Abstract
Here we discuss studies of the structure, folding, oligomerization and amyloid fibril formation of several proline mutants of human stefin B, which is a protein inhibitor of lysosomal cysteine cathepsins and a member of the cystatin family. The structurally important prolines in stefin B are responsible for the slow folding phases and facilitate domain swapping (Pro 74) and loop swapping (Pro 79). Moreover, our findings are compared to β2-microglobulin, a protein involved in dialysis-related amyloidosis. The assessment of the contribution of proline residues to the process of amyloid fibril formation may shed new light on the critical molecular events involved in conformational disorders.
1. Introduction
Proline residues play a prominent role in protein folding [1,2], protein mis-folding, and aggregation [3]. They are key to attaining the functional state of proteins [4]. Prolines also play a role in domain-swapping [5,6] and in protein aggregation to amyloid fibrils [7,8,9]. Peptidyl-prolyl cis/trans isomerases are enzymes that catalyze the cis/trans isomerization of peptide bonds preceding prolines (Figure 1). The cis/trans isomerization of the peptide bond acts as a molecular switch controlling several physiologically important processes, such as opening of the pore of a neurotransmitter-gated ion channel [10] or the formation of α-synuclein inclusions [11] in Parkinson’s disease.
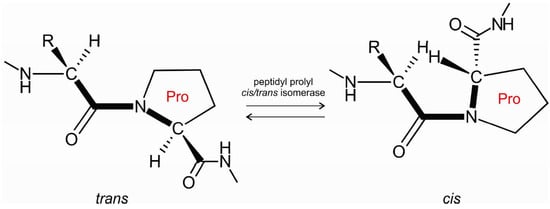
Figure 1.
Peptidyl-prolyl cis/trans isomerase facilitates cis/trans isomerization of the X-Pro peptide bond.
Amyloid fibril formation is a generic property among most proteins [12,13]. The cystatins, which are protein inhibitors of lysosomal cysteine cathepsins, are a model for studies of amyloid fibril formation. The cystatin family consists of three types of inhibitory proteins, namely, stefins (type-1), cystatins (type-2) and kininogens (type-3). Stefins are intracellular proteins present in the cytosol [14,15], including stefins A and B in humans [16] and stefins, A, B and C in bovidae [17,18]. Human stefin B [19,20,21,22,23], chimeric stefins [24] and cystatin C [25,26] have been used as suitable model proteins to study protein folding and amyloid fibril formation. Human stefin B is a small globular protein consisting of 98 amino acids with no disulfide bonds; its native sequence possesses a free Cys residue at position 3. To avoid intermolecular disulfide bridge formation, this Cys is changed into Ser for all in vitro studies (hereafter referred to wild-type protein, wt). This cytoplasmic protein is supposed to act primarily as a cysteine protease inhibitor [15], scavenging and inhibiting accidentally released cathepsins from the lysosome. In addition, stefin B also resides in the nucleus [27] where a number of alternative functions have been proposed. Stefin B (also termed cystatin B) gene mutations, either dodecamer repeats resulting in lower protein production or missense mutations leading to misfolding, cause a progressive myoclonus epilepsy of type 1 (EPM1) with slow signs of neurodegeneration [28,29]. Similarly to cystatin C, stefin B protects neurons from excessive oxidative stress [30,31] and protein misfolding [32]. Alternative functions, such as amateur chaperone function, have also been suggested from both experimental data and bioinformatic analysis [33]. A breakthrough in the understanding of the structure of cystatins and their mechanism of interaction with papain-like cysteine proteases, including lysosomal cathepsins, was provided by the three-dimensional (3D) structures of chicken cystatin monomer [34,35] and human stefin B-papain complex [36].
Our in vitro studies of stefin B folding revealed several slow phases [37,38], which were accompanied by dimerization of the protein. We were able to determine the crystal structure of a stefin B tetramer, which is composed of two domain-swapped dimers [19]. Of note, in stefin B, the proline residue at position 74 in the tetramer is in a cis conformation [19]. These structures were crucial for further development in the study of proteolysis and its inhibition, and represent the basis for understanding the mechanism of amyloid-fibril formation through 3D-domain swapping.
The cis-to-trans proline isomerization [2] is a slow process, dependent on pH. Stefin B has in total five proline residues at positions 6, 11, 36, 74 and 79. We have examined in more detail prolines at positions 74 and 79, and both have proved to be structurally relevant. When Pro 79 was mutated into a Ser in a stefin B-Y31 variant (with Y at site 31), the protein oligomerized predominantly as a tetramer which could be crystallized [19]. If Pro 74 was mutated into a Ser in the same variant, it underwent a transition to an oligomeric molten globule state [19,39]. We also studied the stefin B-Y31 P36G mutant, which rendered the protein less stable [40]. Historically, while the stefin B-Y31 variant [41] was observed and characterized first, the E31 variant is now referred to as wild-type since it is the most abundant.
To put our work in a wider context, we describe another protein where prolines dictate folding and amyloid fibril formation, β2-microglobulin (β2m). Interestingly, β2m shares with stefin B the same number of prolines, at positions 5, 14, 32, 72 and 90, and we compare the two systems in our Discussion and Conclusions sections.
2. Results
2.1. Influence of Prolines on Folding and Stability of Stefin B
In the early folding studies using a stefin B-Y31 variant, we observed that the protein, in contrast to stefin A, undergoes slow folding phases that are a repetition of the fast folding phases [37]. The amplitude of the slow phases is about 25%–30%. This can be explained by the existence of a population of molecules in the denatured state with either one or two non-native (cis) proline isomers that undergo similar, albeit slow, folding as the fast folding molecules with native (trans) proline configuration in the denatured state. However, the final oligomeric state of the slow phase proves to be dimeric, thus the dimer can stabilize a structurally important proline in a trans conformation. If a proline were cis, one would expect a higher amplitude of the slow phases, amounting to 70%, not only 30%. When we used size exclusion chromatography (SEC) to study the Y31 variant and its P36G mutant, we showed that 70% and 75% of molecules were monomers, respectively and the rest were dimers, whereas for the P79S mutant of the same variant, 100% were dimeric. This points to Pro 79 trans to cis isomerization as the very likely cause for the slow phase of folding towards a dimer. Nevertheless, in the tetrameric structure of the stefin B-Y31 P79S mutant, two domain-swapped dimers form the tetramer in which the Pro 74 was found in cis conformation [19].
