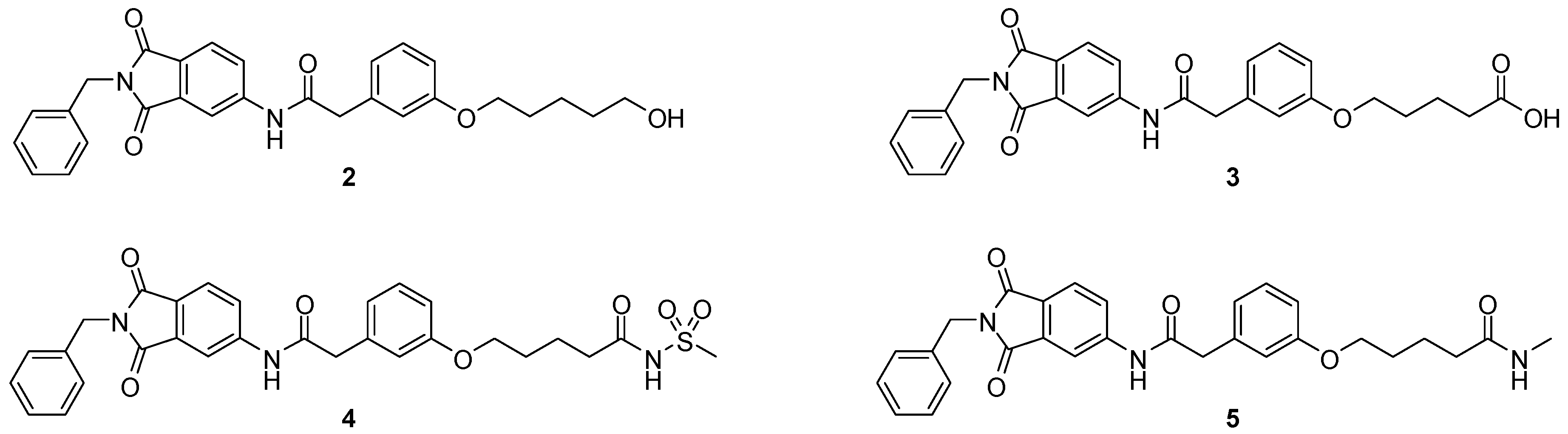
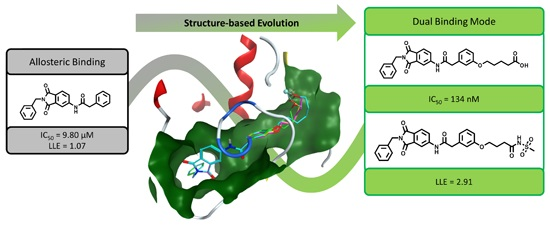
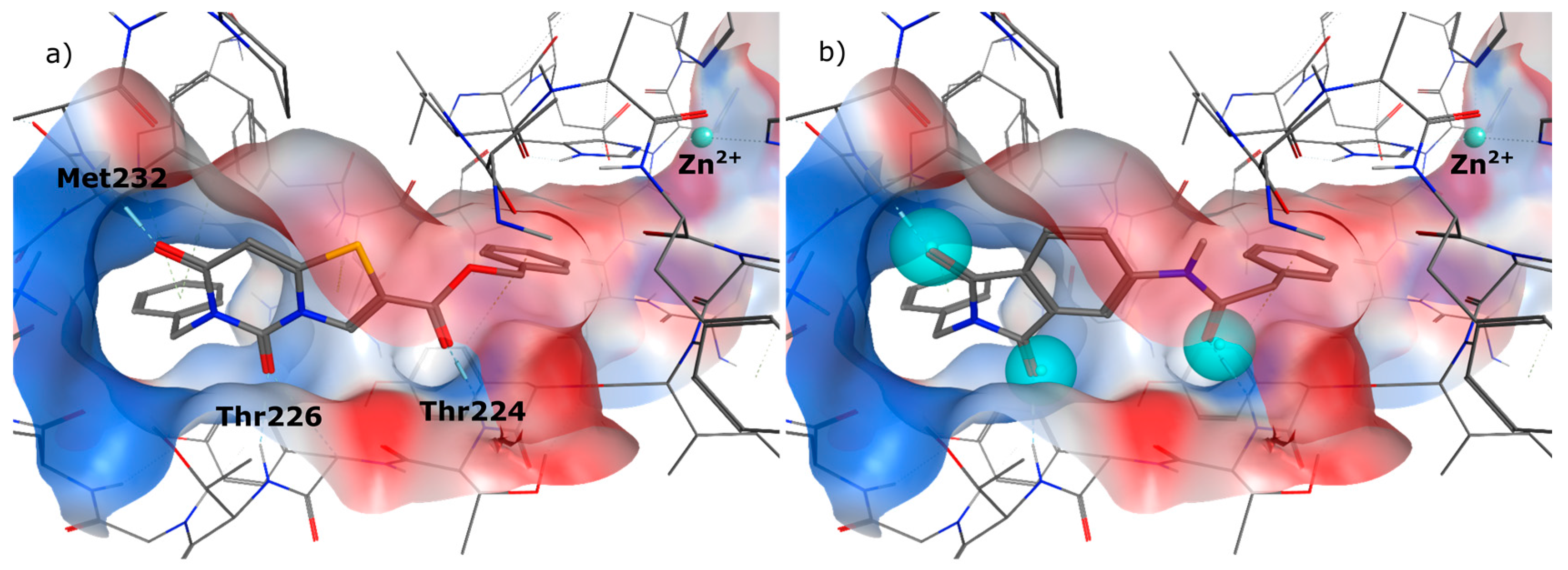
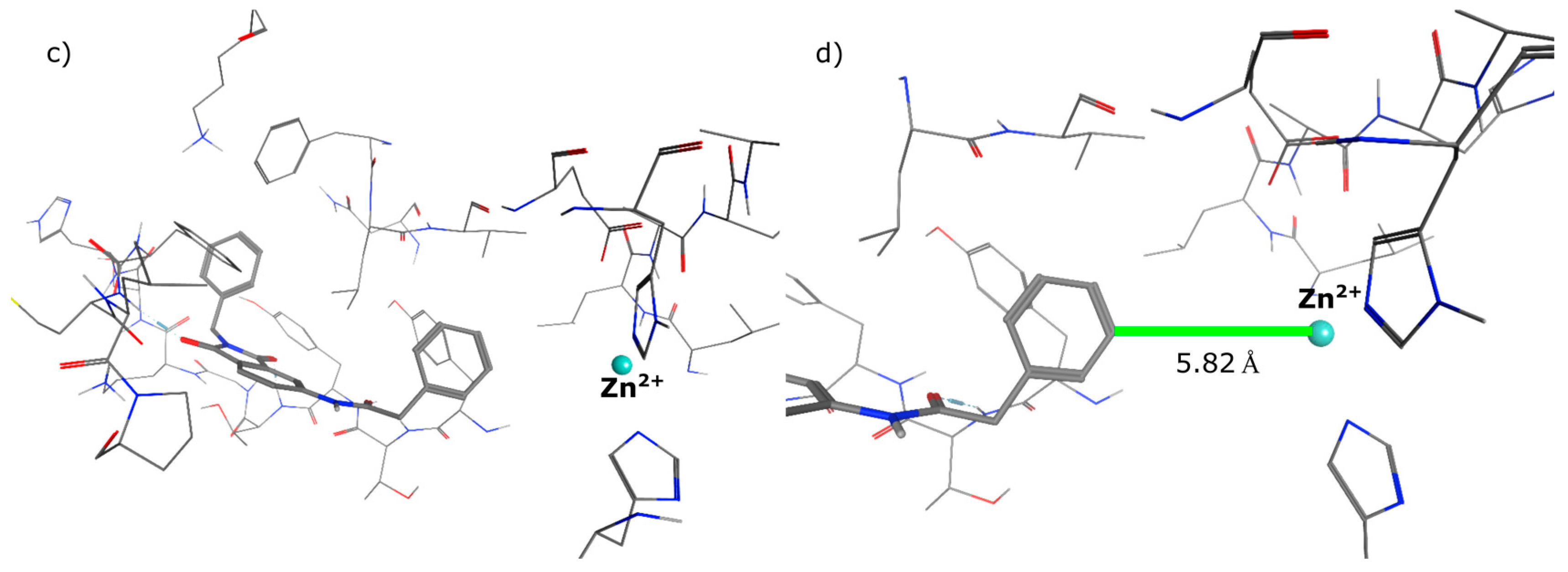
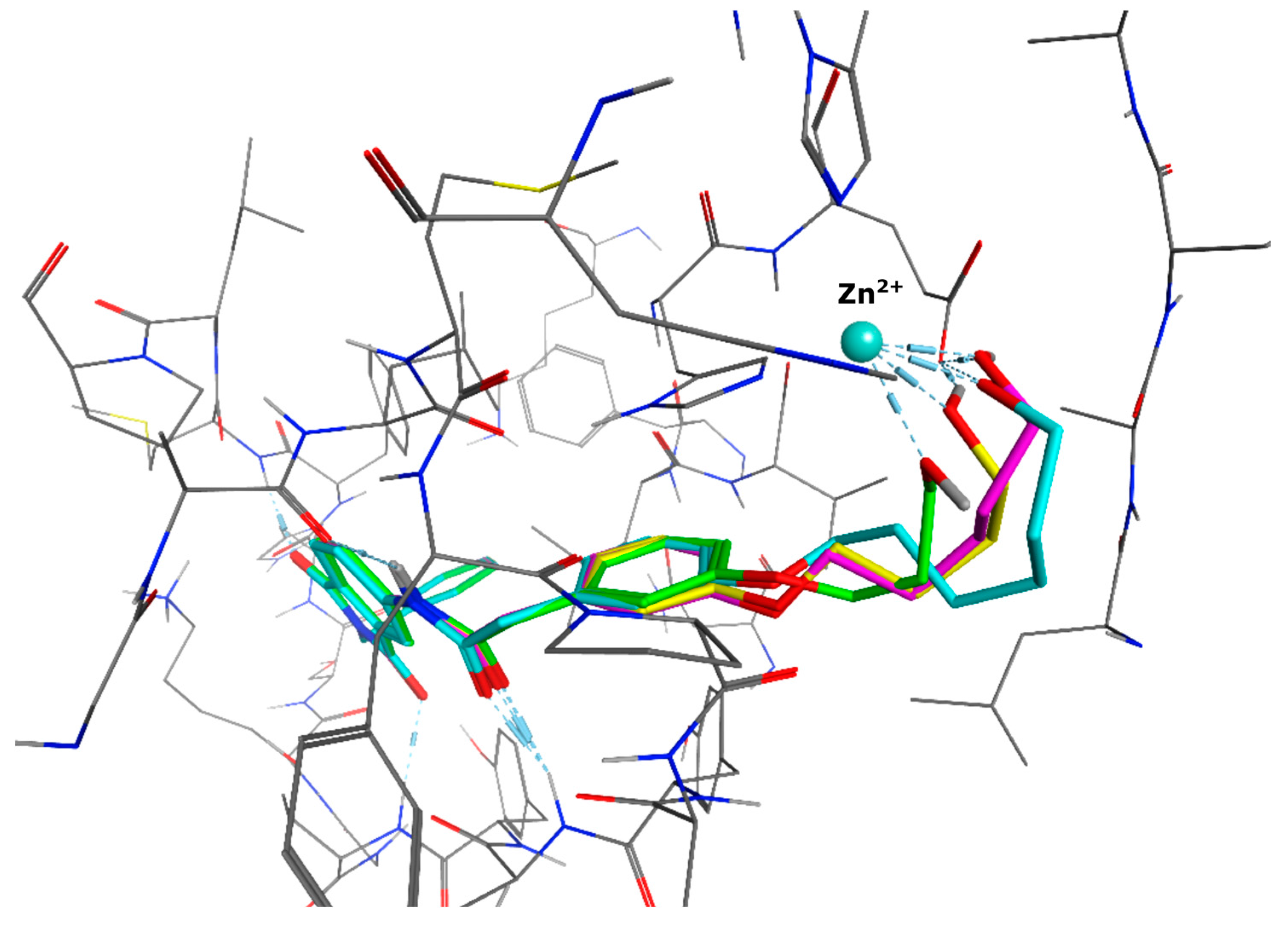
4.2. Chemistry
All reagents and solvents used in the synthesis were purchased from Sigma Aldrich (Buchs, Switzerland) or TCI (Zwijndrecht, Belgium) and used as received. Solvents were stored over molecular sieves 4 Å.
Methyl 2-(3-{[5-(benzyloxy)pentyl]oxy}phenyl)acetate (8c; ZHAWOC4927): under an argon atmosphere, methyl 2-(3-hydroxyphenyl)acetate (7) (2.5 g, 15.05 mmol) and caesium carbonate (9.81 g, 30.11 mmol) were suspended in dimethylformamide (100 mL), the mixture was stirred at ambient temperature for 2 h. Benzyl-5-bromoamylether (4.26 g, 16.56 mmol) was added and it was stirred at ambient temperature for further 12 h. Water (250 mL) and ethyl acetate (250 mL) was added and the resulting phases separated. The organic phase was dried over sodium sulfate and concentrated in vacuum. Purification by chromatography on silica gel (Gradient: 0%–100% ethyl acetate in cyclohexane) afforded the title compound 8c as a white solid (4.30 g, 83% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.31 (4H; m; C-Haromatic), 7.29–7.24 (1H; m; C-Haromatic), 7.20 (1H; t; J = 7.9 Hz; C-Haromatic), 6.85–6.76 (3H; m; C-Haromatic), 4.50 (2H; s; CH2), 3.94 (2H; t; J = 6.5 Hz; CH2), 3.67 (3H; s; CH3), 3.57 (2H; s; CH2), 3.49 (2H; t; J = 6.3 Hz; CH2), 1.82–1.75 (2H; m; CH2), 1.71–1.65 (2H; m; CH2), 1.59–1.51 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.96, 159.28, 138.66, 135.37, 129.56, 128.41, 127.67, 127.55, 121.46, 115.54, 113.19, 72.97, 70.27, 67.77, 52.08, 41.27, 29.57, 29.15, 22.86 ppm. HRMS-TOF: m/z [M + H]+ calculated for C21H26O4: 343.1909, found: 343.1898.
