evaluation of patient 1 showed a learning disability, impairment of adaptive skills and psychopathological findings, such as an anxiety disorder, multiple phobias, bizarre behaviour and emotional dysregulation. A similar behavioural profile was also found in patient KB369, who was the sole patient in our cohort presenting with intellectual disability and autistic-like behaviour. A recent study of the intellectual functioning in 31 children with KTM2D mutations, showed IQs ranging from normal to severe disability, with most of them presented with moderately impaired IQ and a mean TIQ of 57.4 [16]. Another study of nine additional patients disclosed a similar trend, with a better mean TIQ (67) [17]. In the present study, patients GM13-3816 and KB450 showed borderline intellectual functioning (TIQ 87 and 84), while the Banka et al. [6] cases and our patient KB369 presented an obvious mild to moderate developmental delay (Table 2). Interestingly, patients displayed a more severe delay resulting from the deletion of the entire KMT2D gene or mutations causing a premature truncation of KMT2D in the first-half of the protein, predicting a shorter protein lacking the majority of KMT2D functional domains, including WIN and FYRC/N protein–protein interaction domains and the SET catalytic domain, therefore hampering its enzymatic activity.
The clinical examination and neurobehavioral assessment of the few mosaic KS patients did not allow us to draw at present any firm genotype–phenotype correlation. Nevertheless, some distinct facial dysmorphisms overlapping those found in non-mosaic KS patients and mild developmental delays should suggest targeted genetic testing in these subjects.
4. Materials and Methods
Clinical investigations and genetic analyses were approved by the institutional scientific board of the institutes involved, and were conducted in accordance with the Helsinki Declaration. DNA from KB369 and KB450 samples were deposited in the Genomic and Genetic Disorders Biobank [18]. Informed consent was obtained from family members. All children underwent complete physical and dysmorphology evaluations by the referring clinical geneticists. The clinical features of patients are summarized in Table 1.
Genomic DNA was extracted using standard procedures. Mutational analysis was carried out for patient 1 by a customized TruSeq Custom Amplicon panel using MiSeq (Illumina, San Diego, CA, USA) and validated with Sanger Sequencing. Sanger Sequencing was used to screen patient 2 and 3 as reported in [12]. PCR product was pyrosequenced twice on a PyroMark Q24 using Pyromark PCR Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany).
5. Conclusions
KS is a multi-systemic disorder not limited to a single organ or tissue of a common embryonic origin. The present data suggesting that the haploinsufficiency of KMT2D is limited to some cell types may be sufficient to recapitulate the associated phenotype. It is conceivable that screening of a larger cohort will likely identify additional cases of mosaic KS, which is so far underdiagnosed. We are persuaded that the targeted next-generation sequencing panel is the more appropriate technique for analysing this condition, since a single experiment allows full sequencing of the disease genes and the detection of somatic events, often overlooked by Sanger sequencing.
Acknowledgments
We are grateful to the Genomic and Genetic Disorders Biobank, Telethon Network of Genetic Biobanks (Telethon Italy grant GTB12001G), and of the EuroBioBank network for biospecimens banking. The financial support of Telethon—Italy (Grant No. GGP13231), Italian Ministry of Health, Jérôme Lejeune Foundation, and Daunia Plast to GM is gratefully acknowledged.