2.2. Influence of Prolines on Conformation and Oligomerization of Stefin B
As we observed that the wt stefin B (as defined in the Introduction) is more stable and less prone to forming a molten globule, we studied the role of all five single-point proline mutants of stefin B. Using multiple sequence alignment of several stefins, we identified common amino acid substitution of the prolines in human stefin B (Figure 2). The five prolines at positions 6, 11, 36, 74 and 79 were mutated to leucine, serine, aspartic acid, serine and serine, respectively. All mutant proteins were produced in an Escherichia coli (E. coli) expression system (Supplementary Figure S1) and were shown to retain their inhibitory activity (Supplemenatry Figure S2).
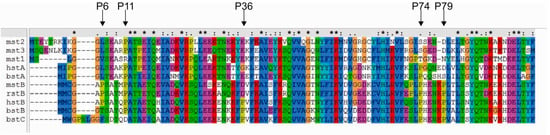
Figure 2.
Sequence alignment of different stefins. Mouse stefin B (mstB), human stefin A (hstA), rat stefin B (rstB), mouse stefin 1 (mst1), mouse stefin 2 (mst2), mouse stefin 3 (mst3), bovine stefin A (bstA), bovine stefin B (bstB) and bovine stefin C (bstC) were compared against human stefin B (hstB). Sequences were retrieved from UniProt database. The multiple sequence alignment was performed with ClustalX [42]. All five proline residues (P6, P11, P36, P74 and P79) are indicated with an arrow.
The stability and exposure of hydrophobic patches were confirmed by measuring ANS (1-anilinonaphthalene-8-sulfonic acid) fluorescence spectra (Figure 3A). Together with far UV-CD (ultraviolet circular dichroism) spectra (Figure 3B) we observed that P6L, P11S, P36D and P79S have hydrophobic exposure and secondary structure similar to the wild-type (wt) protein. The highest ANS binding is observed for the P74S mutant indicating a molten globule-like state. However, this was not observed consistently for the P74S mutant of the wt [20] by measuring CD spectra. It may well be that the molten globule intermediate forms only under destabilizing conditions or upon freeze-thawing cycles.
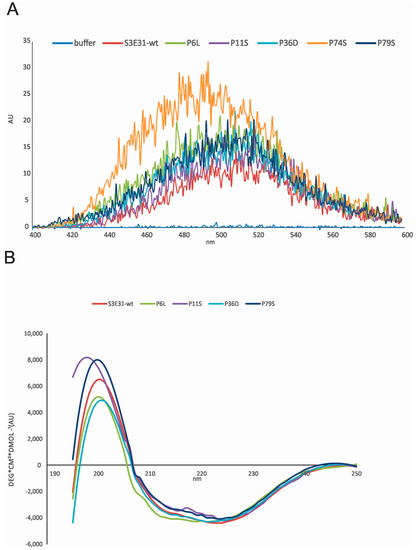
Figure 3.
(A) ANS fluorescence upon binding to wild-type protein (wt) stefin B and proline mutants. The excitation wavelength was 370 nm, and the spectra were recorded from 400 nm to 600 nm on a Perkin Elmer LS50B (Perkin Elmer, Waltham, MA, USA). Slits were open 2.5 nm. ANS was dissolved in 0.01 M phosphate buffer, pH 7, and 0.15 M NaCl. Final concentrations were 1.25 mM ANS and 25 µM proteins; (B) Far UV CD spectra of wt stefin B and proline mutants were recorded from 195 to 250 nm, as indicated. The bandwidth was 1 nm and spectra recording time at each nm was 3 s; the temperature was 25 °C. Final concentrations were 34 µM. Due to high aggregation propensity, the final concentrations varied. Therefore, ellipticity values were normalized to the wt spectrum (i.e., a factor was used to give ellipticity—4200 ± 100 deg·cm2·dmol−1).
SEC data on the wt and its proline mutants show (Supplementary Figure S3) that dimers are the main oligomeric form of the wt stefin B and P36D mutant—when frozen and unfrozen once. The amount of the higher oligomers increases upon freeze-thawing. Then come the tetramers and higher oligomers. An estimate from the surface area of the peaks indicates 65% dimers, 20% tetramers, 5% monomers and 10% of higher oligomers. A similar distribution of the oligomers was obtained for P6L and P11S of the wt stefin B. In the case of P79S, the tetramer peak amounted to a higher percentage of around 30% tetramers, 55% dimers and 15% of higher molecular weight species. The tendency to form oligomers is high for the P74S mutant of the wt (>50%), in accordance with its tendency to transform into an oligomeric molten globule as observed previously for the Y31 variant [19]. The P79S mutant of the the Y31 variant was predominantly in the form of tetramers [19]. Taken together, stabilization of the dimer of stefin B is sensitive to Pro 36, whereas stabilization of the tetramer is sensitive to Pro 79. When cis to trans transition is facilitated by the Pro mutation at the two sites, respectively, dimers and tetramers are populated to a higher amount.
2.3. Prediction of the Effects of Proline Mutations on Human Stefin B Stability
The prediction of human stefin B stability upon single-point mutations of proline residues in the protein sequences (UniProt ID: P04080 and its mutant C3S) [43] as well as the monomeric (1STF:I [36] and 4N6V:chain0 [44] and tetrameric 2OCT:chainA [19]) protein structures, was performed at pH 7.0 and 25 °C using a support vector machine (SVM)-based tool, I-Mutant2.0 [45]. Of note, the protein stability increased only for the P6L mutant, whereas all remaining mutations, namely P36D and three Pro to Ser mutations at positions 11, 74 and 79, decreased protein stability (Table 1).

Table 1.
Prediction of human stefin B stability upon proline mutations.
2.4. Influence of Prolines on Amyloid Fibril Formation of Human Stefin B
Substituting proline at position 74 with a serine in the sequence of the wt stefin B did not affect the protein structure and stability to any significant extent, as shown by urea and thermal denaturation [20]. In fact, the mutant was slightly more stable, which is in contrast to the prediction in Table 1 (one, however, has to bear in mind that the changes in stability in both cases: prediction and experiment, are rather small and within the standard error of 1.4 ± 0.1 kcal/mol of the I-Mutant2.0 program). The exchange of a proline would be expected to lead to a more stable protein, due to higher flexibility—i.e., entropic contribution to stability, however, enthalpic contribution and hydration effects increase or decrease the stability.
When the fibrillation rate of the P74S mutant was compared to the fibrillation rate of the wt-like protein, however, it was shown that P74 is essential not only for stefin B tetramer formation but also for amyloid fibrillation. Indeed, when Pro74 was replaced with Ser, the lag phase was extended up to 10 times with a smaller final yield (Figure 4A,B). CD spectra show that this mutant adopts a folded structure, thus these differences are not the result of a change in the overall fold of the mutant. Transmission electron microscopy (TEM) results (Figure 4C,D) reveals that P74S remains in the form of granular aggregates (Figure 4D), whereas the wt protein formed amyloid fibrils after 7 days of incubation (Figure 4C). Moreover, when the effects of peptidyl-prolyl isomerase cyclophilin A (CypA) were examined, it was shown that CypA prolongs the lag phase and increases the final yield and length of the fibrils. On the other hand, the inactive cyclophilin A R55A caused a prolonged lag phase, but did not lead to an increase in the final fibril yield [20]. Although the fibrils formed in the presence or absence of CypA had the same shape and morphology, the presence of CypA provides a higher yield of stefin B fibrils [20].