In analogy to 8c the following derivatives were synthesized:
Methyl 2-(3-{[3-(benzyloxy)propyl]oxy}phenyl)acetate (8a; ZHAWOC4557): The title compound 8a was obtained as a white solid in 80% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.29 (4H; m; C-Haromatic), 7.29–7.24 (1H; m; C-Haromatic), 7.24–7.18 (1H; t; m; C-Haromatic), 6.86–6.76 (3H; m; C-Haromatic), 4.51 (2H; s; CH2), 4.06 (2H; t; J = 6.3 Hz; CH2), 3.67 (3H; s; CH3), 3.65 (2H; t; J = 6.3 Hz; CH2), 3.57 (2H; s; CH2), 2.07 (2H; quint; J = 6.19 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.93, 159.20, 138.47, 135.39, 129.56, 128.41, 127.65, 127.59, 121.56, 115.63, 113.21, 73.07, 66.87, 64.82, 52.06, 41.25, 29.80 ppm. MS: m/z [M + Na]+ 337.
Methyl 2-(3-{[4-(benzyloxy)butyl]oxy}phenyl)acetate (8b; ZHAWOC4558): The title compound 8b was obtained as a white solid in 85% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.30 (4H; m; C-Haromatic), 7.30–7.23 (1H; m; C-Haromatic), 7.22–7.16 (1H; t; m; C-Haromatic), 6.85–6.74 (3H; m; C-Haromatic), 4.50 (2H; s; CH2), 3.95 (2H; t; J = 6.14 Hz; CH2), 3.66 (3H; s; CH3), 3.56 (2H; s; CH2), 3.52 (2H; t; J = 6.14 Hz; CH2), 1.93–1.72 (4H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.93, 159.26, 138.63, 135.39, 129.56, 128.41, 127.67, 127.57, 121.49, 115.58, 113.20, 72.94, 69.98, 67.60, 52.04, 41.26, 26.42, 26.22 ppm. MS: m/z [M + Na]+ 351.