Author Contributions
Francesca Romana Lepri, Antonio Novelli, and Giuseppe Merla conceived and designed the experiments; Francesca Romana Lepri, Gabriella Maria Squeo, Natascia Malerba, Iolanda Adipietro performed the experiments; Dario Cocciadiferro and Bartolomeo Augello analyzed the data; Valentina Pes, Alessandra Vancini, and Maria Cristina Digilio performed genetic counseling, phenotype assessment, and are following the patients over time; Paolo Alfieri, Cristina Caciolo, Stefano Sotgiu, Renzo Gherardi performed cognitive tests and assessed neurobehavioral profile; Francesca Romana Lepri, Antonio Novelli, Bruno Dallapiccola and Giuseppe Merla wrote the paper with the support of all authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kuroki, Y.; Suzuki, Y.; Chyo, H.; Hata, A.; Matsui, I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 1981, 99, 570–573. [Google Scholar] [CrossRef]
- Niikawa, N.; Matsuura, N.; Fukushima, Y.; Ohsawa, T.; Kajii, T. Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 1981, 99, 565–569. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bogershausen, N.; Gatinois, V.; Riehmer, V.; Kayserili, H.; Becker, J.; Thoenes, M.; Simsek-Kiper, P.O.; Barat-Houari, M.; Elcioglu, N.H.; Wieczorek, D.; et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat. 2016, 37, 847–864. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Howard, E.; Bunstone, S.; Chandler, K.E.; Kerr, B.; Lachlan, K.; McKee, S.; Mehta, S.G.; Tavares, A.L.; Tolmie, J.; et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin. Genet. 2013, 83, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; Orsini, A., Pezzuti, L., Picone, L., Eds.; Italian Edition; Giunti OS: Firenze, Italy, 2012; The Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Harrison, P.L.; Oakland, T. ABAS-II Adaptive Behavior Assessment System, 2nd ed.; Ferrri, R., Orsini, A., Rea, M., Eds.; Italian Edition; Giunti OS: Firenze, Italy, 2014. [Google Scholar]
- Cornoldi, C.; Colpo, G. Prove di Lettura MT-2 per la Scuola Primaria; Giunti OS: Firenze, Italy, 2011. [Google Scholar]
- Tressoldi, P.E.; Cornoldi, C. BVSCO-2 Batteria per la Valutazione della Scrittura e della Competenza Ortografica–2; Giunti OS: Firenze, Italy, 2013. [Google Scholar]
- Kaufman, J.; Birmaher, B.; Rao, U.; Ryan, N. Test K-SADS-PL-Intervista Diagnostica per la Valutazione dei Disturbi Psicopatologici in Bambini e Adolescenti; Edizioni Centro Studi Erickson S.p.A.: Trento, Italy, 2004. [Google Scholar]
- Micale, L.; Augello, B.; Fusco, C.; Selicorni, A.; Loviglio, M.N.; Silengo, M.C.; Reymond, A.; Gumiero, B.; Zucchetti, F.; D’Addetta, E.V.; et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Cornoldi, C.; Lucangeli, D.; Bellina, M. Test AC-MT 6–11—Test di Valutazione Delle Abilità di Calcolo e Soluzione di Problemi; Edizioni Centro Studi Erickson S.p.A.: Trento, Italy, 2002. [Google Scholar]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, A.D.; Stegmann, A.P.; Blok, M.J.; Tserpelis, D.; Posma-Velter, C.; Detisch, Y.; Smeets, E.E.; Wagemans, A.; Schrander, J.J.; van den Boogaard, M.J.; et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011, 32, E2018–E2025. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.; Mazery, A.C.; Visier, A.; Baumann, C.; Lachesnais, D.; Capri, Y.; Toutain, A.; Odent, S.; Mikaty, M.; Goizet, C.; et al. Molecular, clinical and neuropsychological study in 31 patients with Kabuki syndrome and KMT2D mutations. Clin. Genet. 2017, 92, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Caciolo, C.; Alfieri, P.; Piccini, G.; Digilio, M.C.; Lepri, F.R.; Tartaglia, M.; Menghini, D.; Vicari, S. Neurobehavioral features in individuals with Kabuki Syndrome. Mol. Genet. Genom. Med. 2017, in press. [Google Scholar]
- Fusco, C.; Micale, L.; Pellico, M.T.; D’Addetta, E.V.; Augello, B.; Mandriani, B.; De Nittis, P.; Cocciadiferro, D.; Malerba, N.; Sacco, M.; et al. Genomic and Genetic Disorders Biobank. Genetic Biobank from patients with Williams-Beuren syndrome and other genomic and genetic disorders. Open J. Bioresour. 2015, 2. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).