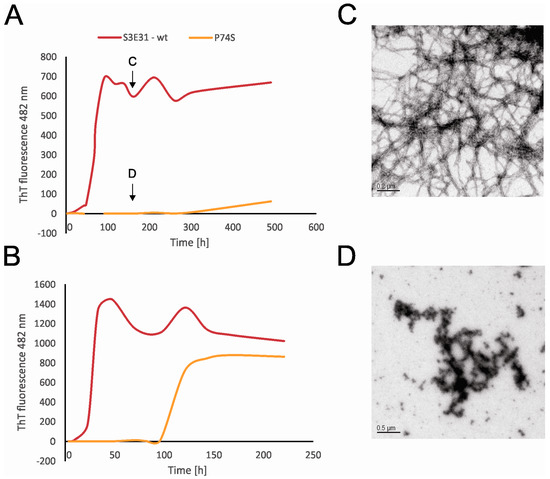
Figure 4.
The time courses of amyloid fibril formation of wild-type stefin B and its site-specific mutant P74S were monitored by ThT fluorescence at 482 nm. The fibrillation reactions took place in 0.015 M acetate buffer, 0.15 M NaCl, pH 4.8, 12% TFE at 25 °C (A) and in the same buffer at 30 °C (B). TEM images taken during the fibrillation reactions. The wild-type stefin B (C) and P74S mutant (D) on the 7th day of fibrillation (see arrows) [20].
2.5. Structure of Monomer and Tetramer Composed of Domain-Swapped Dimers of Stefin B
The crystal structure of the monomer of stefin B (Figure 5A,B) determined in a complex with papain was one of the first structures of cystatins and as such represented a cornerstone in our ability to understand the mechanism of its inhibitory action on proteases of the papain family [36]. The monomer is a typical α/β protein, with a well-formed β-sheet of 5 β-strands and an α-helix (residues 12 to 37). As an interesting point, a monomer of stefin B crystallized at pH 10 [44], showed a 4-dimensional arrangement in the crystal cage, resembling a channel.
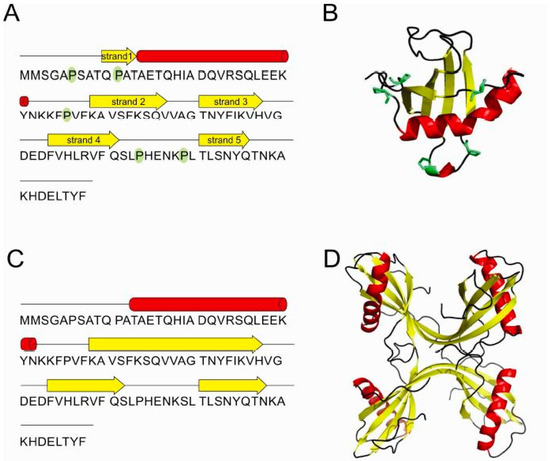
Figure 5.
Stefin B—schematic representation of secondary structure elements (A) and the 3D structure of monomer (PDB id: 1STF) [36] (B). Schematic of secondary structure elements (C) of the two domain-swapped dimers building-up the tetramer (PDB id: 2OCT) [19] (D). All five prolines are highlighted in green in the monomer.
The monomeric structure of stefin B [36] was also crucial for further understanding the mechanism of amyloid-fibril formation through 3D-domain swapping. One mechanism for oligomerization is a 3D domain-swapping mechanism [26,46,47] where an intramolecular interface from one monomer becomes an intermolecular interface between subunits in the oligomers [48]. First, the crystal structure of cystatin C domain-swapped dimer was determined [49], closely followed by the NMR-derived structure of stefin A [26]. In the domain-swapped dimer of stefins, each stefin fold is made of strand 1, the α-helix and strand 2 from one monomer and strands 3–5 from the other monomer. In addition, the stefin B tetramer has been shown to have two domain-swapped dimeric units which interact through loop-swapping, also termed “hand-shaking” (Figure 5C,D).
In the case of stefin B it has been demonstrated that prolines play an important role in domain swapping, as they control the rigidity of loops between secondary structure elements. The trans conformer of Pro 74 is found widely conserved among stefins and cystatins [26,49], whereas the cis conformer is reported in the structure of the stefin B tetramer only [19]. This isomer is particularly important as it brings the Ser 72-Leu 80 loop in the close vicinity of the N-terminal trunk. In the loop swap of two domain-swapped dimer units, the loop position from residues Ser 72 to Leu 80 is provided by Pro 74 and Pro 79. Pro 79 contributes to the rigidity of the loop through its trans conformation.
3. Discussion
For comparison with our model protein stefin B data, we reviewed the literature on the role of proline isomerization in the structure, folding and amyloid fibril formation of β2-microglobulin (β2m). Furthermore, we predicted how chosen proline mutations may influence the stability of this protein.
β2m is a 99 amino acid long protein containing the light-chain of the major histocompatibility complex I (MHC I) [50]. β2m is present at the surface of almost all cells. Upon dissociation from MHC I it is catabolized in the kidneys. Therefore, in patients who suffer from chronic kidney insufficiency and undergo dialysis treatment, the concentration of β2m increases up to 60-fold causing dialysis-related amyloidosis (DRA), i.e., insoluble β2m amyloids form and accumulate in the joints and in connective tissues [51,52]. Intriguingly, high concentrations of β2m cannot completely clarify the onset of amyloid precipitation, as in vitro studies have shown that this protein stays soluble and monomeric at neutral pH even when concentrations are more than 100 times higher than in patients exposed to dialysis [52,53].
3.1. Influence of Prolines in β2-Microglobulin: Folding and Oligomerization
Chiti et al. [54] have shown that β2m folds via two structurally different intermediates on its way to the globular native state. One of these, termed I1, is populated within 5 ms and contains a disorganized hydrophobic core with several hydrophobic residues exposed to solvent. The other one, termed I2, forms within ms from the I1 species and shows a native-like secondary structure with side chains packed in the hydrophobic core. [54] This species further folds to a globular native state within an interval ranging from seconds to minutes at 30 °C. Further studies demonstrated that the slow folding of I2 which precedes the native state is rate limited by trans to cis isomerization of the His 31-Pro 32 peptide bond [55].