Methyl 2-(3-{[6-(benzyloxy)hexyl]oxy}phenyl)acetate (8d; ZHAWOC4928): The title compound 8d was obtained as a white solid in 84% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.31 (4H; m; C-Haromatic), 7.29–7.25 (1H; m; C-Haromatic), 7.21 (1H; t; J = 7.88 Hz; C-Haromatic), 6.85–6.77 (3H; m; C-Haromatic), 4.50 (2H; s; CH2), 3.93 (2H; t; J = 6.5 Hz; CH2), 3.68 (3H; s; CH3), 3.58 (2H; s; CH2), 3.48 (2H; t; J = 6.5 Hz; CH2), 1.81–1.74 (2H; m; CH2), 1.68–1.62 (2H; m; CH2), 1.51–1.42 (4H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.97, 159.29, 138.69, 135.34, 129.54, 128.38, 127.66, 127.52, 121.43, 115.53, 113.19, 72.92, 70.35, 67.81, 52.07, 41.27, 29.74, 29.27, 26.03, 25.96 ppm. MS: m/z [M + Na]+ 379.
2-(3-{[5-(benzyloxy)pentyl]oxy}phenyl)acetic acid (9c; ZHAWOC4929): The ester (8c) (4.30 g, 12.57 mmol) was dissolved in methanol (220 mL) and stirred at ambient temperature. Potassium hydroxide 10% in water (220 mL) was added over 10 min. and the mixture was stirred for another 20 min. Methanol was removed in vacuum and the aqueous phase extracted with diethyl ether (200 mL). The aqueous phase was acidified with concentrated hydrochloric acid and extracted with diethyl ether (200 mL). The second organic phase was dried over sodium sulfate and concentrated in vacuum to obtain the title compound 9c as a white solid (4.13 g, 100% yield) : 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.31 (4H; m; C-Haromatic), 7.30–7.25 (1H; m; C-Haromatic), 7.21 (1H; t; J = 7.9 Hz; C-Haromatic), 6.86–6.77 (3H; m; C-Haromatic), 4.51 (2H; s; CH2), 3.94 (2H; t; J = 6.6 Hz; CH2), 3.59 (2H; s; CH2), 3.50 (2H; t; J = 6.6 Hz; CH2), 1.82–1.75 (2H; m; CH2), 1.72–1.65 (2H; m; CH2), 1.58–1.50 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 177.07, 159.27, 138.54, 134.73, 129.61, 128.40, 127.70, 127.57, 121.56, 115.66, 113.37, 72.94, 70.24, 67.77, 41.09, 29.50, 29.10, 22.80 ppm. HRMS-TOF: m/z [M + H]+ calculated for C20H24O4: 329.1753, found: 329.1748.
In analogy to 9c the following derivatives were synthesized:
2-(3-{[3-(benzyloxy)propyl]oxy}phenyl)acetic acid (9a; ZHAWOC4559): The title compound 9a was obtained as a white solid in 89% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.46 (1H; br. s.; COOH), 7.33–7.17 (6H; m; C-Haromatic), 6.87–6.77 (3H; m; C-Haromatic), 4.51 (2H; s; CH2), 4.05 (2H; t; J = 6.2 Hz; CH2), 3.64 (2H; t; J = 6.2 Hz; CH2), 3.57 (2H; s; CH2), 2.06 (2H; quint; J = 6.2 Hz ; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 177.35, 159.20, 138.33, 134.79, 129.63, 128.43, 127.72, 127.65, 121.70, 115.82, 113.40, 73.06, 66.87, 64.84, 41.13, 29.73 ppm. MS: m/z [M − H]− 299.
2-(3-{[4-(benzyloxy)butyl]oxy}phenyl)acetic acid (9b; ZHAWOC4560): The title compound 9b was obtained as a white solid in quantitative yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.31 (4H; m; C-Haromatic), 7.30–7.25 (1H; m; C-Haromatic), 7.21 (1H; t; J = 7.8 Hz; C-Haromatic), 6.86–6.77 (3H; m; C-Haromatic), 4.51 (2H; s; CH2), 3.96 (2H; t; J = 6.4 Hz; CH2), 3.59 (2H; s; CH2), 3.54 (2H; t; J = 6.4 Hz; CH2), 1.90–1.83 (2H; m; CH2), 1.82–1.75 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 177.27, 159.24, 138.50, 134.69, 129.62, 128.41, 127.71, 127.59, 121.59, 115.68, 113.39, 72.92, 69.93, 67.59, 41.08, 26.35, 26.15 ppm. MS: m/z [M − H]− 313.
2-(3-{[6-(benzyloxy)hexyl]oxy}phenyl)acetic acid (9d; ZHAWOC4930): The title compound 9d was obtained as a white solid in 94% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.35–7.31 (4H; m; C-Haromatic), 7.29–7.24 (1H; m; C-Haromatic), 7.21 (1H; t; J = 7.8 Hz; C-Haromatic), 6.85–6.78 (3H; m; C-Haromatic), 4.50 (2H; s; CH2), 3.92 (2H; t; J = 6.6 Hz; CH2), 3.58 (2H; s; CH2), 3.47 (2H; t; J = 6.6 Hz; CH2), 1.80-1.73 (2H; m; CH2), 1.67–1.61 (2H; m; CH2), 1.50–1.38 (4H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 177.17, 159.30, 138.55, 134.80, 129.61, 128.41, 127.73, 127.59, 121.57, 115.68, 113.40, 72.89, 70.34, 67.84, 41.15, 29.67, 29.24, 26.00, 25.94 ppm. MS: m/z [M − H]− 341.
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-(3-{[5-(benzyloxy)pentyl]oxy}phenyl)acetamide (10c; ZHAWOC5606): The acid (9c) (4.10 g, 12.49 mmol) was stirred in an excess of thionylchloride at 55 °C for 1 h. After removal of excess thionylchloride in vacuum the acid chloride was dissolved in tetrahydrofuran (10 mL) and added to a solution of 5-amino-2-benzylisoindoline-1,3-dione (6) (2.42 g, 9.60 mmol) in tetrahydrofuran (200 mL) under argon at ambient temperature. Diisopropylethylamine (1.90 g, 14.70 mmol) was added and the mixture was stirred at ambient temperature for 2 h. After removal of the tetrahydrofuran in vacuum, ethyl acetate (200 mL) and 10% citric acid (200 mL) were added and the resulting phases were separated. The organic phase was washed with 10% sodium bicarbonate (200 mL) and brine (200 mL), dried over sodium sulfate and concentrated in vacuum. Purification by chromatography on silica gel (Gradient: 0%–100% ethyl acetate in cyclohexane) afforded the title compound 10c as a yellow solid (3.56 g, 66% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 7.89 (1H; d; J = 2.1 Hz; C-Haromatic), 7.84 (1H; dd; J = 8.2 Hz, 2.1 Hz; C-Haromatic), 7.74 (1H; d; J = 8.2 Hz; C-Haromatic), 7.71 (1H; br. s; N-H), 7.43–7.39 (2H; m; C-Haromatic), 7.37–7.24 (9H; m; C-Haromatic), 6.93–6.81 (3H; m; C-Haromatic), 4.82 (2H; s; CH2), 4.53 (2H; m; CH2), 3.98 (2H; t; J = 6.5 Hz; CH2), 3.74 (2H; s; CH2), 3.53 (2H; t; J = 6.5 Hz; CH2), 1.87–1.77 (2H; m; CH2), 1.76–1.67 (2H; m; CH2), 1.63––.53 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.22, 169.33, 167.53, 159.83, 143.21, 136.35, 130.47, 128.66, 128.49, 128.38, 127.79, 127.65, 127.56, 124.45, 121.41, 115.83, 114.08, 113.76, 72.97, 70.23, 67.88, 44.88, 41.64, 29.52, 29.06, 22.84 ppm. HRMS-TOF: m/z [M + H]+ calculated for C35H34N2O5: 563.2546, found: 563.2542.
In analogy to 10c the following derivatives were synthesized:
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-(3-{[3-(benzyloxy)propyl]oxy}phenyl)acetamide (10a; ZHAWOC4754): The title compound 10a was obtained as a yellow solid in 74% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.08 (1H; s; NH), 7.90 (1H; d; J = 1.8 Hz; C-Haromatic), 7.82 (1H; dd; J = 8.2 Hz, 1.8 Hz; C-Haromatic), 7.68 (1H; d; J = 8.2 Hz; C-Haromatic), 7.39–7.19 (11H; m; C-Haromatic), 6.89–6.76 (3H; m; C-Haromatic), 4.78 (2H; s; CH2), 4.50 (2H; m; CH2), 4.05 (2H; t; J = 6.2 Hz; CH2), 3.67 (2H; s; CH2), 3.64 (2H; t; J = 6.2 Hz; CH2), 2.11–2.02 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 171.27, 169.55, 167.62, 167.57, 159.62, 143.46, 138.33, 136.36, 135.22, 133.51, 130.29, 128.66, 128.43, 128.39, 127.79, 127.61, 126.78, 124.42, 123.85, 121.47, 115.82, 114.14, 113.64, 73.04, 66.78, 64.92, 44.72, 41.62, 29.68 ppm. MS: m/z [M − H]− 533; [M + Na]+ 557.
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-(3-{[4-(benzyloxy)butyl]oxy}phenyl)acetamide (10b; ZHAWOC4755): The title compound 10b was obtained as a yellow solid in 64% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.21 (1H; s; NH), 7.91 (1H; d; J = 1.9 Hz; C-Haromatic), 7.83 (1H; dd; J = 8.2 Hz, 1.9 Hz; C-Haromatic), 7.68 (1H; d; J = 8.2 Hz; C-Haromatic), 7.40–7.19 (11H; m; C-Haromatic), 6.87–6.77 (3H; m; C-Haromatic), 4.78 (2H; s; CH2), 4.50 (2H; m; CH2), 3.94 (2H; t; J = 6.0 Hz; CH2), 3.66 (2H; s; CH2), 3.53 (2H; t; J = 6.0 Hz; CH2), 1.92–1.71 (4H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 169.63, 167.63, 167.58, 159.64, 143.53, 138.48, 136.37, 135.26, 133.49, 130.22, 128.66, 128.42, 128.38, 127.78, 127.64, 127.57, 126.74, 124.40, 123.86, 121.38, 115.79, 114.15, 113.51, 72.92, 69.93, 47.66, 44.68, 41.61, 26.34, 26.15 ppm. MS: m/z [M − H]− 547; [M + Na]+ 571.
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-(3-{[4-(benzyloxy)butyl]oxy}phenyl)acetamide (10d): The title compound 10d was prepared from the acid 9d and used directly for further synthesis without purification.
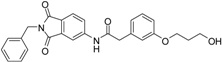
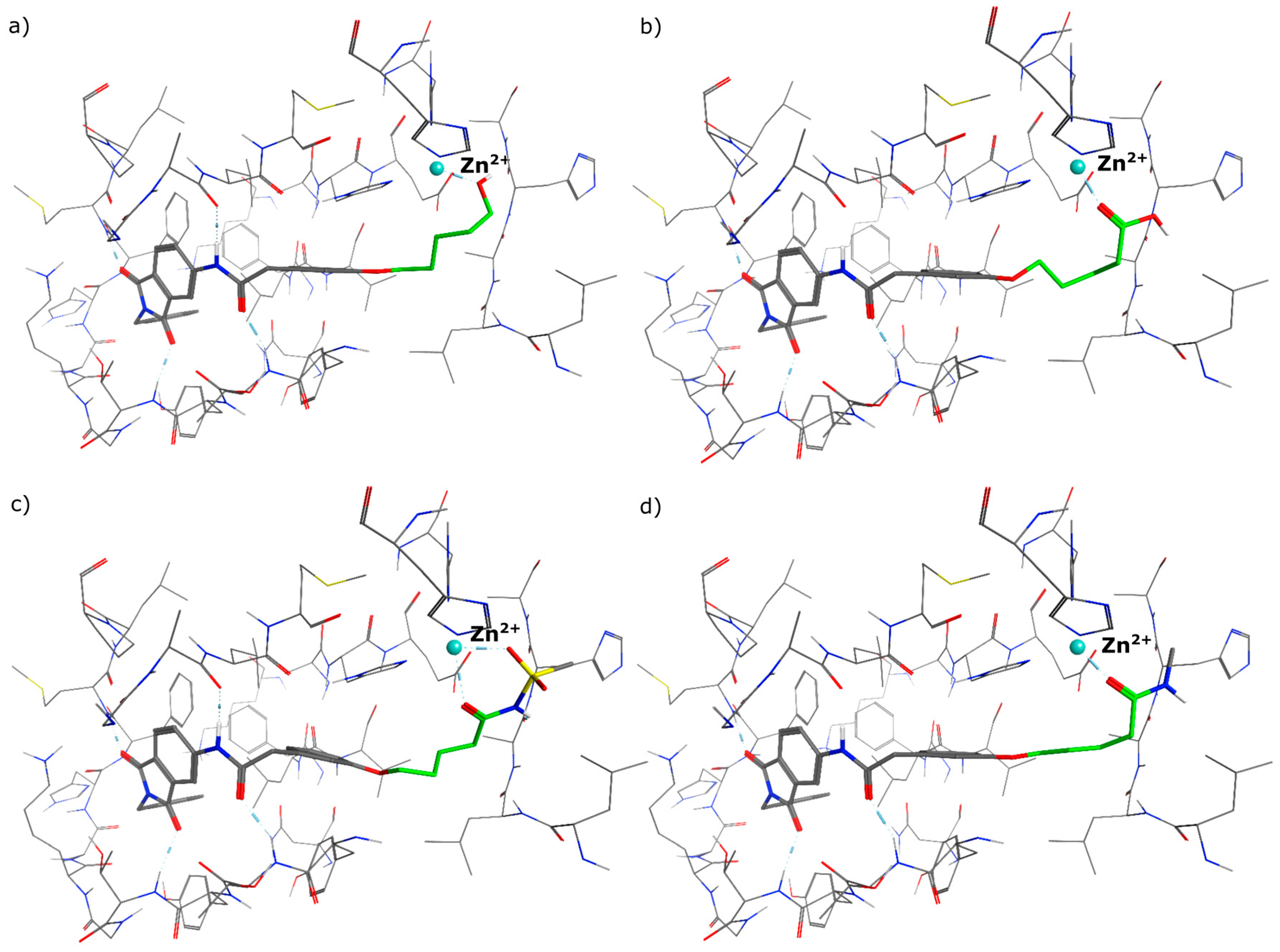
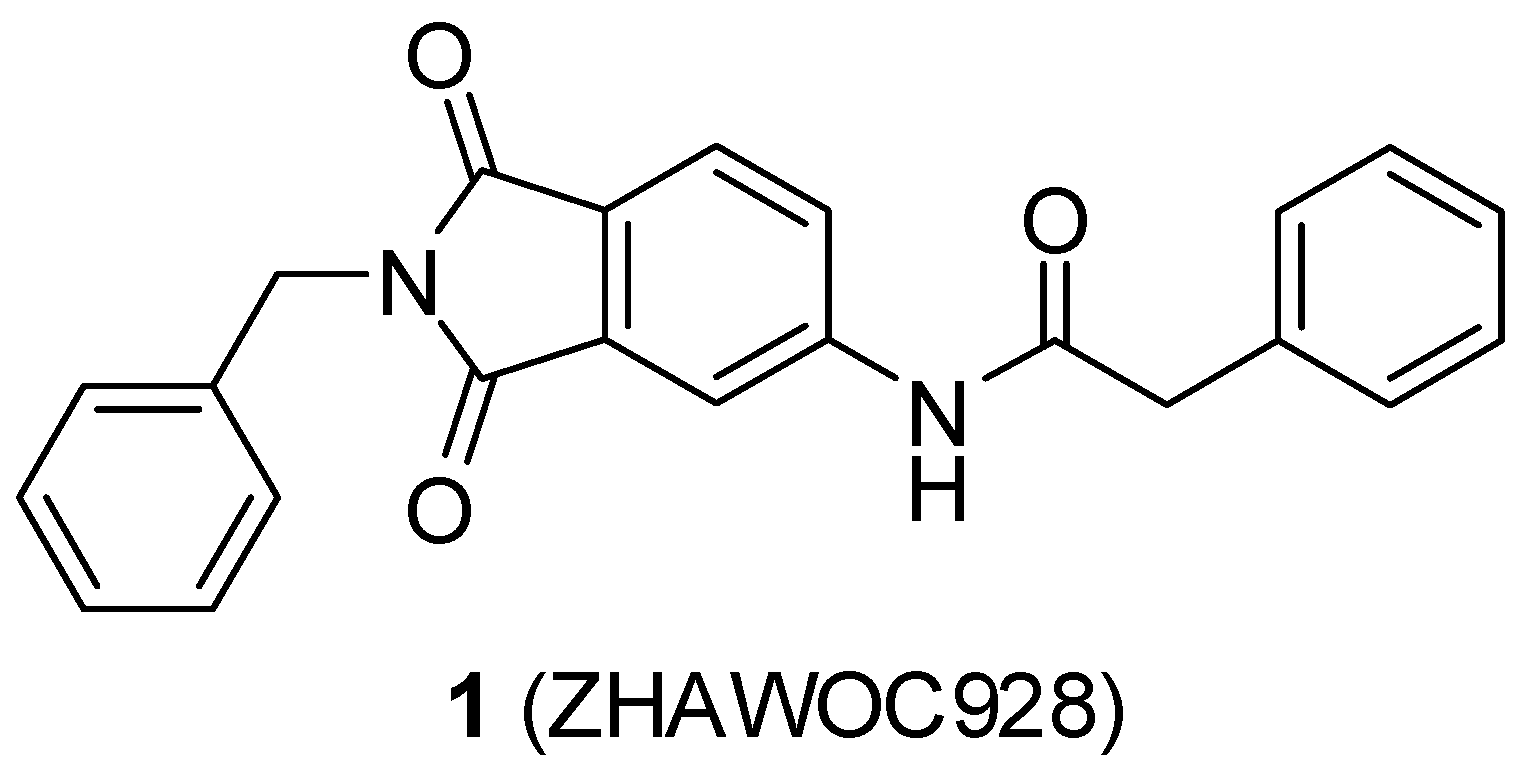