β2m fibril formation starts rapidly at low pH with lag-dependent kinetics where dimers, trimers and tetramers are formed [56,57]. Studies of the kinetics of fibril formation have shown that monomers form a nucleus consisting of six β2m polypeptide chains, whereas fibrils are formed in the elongation phase by adding monomers [56]. Even though several β2m oligomeric species have been characterized [58] the linkage between oligomers and fibrils remains unknown.
Oligomerization is considered as a crucial step towards self-association of proteins into amyloid fibrils. Moreover, oligomers are believed to be toxic in several types of amyloid-related neurodegenerative diseases [59,60,61,62]. Exploring the molecular mechanisms leading to the formation of oligomers is a great challenge, as it would help in developing strategies to suppress amyloid-related diseases. Toxicity is not restricted to pathological proteins alone, it is instead related to a common structural/conformational property of the prefibrillar oligomers [61,62]. The mechanism through which β2m causes DRA remains poorly understood. It has been reported that β2m forms nonselective, long-lived and voltage-independent ion channels in phospholipid bilayers and that their appearance is tightly correlated with DRA [63]. These channels can bind Congo red and zinc, hence it was suggested by the authors that their structure includes β-sheets [63]. On a separate note, it is also not clear whether it takes the full-length protein to develop the pathophysiology or whether fragments can cause it. In order to clarify this issue, Mustata and coworkers designed K3, which is a digestion fragment of the full length β2m (Ser 20-Lys 41) [64]. It is known that this peptide forms amyloid fibrils under a wide range of conditions [64]. Combining solid state NMR, atomic force microscopy and X-ray diffraction, the characteristic amyloid conformation was elucidated; thus showing that K3 has adopted a U-shaped β-strand-turn-β-strand motif [64]. Interestingly, this motif had already been reported as a universal amyloid feature and hence it was speculated that it might play a role in toxicity [65,66,67]. Moreover, the same authors have proven by channel modelling that this K3 oligomer can constitute the structure of the channel. These results, together with fluorescence measurements in kidney cells which have shown channel-mediated calcium uptake, indicate that the β2m related DRA can be mediated by ion channels formed by the K3 fragment [64]. These data add weight to the so called “channel hypothesis”; these channels lead to Ca2+ influx which can cause apoptosis and alter signaling, hence changing the plasma membrane and electrical properties of the neuron.
3.2. Aggregation and Amyloid-Fibril Formation of β2-Microglobulin
β2m has been widely used as a powerful model for exploration of the structural molecular mechanisms of fibril formation from a full-length protein in vitro. Natively, this protein folds into a β-sandwich fold consisting of 2 β-sheets, one containing 4 strands and the other 3, which are covalently linked by a disulphide bond between 2 cysteines (residues 26 and 81) [68]. It contains five peptidyl-prolyl bonds and one of them (His 31-Pro 32) exhibits a thermodynamically unfavorable cis-isomer conformation in the native state [52].
A huge body of evidence has shown that ~60% of the sequence of β2m is highly amyloidogenic [69,70,71]. Nevertheless, the natively folded protein is not prone to aggregation [72,73] which implies that the folded structure strongly affects its amyloidogenic potential. The partial unfolding in vivo therefore appears to be a mandatory step leading to aggregation as it provides the exposure of aggregation-prone regions of the sequence. β2m spontaneously forms fibrils in vitro at pH < 3.0 with low ionic strength (<50 mM NaCl) when stimulated by agitation [57]. In addition to setting amyloid fibrillation at low pH conditions, in order to cause partial unfolding and drive amyloid fibrillation of β2m at neutral pH, a plethora of conditions has been suggested, such as adding glycosaminoglycans, detergents, denaturants or by using ultrasonication and elevated temperature [53,74,75,76,77,78]. These intrinsic and extrinsic factors increase the concentration of a partially unfolded intermediate in which the natively cis-configured proline 32 in the polypeptide chain is isomerized to a trans isomer [53,79]. In addition, solid-state NMR studies have shown that amyloid fibrils which form from acid-denaturated β2m at pH < 3 contain a trans Pro 32 as well [46,80]. It should be noted that this is not the only structural change reported to be associated with amyloid formation as β-sheets in the protein turn from antiparallel in native β2m to parallel in the amyloid [46]. However, cis-trans isomerization of Pro 32 is considered as a crucial trigger for the transition of soluble monomeric β2m to its misfolded amyloidogenic species [7,8]. This hypothesis is supported by the observation that in the ΔN7 variant of β2m, where the first 7 N-terminal residues are truncated, the cis-Pro 32 conformer is destabilized in such a manner that only the trans-Pro 32 exists at neutral pH [47].
So far, it has been proven that a single region, approximately 10 residues long (60–70), is crucial for elongation of the full-length protein under certain conditions [81]. Aromatic residues are widely present in this region, which most probably contributes to the propensity of β2m to aggregate [70]. Studies of the full-length protein sequence at low pH have shown that shifting certain residues, especially Leu 23, His 51 and Val 82 with Pro which acts as β-sheet breaker, causes a lowering in fibril elongation kinetics. Moreover, when comparing intact protein at low pH and peptide studies in the context of the effects of sequence alteration on the fibril growth kinetics, results are surprising. Namely, isolated fragments including residues 20–40, 60–70 and ~80–99 all form amyloid fibrils [71,82], whereas in the full-length protein chain mutation of residues, only the ~60–70 region has altered fibril formation kinetics [81,83]. Results of NMR studies explain this observation; the acid-unfolded non-native structure of β2m is stabilized by the disulphide bond and includes gathering of hydrophobic residues in two regions (29–51 and 58–78) [8], meaning that a single strain of 10 residues might have a strong impact on the aggregation potential of the entire protein. It is speculated that this might be a result of an evolutive twist [79]. Namely, this sequence includes aromatic residues such as Phe 55, Trp 60, Phe 62, Tyr 63 and Leu 65, which are important for interaction with the MHC I heavy chain [79] and hence for regulation of immune system.
As mentioned above, the cis Pro 32 conformer is proposed as an essential residue for β2m nucleation at neutral pH and P32G and P32V mutants have been used to show this. Namely, both mutants adopt trans Gly or Val 32, respectively, but cannot form amyloid-like fibrils spontaneously, even though P32G can elongate preformed seeds more efficiently than wt β2m [8,55]. These acyclic amino acids favor the trans conformation at the peptide bond, but it is obvious that they cannot completely imitate the unique conformation of Pro 32 [79]. Moreover, variants such as P5G and ΔN7 also affect isomerization of the Pro 32 peptide bond, facilitating fibril nucleation at pH 7.0 [53,84]. On the other hand, β2m can form oligomers and fibrils at neutral pH by addition of Cu2+ and 1 M urea [7]. Namely, peptide bond isomerization at Pro 32 can be initiated by the coordination of a metal ion causing the rapid formation of oligomers [7,53]. Therefore, the isomerization of Pro 32 has been constantly shown as a key initial step in β2m amyloid fibrillation [85].
In summary, aggregation of β2m into amyloid structures may be achieved via a numerous routes as β2m forms amyloid fibrils at both pH 2.5 and 7.0 [53]. There are many avenues that might finally lead to a better understanding of the assembly pathways in different conditions. In both cases, interactions between specific hydrophobic and aromatic residues may lead to fibrillation. However, fibrils formed at neutral pH contain a highly-charged surface [86], which could be neutralized at low pH. This might explain the fact that fibrils form much more rapidly under acidic conditions and provides support for a convergent mechanism of assembly at acidic and neutral pH. Another important hallmark of amyloid fibrillation which is considered as a key to amyloid formation is the destabilization of the N-terminal region; a double variant P32G/17A which combines a trans peptide bond at Pro 32 with the destabilization of the N-terminal region forming fibrils spontaneously at pH 7 [86]. It remains to be elucidated whether the assembly pathways are similar and how they converge in the form of a common fibrillar structure.
3.3. Prediction of Stability of β2-Microglobulin and Its Proline Mutants
β2m stability was assessed using I-Mutant2.0 [45] upon Pro to Ser mutations in positions 5, 14, 32, 72 and 90 (numbering according the processed form of the protein—UniProt ID: P61769) and mutations P32G [53] and P32L on the primary and tertiary structures [87,88], respectively. Of note, all five Pro to Ser mutations destabilize the protein (−2.24 to −0.99 Kcal/mol) as well as the β2m-P32G mutation (−2.62 to −1.74 Kcal/mol) (Table 2). On the other hand, the β2m P32L mutant exhibits a destabilizing effect on its primary structure (−2.05 Kcal/mol) and a stabilizing effect on the tertiary structure of its monomeric [88] (1LDS:A, 0.79 Kcal/mol) and dimeric (3LOW:A [87], 1.05 Kcal/mol) forms, respectively (Table 2).
Table 2.
Prediction of stability of human β2-microglobulin proline mutations.
3.4. Structures of β2-Microglobulin Monomer and Domain-Swapped Dimer
3D structures of the β2m monomer, dimer and tetramer are also known (Figure 6A–D). Some studies suggest that different reagents can trigger different oligomerization pathways [53]. Namely, the crystallographic structure of the reductant-triggered β2m dimer [87] was different from the dimer and hexamer triggered by copper [7,89] suggesting that different conditions alter the protein structure in different ways, leading to different results. Liu et al. [87] suggested that dimerization of β2m may occur via a relatively uncommon run-away domain swap with a covalent linkage where β strands are exchanged between two subunits, creating two interfaces. One is called the closed interface, and the other the open interface due to a new β-sheet that contributes to the stability of dimer. Another hallmark of this phenomenon is the rearrangement of the disulfide bonds as they serve as an intermolecular bond to stabilize the dimer. Moreover, in cases where natively-folded proteins form amyloids, a newly formed cross-β spine is required for the fibril ensemble and in the cases of domain-swapping, studies have shown that the hinge loop is essential for forming the cross-β spine. The LSFSKD structure (residues 53–58 of human β2m) acts as a typical steric zipper structure. Upon reduction of the intramolecular disulfide bond, the β2m monomer can assemble as “closed-ended” oligomers or “open-ended” runaway domain-swapped oligomers. The formation of intermolecular disulfide bonds stabilizes the domain-swapped oligomers. The self-association of hinge loops into a zipper spine allows the transformation from oligomers into fibrils; as the oligomer grows, the loop regions between swapped domains can slide slightly to fit into a particular frame. Based on these findings they postulated the so-called “domain-swapped zipper-spine model” of a β2m fibril [87].
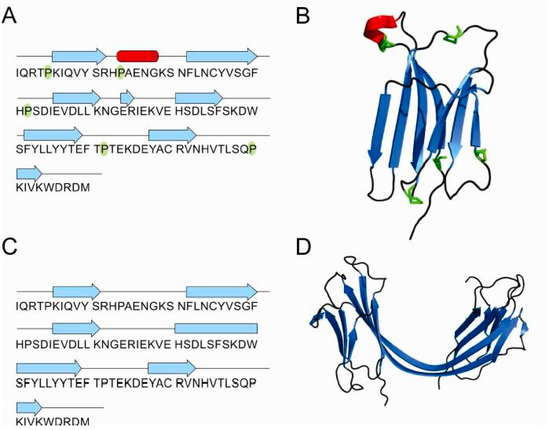
Figure 6.
β2-microglobulin schematic presentation of secondary structure elements and the 3D structure of monomer (PDB id: 1LDS) [88] (A,B) and domain-swapped dimer (PDB id: 3LOW) [87] (C,D). All five prolines are highlighted in green in the monomer.
4. Materials and Methods
4.1. Protein Isolation
In brief: the recombinant wild-type like, C3S E31-stefin B and its corresponding proline mutants were expressed in E. coli and purified by carboxymethylated (CM) papain Sepharose affinity chromatography. The unbound material was eluted with 0.01 M Tris-HCl containing 0.5 M NaCl at pH 8.0. Stefin B protein was eluted with 0.02 M triethanolamine (TEA) buffer at pH 10.5 and was fast refolded into a stronger buffer of a neutral pH. Furthermore, in the cold room, gel-filtration on Sephacryl S-200 was performed using phosphate 0.01 M buffer pH 7.5., 0.12 M NaCl. For analytical purposes, size-exclusion chromatography was used. Using a Superdex 75 column (Pharmacia, Uppsala, Sweden), stefin B eluted as a set of oligomers: monomers, dimers, tetramers and higher oligomers.
All other methods: expression, isolation and purification of stefin B wt and mutants as well as the conditions to follow fibril fluorescence by ThT fluorescence, were the same as previously described [20,90].
4.2. Fluorescence Spectra
Fluorescence was measured using a model 1.2× fluorimeter from PTI-Photon Technology International (Birmingham, NJ, USA) with a thermo unit for temperature control. 1-anilinonaphthalene-8-sulfonic acid (ANS) fluorescence was measured using an excitation wavelength of 370 nm and spectra were recorded from 400 to 600 nm. The entrance and exit slits for the excitation light-beam were 3 nm, 2 nm and 2 nm, respectively. Measurements were made in a 10-mm micro-cuvette at 25 °C. Thioflavin T (ThT) fluorescence was measured using an excitation wavelength of 440 nm and emission wavelength of 482 nm.