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-{3-[(5-hydroxypentyl)oxy]phenyl}acetamide (2; ZHAWOC5041): The benzyl ether (10c) (3.30 g, 5.87 mmol) was dissolved in ethanol (300 mL). Palladium 10% on activated charcoal (0.70 g, 0.59 mmol) was added and a hydrogen atmosphere was applied at 1 bar. After stirring at ambient temperature for 2 h the mixture was filtered over celite and concentrated in vacuum. Purification by chromatography on silica gel (Gradient: 0%–100% ethyl acetate in cyclohexane) afforded the title compound 2 as a white solid (2.30 g, 83% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.99 (1H; s; N-H), 7.98 (1H; d; J = 2 Hz; C-Haromatic), 7.83 (1H; dd; J = 8.2 Hz, 2 Hz; C-Haromatic), 7.64 (1H; d; J = 8.2 Hz; C-Haromatic), 7.37–7.32 (2H; m; C-Haromatic), 7.28–7.12 (4H; m; C-Haromatic), 6.84–6.69 (3H; m; C-Haromatic), 4.76 (2H; s; CH2), 3.85 (2H; t; J = 6.3 Hz; CH2), 3.64 (4H; br. s; 2 x CH2), 2.21 (1H; br. s; O-H), 1.77–1.69 (2H, m; CH2), 1.63–1.55 (2H; m; CH2), 1.53–1.44 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.14, 167.68, 167.65, 159.38, 143.87, 136.33, 135.48, 133.34, 129.90, 128.61, 128.33, 127.74, 124.28, 123.93, 121.28, 112.72, 114.22, 113.08, 67.69, 62.48, 44.41, 41.54, 32.31, 28.94, 22.40 ppm. HRMS-TOF: m/z [M + H]+ calculated for C28H28N2O5: 473.2076, found: 473.2076.
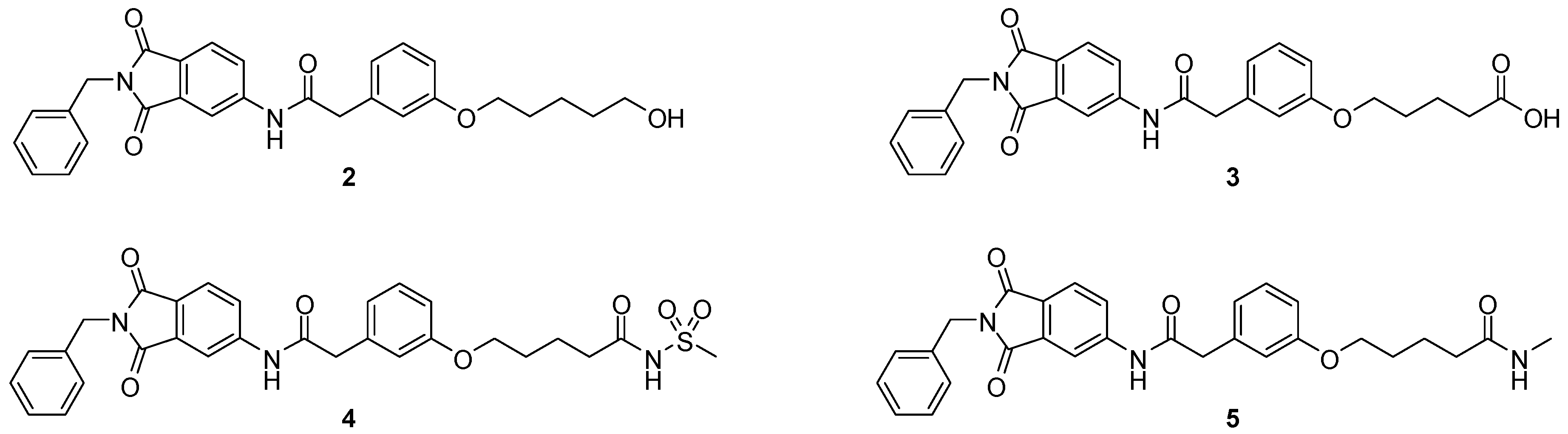
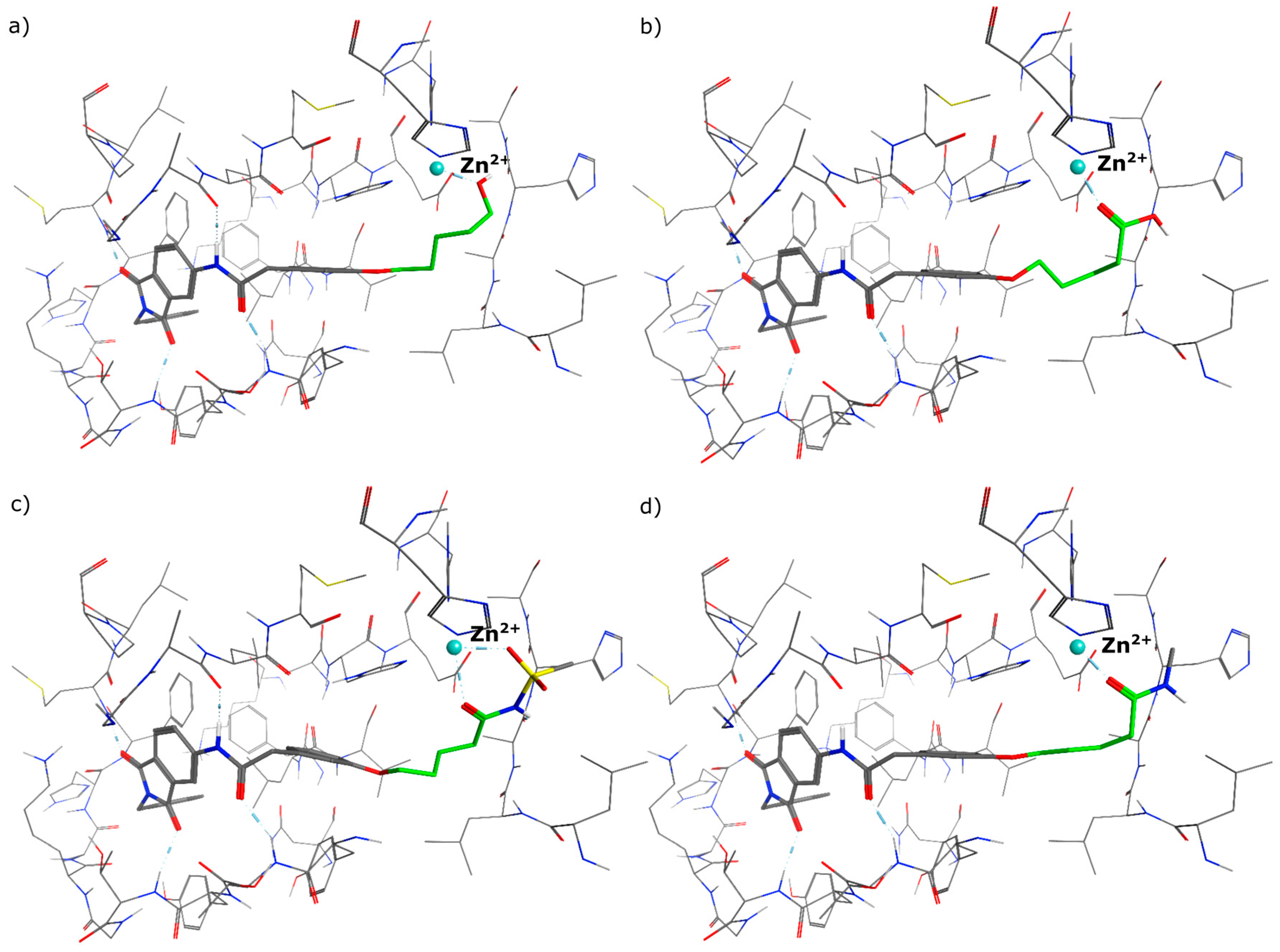
5-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)pentanoic acid (3; ZHAWOC5077): The alcohol (2) (2.20 g, 4.66 mmol), TEMPO (0.07 g, 0.352 mmol) and 0.67 M aqueous sodium phosphate (22 mL) in acetonitrile (26.4 mL) were stirred and heated to 40 °C. NaClO2 (1.12 g, 9.90 mmol in 5 mL water) and 10% NaOCl (2.2 mL) were added in parallel dropwise and it was stirred for 4 h. After cooling to ambient temperature water (20 mL) was added and the mixture was poured in an ice could solution of Na2SO3 (1.8 g in 30 mL water). The aqueous phase was extracted with diethylether (80 mL), acidified with 10% citric acid and again extracted with diethylether (2 × 100 mL). The organic phases were dried over sodium sulfate and concentrated in vacuum to obtain the title compound 3 as a white solid (1.20 g, 53% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 12.05 (1H; br. s; COOH), 10.77 (1H; s; N-H), 8.22 (1H; d; J = 1.6 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.2 Hz, 1.6 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.35–7.21 (6H; m; C-Haromatic), 6.93–6.80 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.96 (2H; t; J = 6.2 Hz; CH2), 3.69 (2H; s; CH2), 2.28 (2H; t; J = 7.5 Hz; CH2), 1.76–1.69 (2H; m; CH2), 1.68–1.60 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 174.82, 170.44, 167.93, 167.76, 159.08, 145.23, 137.21, 137.14, 129.85, 129.04, 127.85, 127.82, 125.78, 124.93, 123.86, 121.75, 116.00, 113.36, 113.04, 67.48, 43.86, 41.31, 33.79, 28.61, 21.70 ppm. HRMS-TOF: m/z [M + H]+ calculated for C28H26N2O6: 487.1869, found: 487.1850.