4.3. Circular Dicroism Specta
CD spectra were measured using an Aviv model 62A DS CD spectropolarimeter (AVIV, Lakewood, NJ, USA). Far-UV CD spectra were recorded in a 0.1 cm cell. Protein concentration was 34 μM or lower for the far-UV CD. For the far UV CD the bandwidth was 1 nm, and the step of measurement was 1 nm, with data integration time 4 s. Measurements were performed at 25 °C.
4.4. Size-Exclusion Chromatography (SEC)
The oligomeric state and purity of the protein samples was determined by size-exclusion chromatography (SEC) using a Superdex 75 FPLC column (Pharmacia, Uppsala, Sweden). The flow rate was 0.5 mL/min and typically a 100 μL of 50 μM sample of the protein was applied. Buffer was 10 mM potassium phosphate, pH 7.0, with 0.15 M NaCl added—if not otherwise specified.
5. Conclusions
Studies on both stefin B and β2m indicate that there is a link between oligomerization and cis to trans isomerization of certain Pro residues. For β2m, in the monomer, Pro 32 is found in a cis conformation. In this case, cis to trans isomerism leads directly to fibril formation, whereas in stefin B the trans to cis isomerization leads to the off-pathway tetramer [91] so that yet another transition from cis to trans is needed for fibril elongation [91]. In conclusion, cis to trans isomerization of a critical proline may act as a switch towards amyloid fibrils, starting with domain-swapping. Neighboring residues of the proline undergoing cis/trans isomerism are also important for the regulatory switch, such as lysine or serine/threonine residues, in prion and phosphorylated Tau, respectively [92,93].
Supplementary Materials
Supplementary materials can be found at www.mdpi.com/1422-0067/18/3/549/s1.
Acknowledgments
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding No. P1-0048 to Dušan Turk, and P1-0140 to Boris Turk and the project P1-0048 to Eva Žerovnik). Samra Hasanbašić was given a Fellowship via CMEPIUS (for student exchange at the Jožef Stefan International Postgraduate School).
Author Contributions
Eva Žerovnik and Samra Hasanbašić reviewed literature data and wrote the paper, Ajda Taler-Verčič, Veronika Stoka and Samra Hasanbašić performed experiments on stefin B proline mutants and wrote their parts; Dušan Turk, Ajda Taler-Verčič, Veronika Stoka, Selma Berbić and Eva Žerovnik contributed to the Discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef]
- Wedemeyer, W.J.; Welker, E.; Scheraga, H.A. Proline cis-trans isomerization and protein folding. Biochemistry 2002, 41, 14637–14644. [Google Scholar] [CrossRef]
- Borgia, A.; Kemplen, K.R.; Borgia, M.B.; Soranno, A.; Shammas, S.; Wunderlich, B.; Nettels, D.; Best, R.B.; Clarke, J.; Schuler, B. Transient misfolding dominates multidomain protein folding. Nat. Commun. 2015, 6, 8861. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.; Itzhaki, L.S. Implications of 3D domain swapping for protein folding, misfolding and function. Adv. Exp. Med. Biol. 2012, 747, 137–152. [Google Scholar]
- Rousseau, F.; Schymkowitz, J.W.; Itzhaki, L.S. The unfolding story of three-dimensional domain swapping. Structure 2003, 11, 243–251. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.W.; Wilkinson, H.R.; Itzhaki, L.S. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc. Natl. Acad. Sci. USA 2001, 98, 5596–5601. [Google Scholar] [CrossRef]
- Eakin, C.M.; Berman, A.J.; Miranker, A.D. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 2006, 13, 202–208. [Google Scholar] [CrossRef]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Christensen, G.; Otzen, D.E. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol. 2004, 341, 575–588. [Google Scholar] [CrossRef]
- Lummis, S.C.; Beene, D.L.; Lee, L.W.; Lester, H.A.; Broadhurst, R.W.; Dougherty, D.A. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef]
- Ryo, A.; Togo, T.; Nakai, T.; Hirai, A.; Nishi, M.; Yamaguchi, A.; Suzuki, K.; Hirayasu, Y.; Kobayashi, H.; Perrem, K.; et al. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of alpha-synuclein inclusions. J. Biol. Chem. 2006, 281, 4117–4125. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein Folding and its Links with Human Disease. Biochem. Soc. Symp. 2001, 68, 1–26. [Google Scholar]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid Fibril Formation by an SH3 Domain. Proc. Natl. Acad. Sci USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef]
- Turk, B.; Bieth, J.G.; Bjork, I.; Dolenc, I.; Turk, D.; Cimerman, N.; Kos, J.; Colic, A.; Stoka, V.; Turk, V. Regulation of the Activity of Lysosomal Cysteine Proteinases by Ph-Induced Inactivation and/or Endogenous Protein Inhibitors, Cystatins. Biol. Chem. Hoppe Seyler 1995, 376, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Colic, A.; Stoka, V.; Turk, V. Kinetics of Inhibition of Bovine Cathepsin-S by Bovine Stefin-B. FEBS Lett. 1994, 339, 155–159. [Google Scholar] [CrossRef]
- Turk, B.; Krizaj, I.; Kralj, B.; Dolenc, I.; Popovic, T.; Bieth, J.G.; Turk, V. Bovine Stefin-C, a New Member of the Stefin Family. J. Biol. Chem. 1993, 268, 7323–7329. [Google Scholar] [PubMed]
- Jenko Kokalj, S.; Guncar, G.; Stern, I.; Morgan, G.; Rabzelj, S.; Kenig, M.; Staniforth, R.A.; Waltho, J.P.; Zerovnik, E.; Turk, D. Essential role of proline isomerization in stefin B tetramer formation. J. Mol. Biol. 2007, 366, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Smajlovic, A.; Berbic, S.; Schiene-Fischer, C.; Tusek-Znidaric, M.; Taler, A.; Jenko-Kokalj, S.; Turk, D.; Zerovnik, E. Essential role of Pro 74 in stefin B amyloid-fibril formation: Dual action of cyclophilin A on the process. FEBS Lett. 2009, 583, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Taler-Vercic, A.; Kirsipuu, T.; Friedemann, M.; Noormagi, A.; Polajnar, M.; Smirnova, J.; Znidaric, M.T.; Zganec, M.; Skarabot, M.; Vilfan, A.; et al. The role of initial oligomers in amyloid fibril formation by human stefin B. Int. J. Mol. Sci. 2013, 14, 18362–18384. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E.; Pompe-Novak, M.; Skarabot, M.; Ravnikar, M.; Musevic, I.; Turk, V. Human stefin B readily forms amyloid fibrils in vitro. Biochim. Biophys. Acta 2002, 1594, 1–5. [Google Scholar] [CrossRef]
- Zerovnik, E.; Virden, R.; Jerala, R.; Kroon- Zitko, L.; Turk, V.; Waltho, J.P. Differences in the effects of TFE on the folding pathways of human stefins A and B. Proteins Struct. Funct. Bioinf. 1999, 36, 205–216. [Google Scholar] [CrossRef]