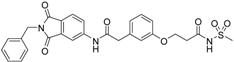
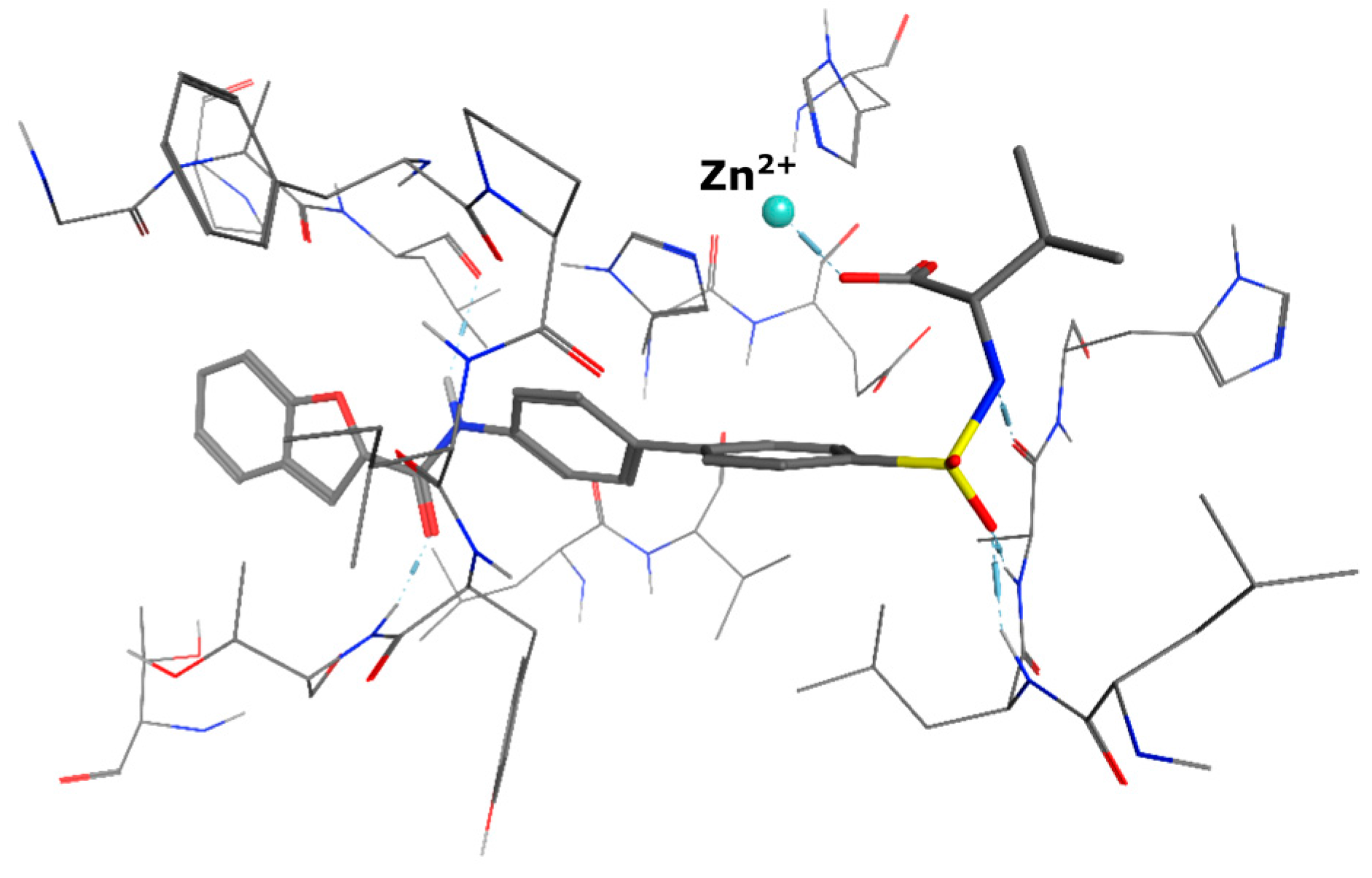
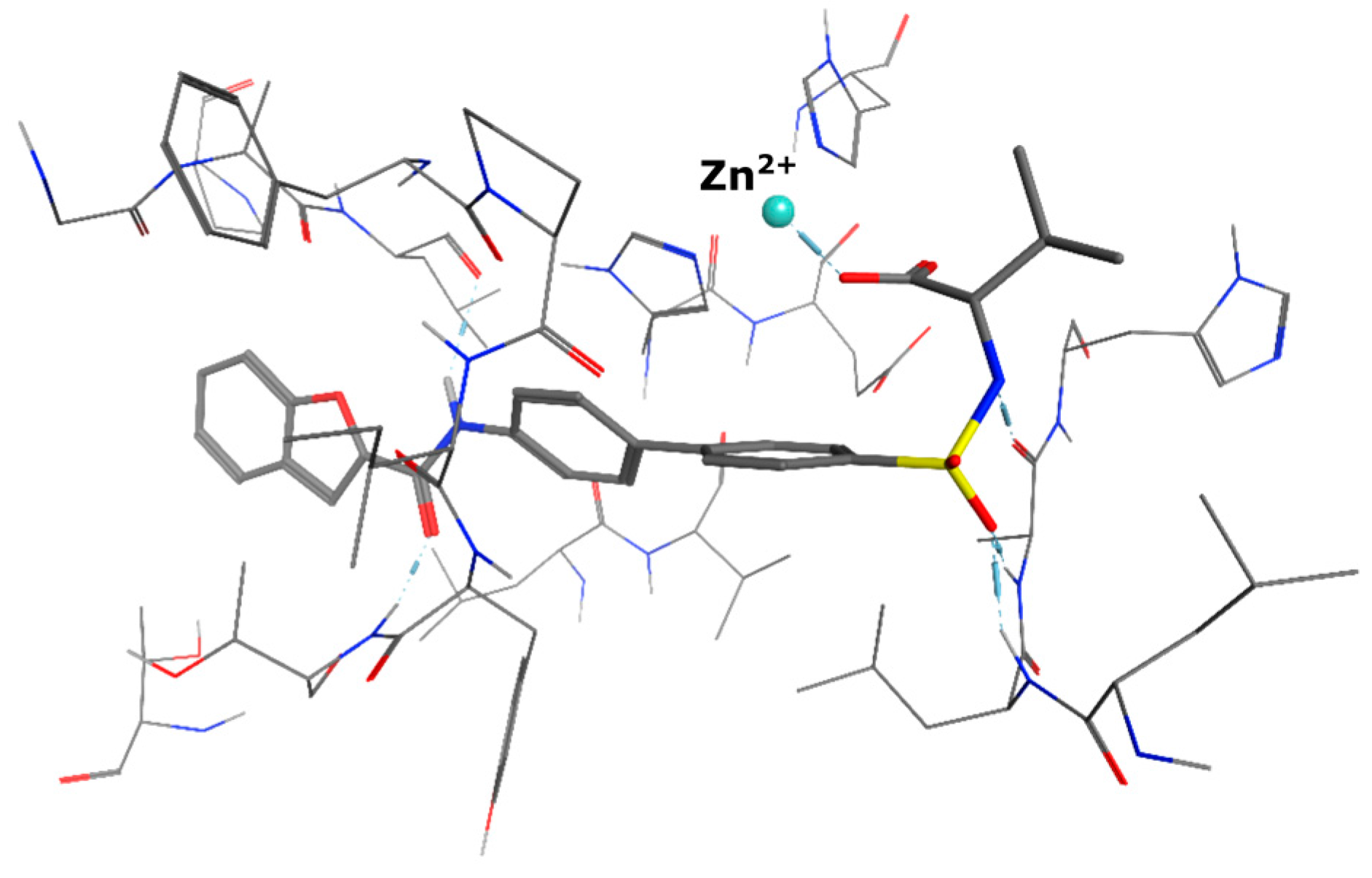
5-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)-N-methanesulfonylpentanamide (4; ZHAWOC5079): The acid (3) (106 mg, 0.218 mmol) was dissolved in dichloromethane (5 mL) and cooled to 0 °C. EDC·HCl (63 mg, 0.327 mmol), methansulfonamide (23 mg, 0.240 mmol) and DMAP (40 mg, 0.327 mmol) were added and stirred at 0 °C for further 20 min. After warming up to ambient temperature it was stirred for another 12 h. The reaction was quenched by adding 10% citric acid (5 mL). The precipitate was filtered off and dissolved in DMSO (1 mL). Purification by reversed phase chromatography on silica gel C18 (Gradient: 0%–100% methanol in water) afforded the title compound 4 as a white solid (50 mg, 41% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 11.68 (1H; s; N-H), 10.76 (1H; s; N-H), 8.22 (1H; d: J = 1.9 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.5 Hz, 1.9 Hz; C-Haromatic), 7.85 (1H; d; J = 8.5 Hz; C-Haromatic), 7.35–7.21 (6H; m; C-Haromatic), 6.94–6.81 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.96 (2H; t; J = 5.7 Hz; CH2), 3.69 (2H; s; CH2), 3.22 (3H; s ; CH3), 2.35 (2H; t; J = 7.2 Hz; CH2), 1.76–1.60 (4H; m; 2 × CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 172.94, 170.43, 167.93, 167.76, 159.05, 145.22, 137.21, 137.15, 133.55, 129.85, 129.04, 127.86, 127.83, 125.79, 124.94, 123.85, 121.80, 115.99, 113.35, 113.06, 67.37, 43.84, 41.45, 41.32, 35.47, 28.42, 21.29 ppm. HRMS-TOF: m/z [M + H]+ calculated for C29H29N3O7S: 564.1804, found: 564.1808.
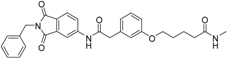
5-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)-N-methylpentanamide (5; ZHAWOC5080): The acid (3) (100 mg, 0.206 mmol) was dissolved in tetrahydrofuran (3 mL) and diisopropylethylamine (138 mg, 1.070 mmol), HATU (215 mg, 0.565 mmol) such as methylamine 2 M in THF (0.206 mL, 0.412 mmol) were added. The mixture was stirred at ambient temperature for 18 h before it was concentrated in vacuum and purified by chromatography on silica gel (Gradient: 0%–100% ethyl acetate in Cyclohexane) to obtain the title compound 5 as a white solid (40 mg, 39% yield): 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.76 (1H; s; N-H), 8.21 (1H; d; J = 1.9 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.2 Hz, 1.9 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.71 (1H; br. q; J = 4.5 Hz; N-H), 7.35–7.20 (6H; m; C-Haromatic), 6.92–6.79 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.94 (2H; t; J = 6 Hz; CH2), 3.68 (2H; s; CH2), 2.55 (3H; d; J = 4.5 Hz; CH3), 2.11 (2H; t; J = 7.3 Hz; CH2), 1.72–1.58 (4H; m; 2 × CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 172.71, 170.44, 167.93 167.76, 159.09, 145.23, 137.21, 137.14, 129.84, 129.04, 127.86, 127.83, 125.79, 124.93, 123.85, 121.74, 115.98, 113.36, 113.05, 67.46, 43.85, 41.32, 35.38, 28.78, 25.87, 22.36 ppm. HRMS-TOF: m/z [M + H]+ calculated for C29H29N3O5: 500.2185, found: 500.2181.
The following alcohols were synthesized according to compound (2):
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-[3-(3-hydroxypropoxy)phenyl]acetamide (11; ZHAWOC4756): The title compound was isolated as a white solid in 57% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.95 (1H; s; N-H), 7.95 (1H; d; J = 1.7 Hz; C-Haromatic), 7.77 (1H; dd; J = 8.2 Hz, 1.7 Hz; C-Haromatic), 7.60 (1H; d; J = 8.2 Hz; C-Haromatic), 7.38–7.05 (6H; m; C-Haromatic), 6.82–6.64 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.97 (2H; t; J = 6 Hz; CH2), 3.77 (2H; t; J = 5.9 Hz; CH2), 3.58 (2H; s; CH2), 2.95 (1H; br. s; O-H), 1.94 (2H; quint.; J = 5.9 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.31, 167.82, 167.73, 159.19, 143.74, 136.27, 135.46, 133.32, 130.02, 128.69, 128.34, 127.84, 126.55, 124.36, 124.08, 121.57, 115.70, 114.32, 113.20, 65.09, 59.68, 44.37, 41.62, 31.97 ppm. HRMS-TOF: m/z [M + H]+ calculated for C26H24N2O5: 445.1763, found: 445.1776.
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-[3-(4-hydroxybutoxy)phenyl]acetamide (12; ZHAWOC4757): The title compound was isolated as a white solid in 48% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.93 (1H; s; N-H), 7.96 (1H; d; J = 1.75 Hz; C-Haromatic), 7.80 (1H; dd; J = 8.2 Hz, 1.75 Hz; C-Haromatic), 7.62 (1H; d; J = 8.2 Hz; C-Haromatic), 7.38–7.10 (6H; m; C-Haromatic), 6.85–6.66 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.87 (2H; t; J = 6 Hz; CH2), 3.66 (2H; t; J = 6.3 Hz; CH2), 3.61 (2H; s; CH2), 2.84 (1H; br. s; O-H), 1.85–1.73 (2H; m; CH2), 1.73–1.60 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.26, 167.82, 167.72, 159.27, 143.78, 136.28, 135.47, 133.35, 130.04, 128.68, 128.34, 127.83, 126.55, 124.38, 124.06, 121.50, 115.83, 114.32, 113.18, 67.70, 62.35, 44.42, 41.61, 29.30, 25.76 ppm. HRMS-TOF: m/z [M + H]+ calculated for C27H26N2O5: 459.1920, found: 459.1922.