- Kenig, M.; Jenko-Kokalj, S.; Tusek-Znidaric, M.; Pompe-Novak, M.; Guncar, G.; Turk, D.; Waltho, J.P.; Staniforth, R.A.; Avbelj, F.; Zerovnik, E. Folding and amyloid-fibril formation for a series of human stefins’ chimeras: Any correlation? Proteins 2006, 62, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Staniforth, R.A.; Dean, J.L.; Zhong, Q.; Zerovnik, E.; Clarke, A.R.; Waltho, J.P. The major transition state in folding need not involve the immobilization of side chains. Proc. Natl. Acad. Sci. USA 2000, 97, 5790–5795. [Google Scholar] [CrossRef] [PubMed]
- Staniforth, R.A.; Giannini, S.; Higgins, L.D.; Conroy, M.J.; Hounslow, A.M.; Jerala, R.; Craven, C.J.; Waltho, J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001, 20, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Ceru, S.; Konjar, S.; Maher, K.; Repnik, U.; Krizaj, I.; Bencina, M.; Renko, M.; Nepveu, A.; Zerovnik, E.; Turk, B.; et al. Stefin B interacts with histones and cathepsin L in the nucleus. J. Biol. Chem. 2010, 285, 10078–10086. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, T.; Lehesjoki, A.E.; Kopra, O. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 2008, 49, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Korja, M.; Kaasinen, V.; Lamusuo, S.; Parkkola, R.; Nagren, K.; Marttila, R.J. Substantial thalamostriatal dopaminergic defect in Unverricht-Lundborg disease. Epilepsia 2007, 48, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N.; Schweiger, A.; Myers, R.M.; Turk, V.; Turk, B. Sensitization of stefin B-deficient thymocytes towards staurosporin-induced apoptosis is independent of cysteine cathepsins. FEBS Lett. 2005, 579, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009, 29, 5910–5915. [Google Scholar] [CrossRef] [PubMed]
- Polajnar, M.; Zavasnik-Bergant, T.; Skerget, K.; Vizovisek, M.; Vidmar, R.; Fonovic, M.; Kopitar-Jerala, N.; Petrovic, U.; Navarro, S.; Ventura, S.; et al. Human Stefin B Role in Cell’s Response to Misfolded Proteins and Autophagy. PLoS ONE 2014, 9, e102500. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E. Putative alternative functions of human stefin B (cystatin B): Binding to amyloid-β, membranes, and copper. J. Mol. Recognit. 2017, 30, e2562. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [PubMed]
- Engh, R.A.; Dieckmann, T.; Bode, W.; Auerswald, E.A.; Turk, V.; Huber, R.; Oschkinat, H. Conformational variability of chicken cystatin. Comparison of structures determined by X-ray diffraction and NMR spectroscopy. J. Mol. Biol. 1993, 234, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: A novel type of proteinase inhibitor interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [PubMed]
- Zerovnik, E.; Jerala, R.; Virden, R.; Kroon Zitko, L.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: II. Folding from GuHCl unfolded, TFE denatured, acid denatured, and acid intermediate states. Proteins 1998, 32, 304–313. [Google Scholar] [CrossRef]
- Zerovnik, E.; Virden, R.; Jerala, R.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: I. Comparison to homologous stefin A. Influence of pH and trifluoroethanol on the fast and slow folding phases. Proteins 1998, 32, 296–303. [Google Scholar] [CrossRef]
- Ceru, S.; Zerovnik, E. Similar toxicity of the oligomeric molten globule state and the prefibrillar oligomers. FEBS Lett. 2008, 582, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kenig, M.; Berbic, S.; Krijestorac, A.; Kroon-Zitko, L.; Tusek, M.; Pompe-Novak, M.; Zerovnik, E. Differences in aggregation properties of three site-specific mutants of recombinant human stefin B. Protein Sci. 2004, 13, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R.; Trstenjak, M.; Lenarcic, B.; Turk, V. Cloning a synthetic gene for human stefin B and its expression in E. coli. FEBS Lett. 1988, 239, 41–44. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.Z.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Ritonja, A.; Machleidt, W.; Barrett, A.J. Amino-Acid Sequence of the Intracellular Cysteine Proteinase-Inhibitor Cystatin-B from Human-Liver. Biochem. Biophys. Res. Commun. 1985, 131, 1187–1192. [Google Scholar] [CrossRef]
- Renko, M.; Taler-Vercic, A.; Mihelic, M.; Zerovnik, E.; Turk, D. Partial rotational lattice order-disorder in stefin B crystals. Acta Crystallogr. D Biol Crystallogr. 2014, 70, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Debelouchina, G.T.; Platt, G.W.; Bayro, M.J.; Radford, S.E.; Griffin, R.G. Intermolecular alignment in β2-microglobulin amyloid fibrils. J. Am. Chem. Soc. 2010, 132, 17077–17079. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M. Protein acrobatics in Pairs—Dimerization via domain swapping. Curr. Opin. Struct. Biol. 2009, 19, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J.; Luoma, A.M. The adaptable major histocompatibility complex (MHC) fold: Structure and function of nonclassical and MHC class I-like molecules. Annu. Rev. Immunol. 2013, 31, 529–561. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Ketteler, M. B2-microglobulin-derived amyloidosis: An update. Kidney Int. Suppl. 2001, 78, S164–S171. [Google Scholar] [CrossRef] [PubMed]
- Verdone, G.; Corazza, A.; Viglino, P.; Pettirossi, F.; Giorgetti, S.; Mangione, P.; Andreola, A.; Stoppini, M.; Bellotti, V.; Esposito, G. The solution structure of human β2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002, 11, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Radford, S.E. Understanding the complex mechanisms of β2-microglobulin amyloid assembly. FEBS J. 2011, 278, 3868–3883. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Mangione, P.; Andreola, A.; Giorgetti, S.; Stefani, M.; Dobson, C.M.; Bellotti, V.; Taddei, N. Detection of two partially structured species in the folding process of the amyloidogenic protein β 2-microglobulin. J. Mol. Biol. 2001, 307, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Kameda, A.; Hoshino, M.; Higurashi, T.; Takahashi, S.; Naiki, H.; Goto, Y. Nuclear magnetic resonance characterization of the refolding intermediate of β2-microglobulin trapped by non-native prolyl peptide bond. J. Mol. Biol. 2005, 348, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.F.; Homans, S.W.; Radford, S.E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. USA 2008, 105, 8926–8931. [Google Scholar] [CrossRef] [PubMed]
- Kad, N.M.; Thomson, N.H.; Smith, D.P.; Smith, D.A.; Radford, S.E. B2-microglobulin and its deamidated variant, N17D form amyloid fibrils with a range of morphologies in vitro. J. Mol. Biol. 2001, 313, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Hagihara, Y.; Higurashi, T.; Hasegawa, K.; Naiki, H.; Goto, Y. Amyloid fibril formation in the context of full-length protein: Effects of proline mutations on the amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 2003, 278, 47016–47024. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Hirakura, Y.; Kagan, B.L. Pore formation by β2-microglobulin: A mechanism for the pathogenesis of dialysis associated amyloidosis. Amyloid 2001, 8, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Mustata, M.; Capone, R.; Jang, H.; Arce, F.T.; Ramachandran, S.; Lal, R.; Nussinov, R. K3 fragment of amyloidogenic β(2)-microglobulin forms ion channels: Implication for dialysis related amyloidosis. J. Am. Chem. Soc. 2009, 131, 14938–14945. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.; Becker, J.; Tidow, H.; Tremmel, S.; Sharpe, T.D.; Krause, G.; Flinders, J.; Petrovich, M.; Berriman, J.; Oschkinat, H.; et al. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA 2006, 103, 16248–16253. [Google Scholar] [CrossRef] [PubMed]
- Saper, M.A.; Bjorkman, P.J.; Wiley, D.C. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J. Mol. Biol. 1991, 219, 277–319. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ohhashi, Y.; Yamaguchi, I.; Takahashi, N.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Amyloidogenic synthetic peptides of β2-microglobulin—A role of the disulfide bond. Biochem. Biophys. Res. Commun. 2003, 304, 101–106. [Google Scholar] [CrossRef]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Manning, J.; Kad, N.M.; Radford, S.E. Amyloid-forming peptides from β2-microglobulin-Insights into the mechanism of fibril formation in vitro. J. Mol. Biol. 2003, 325, 249–257. [Google Scholar] [CrossRef]
- Eakin, C.M.; Miranker, A.D. From chance to frequent encounters: Origins of β2-microglobulin fibrillogenesis. Biochim. Biophys. Acta 2005, 1753, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Thompson, M.J.; Eisenberg, D. A systematic screen of β2-microglobulin and insulin for amyloid-like segments. Proc. Natl. Acad. Sci. USA 2006, 103, 4079–4082. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Hasegawa, K.; Yamaguchi, I.; Tsutsumi, S.; Kardos, J.; Goto, Y.; Gejyo, F.; Naiki, H. Low concentrations of sodium dodecyl sulfate induce the extension of β 2-microglobulin-related amyloid fibrils at a neutral pH. Biochemistry 2004, 43, 11075–11082. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yamaguchi, I.; Hasegawa, K.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of β 2-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 2004, 15, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, Y.; Kihara, M.; Naiki, H.; Goto, Y. Ultrasonication-induced amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 2005, 280, 32843–32848. [Google Scholar] [CrossRef] [PubMed]
- Sasahara, K.; Yagi, H.; Sakai, M.; Naiki, H.; Goto, Y. Amyloid nucleation triggered by agitation of β2-microglobulin under acidic and neutral pH conditions. Biochemistry 2008, 47, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Rennella, E.; Corazza, A.; Giorgetti, S.; Fogolari, F.; Viglino, P.; Porcari, R.; Verga, L.; Stoppini, M.; Bellotti, V.; Esposito, G. Folding and fibrillogenesis: Clues from β2-microglobulin. J. Mol. Biol. 2010, 401, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.W.; Radford, S.E. Glimpses of the molecular mechanisms of β2-microglobulin fibril formation in vitro: Aggregation on a complex energy landscape. FEBS Lett. 2009, 583, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Barbet-Massin, E.; Ricagno, S.; Lewandowski, J.R.; Giorgetti, S.; Bellotti, V.; Bolognesi, M.; Emsley, L.; Pintacuda, G. Fibrillar vs. crystalline full-length β-2-microglobulin studied by high-resolution solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 5556–5557. [Google Scholar] [CrossRef] [PubMed]
- Routledge, K.E.; Tartaglia, G.G.; Platt, G.W.; Vendruscolo, M.; Radford, S.E. Competition between intramolecular and intermolecular interactions in an amyloid-forming protein. J. Mol. Biol. 2009, 389, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Sawaya, M.R.; Gingery, M.; Attinger, A.; Eisenberg, D. An amyloid-forming segment of β2-microglobulin suggests a molecular model for the fibril. Proc. Natl. Acad. Sci. USA 2004, 101, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.W.; Routledge, K.E.; Homans, S.W.; Radford, S.E. Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J. Mol. Biol. 2008, 378, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Estacio, S.G.; Krobath, H.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F. A simulated intermediate state for folding and aggregation provides insights into DeltaN6 β2-microglobulin amyloidogenic behavior. PLoS Comput. Biol. 2014, 10, e1003606. [Google Scholar] [CrossRef] [PubMed]
- Torbeev, V.; Ebert, M.O.; Dolenc, J.; Hilvert, D. Substitution of proline32 by alpha-methylproline preorganizes β2-microglobulin for oligomerization but not for aggregation into amyloids. J. Am. Chem. Soc. 2015, 137, 2524–2535. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Tennent, G.A.; Radford, S.E. A common β-sheet architecture underlies in vitro and in vivo β2-microglobulin amyloid fibrils. J. Biol. Chem. 2008, 283, 17279–17286. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sawaya, M.R.; Eisenberg, D. β2-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat. Struct. Mol. Biol. 2011, 18, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Trinh, C.H.; Smith, D.P.; Kalverda, A.P.; Phillips, S.E.V.; Radford, S.E. Crystal structure of monomeric human β2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. USA 2002, 99, 9771–9776. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.F.; Eakin, C.M.; Wang, J.M.; Miranker, A.D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol. 2008, 15, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Rabzelj, S.; Turk, V.; Zerovnik, E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with EPM1. Protein Sci. 2005, 14, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Skerget, K.; Vilfan, A.; Pompe-Novak, M.; Turk, V.; Waltho, J.P.; Turk, D.; Zerovnik, E. The mechanism of amyloid-fibril formation by stefin B: Temperature and protein concentration dependence of the rates. Proteins 2009, 74, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A. Proline and lysine residues provide modulatory switches in amyloid formation: Insights from prion protein. Prion 2016, 10, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline Isomer-Specific Antibodies Reveal the Early Pathogenic Tau Conformation in Alzheimer’s Disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).