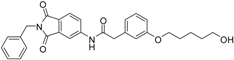
N-(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)-2-{3-[(6-hydroxyhexyl)oxy]phenyl}acetamide (13; ZHAWOC5042): The title compound was isolated as a white solid in 77% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.74 (1H; s; N-H), 7.94 (1H; d; J = 1.8 Hz; C-Haromatic), 7.82 (1H; dd; J = 8.2 Hz, 1.8 Hz; C-Haromatic), 7.65 (1H; d; J = 8.2 Hz; C-Haromatic), 7.38–7.32 (2H; m; C-Haromatic), 7.29–7.15 (4H; m; C-Haromatic), 6.85–6.72 (3H; m; C-Haromatic), 4.76 (2H; s; CH2), 3.86 (2H; t; J = 6.4 Hz; CH2), 3.64 (2H; s; CH2), 3.63 (2H; t; J = 6.5 Hz; CH2), 2.41 (1H; br. s; O-H), 1.76–1.67 (2H; m; ; CH2), 1.60–1.53 (2H; m; ; CH2), 1.47–1.33 (4H; m; ; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.08, 167.74, 167.67, 159.51, 143.74, 136.30, 135.38, 133.38, 130.06, 128.67, 128.36, 127.80, 126.58, 124.38, 121.33, 115.76, 114.25, 113.30, 67.78, 62.71, 44.53, 41.61, 32.58, 29.13, 25.82, 25.53 ppm. HRMS-TOF: m/z [M + H]+ calculated for C29H30N2O5: 487.2233, found: 487.2211.
The following acids were synthesized according to compound (3):
3-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)propanoic acid (14; ZHAWOC4767): The title compound was isolated as a white solid in 51% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 12.35 (1H; br. s; COOH), 10.76 (1H; s; N-H), 8.21 (1H; d; J = 1.7 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.2 Hz, 1.7 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.36–7.19 (6H; m; C-Haromatic), 6.96–6.78 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 4.15 (2H; t; J = 6 Hz; CH2), 3.69 (2H; s; CH2), 2.69 (2H; t; J = 6 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 172.71, 170.42, 167.93, 167.76, 158.80, 145.23, 137.21, 133.55, 129.89, 129.04, 127.83, 125.79, 124.93, 123.86, 122.00, 115.91, 113.37, 113.10, 63.95, 43.83, 41.31, 34.56 ppm. HRMS-TOF: m/z [M + H]+ calculated for C26H22N2O6: 459.1556, found: 459.1557.
4-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)butanoic acid (15; ZHAWOC4768): The title compound was isolated as a white solid in 84% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 12.12 (1H; br. s; COOH), 10.76 (1H; s; N-H), 8.21 (1H; d; J = 1.7 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.2 Hz, 1.7 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.37–7.18 (6H; m; C-Haromatic), 6.95–6.78 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.97 (2H; t; J = 6.4 Hz; CH2), 3.68 (2H; s; CH2), 2.38 (2H; t; J = 7.4 Hz; CH2), 1.93 (2H; quint.; J = 6.7 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 174.62, 170.44, 167.93, 167.76, 158.97, 145.24, 137.21, 137.17, 129.86, 129.04, 127.85, 127.82, 124.92, 123.86, 121.83, 115.95, 113.37, 113.11, 66.97, 43.84, 41.31, 30.72, 24.80 ppm. HRMS-TOF: m/z [M + H]+ calculated for C27H24N2O6: 473.1712, found: 473.1709.
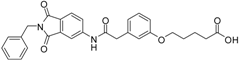
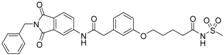
6-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)hexanoic acid (16; ZHAWOC5078): The title compound was isolated as a white solid in 87% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 11.82 (1H; br. s; COOH), 10.78 (1H; s; N-H), 8.21 (1H; d; J = 1.8 Hz; C-Haromatic), 7.90 (1H; dd; J = 8.2 Hz, 1.8 Hz; C-Haromatic), 7.83 (1H; d; J = 8.2 Hz; C-Haromatic), 7.35–7.19 (6H; m; C-Haromatic), 6.93–6.78 (3H; m; C-Haromatic), 4.73 (2H; s; CH2), 3.93 (2H; t; J = 6.3 Hz; CH2), 3.68 (2H; s; CH2), 2.22 (2H; t; J = 7.3 Hz; CH2), 1.73–1.66 (2H; m; CH2), 1.59–1.51 (2H; m; CH2), 1.45–1.37 (2H; m; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 174.94, 170.45, 167.92, 167.75, 159.11, 145.24, 137.20, 137.14, 129.84, 129.04, 127.85, 127.82, 125.77, 124.91, 123.85, 121.72, 115.96, 113.36, 113.05, 67.67, 43.86, 41.31, 34.19, 28.93, 25.65, 24.79 ppm. HRMS-TOF: m/z [M + H]+ calculated for C29H28N2O6: 501.2025, found: 501.2022.
The following acyl sulfonamides were synthesized according to compound (4):
3-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)-N-methanesulfonylpropanamide (17; ZHAWOC5136): The title compound was isolated as a white solid in 19% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 11.86 (1H; s; N-H), 10.77 (1H; s; N-H), 8.21 (1H; d: J = 1.9 Hz; C-Haromatic), 7.89 (1H; dd; J = 8.2 Hz, 1.9 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.35–7.21 (6H; m; C-Haromatic), 6.94–6.81 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 4.18 (2H; t; J = 6 Hz; CH2), 3.69 (2H; s; CH2), 3.22 (3H; s ; CH3), 2.75 (2H; t; J = 6 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.90, 170.41, 167.93, 167.76, 158.72, 145.22, 137.21, 133.55, 129.90, 129.04, 127.86, 127.82, 125.80, 124.94, 123.86, 122.10, 115.99, 113.16, 63.33, 43.82, 41.46, 41.32, 36.15 ppm. HRMS-TOF: m/z [M + H]+ calculated for C27H25N3O7S: 536.1491, found: 536.1483.
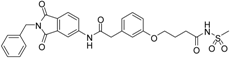
4-(3-{[(2-benzyl-1,3-dioxo-2,3-dihydro-1H-isoindol-5-yl)carbamoyl]methyl}phenoxy)-N-methanesulfonylbutanamide (18; ZHAWOC5135): The title compound was isolated as a white solid in 26% yield: 1H-NMR (500 MHz, [D6]DMSO, 25 °C, TMS): δ = 11.71 (1H; s; N-H), 10.77 (1H; s; N-H), 8.21 (1H; d: J = 1.8 Hz; C-Haromatic), 7.89 (1H; dd; J = 8.2 Hz, 1.8 Hz; C-Haromatic), 7.84 (1H; d; J = 8.2 Hz; C-Haromatic), 7.35–7.21 (6H; m; C-Haromatic), 6.93–6.80 (3H; m; C-Haromatic), 4.74 (2H; s; CH2), 3.96 (2H; t; J = 6.2 Hz; CH2), 3.68 (2H; s; CH2), 3.21 (3H; s ; CH3), 2.44 (2H; t; J = 7.4 Hz; CH2), 1.95 (2H; quint.; J = 6.7 Hz; CH2) ppm. 13C-NMR (125 MHz, [D6]DMSO, 25 °C, TMS): δ = 172.77, 170.42, 167.93, 167.76, 158.92, 145.22, 137.21, 137.18, 133.55, 129.87, 129.04, 127.86, 127.83, 125.80, 124.94, 123.86, 121.90, 115.93, 113.36, 113.13, 66.89, 43.83, 41.46, 41.32, 32.55, 24.15 ppm. HRMS-TOF: m/z [M + H]+ calculated for C28H27N3O7S: 550.1648, found: 550.1640.




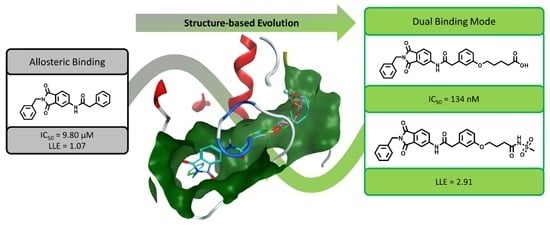
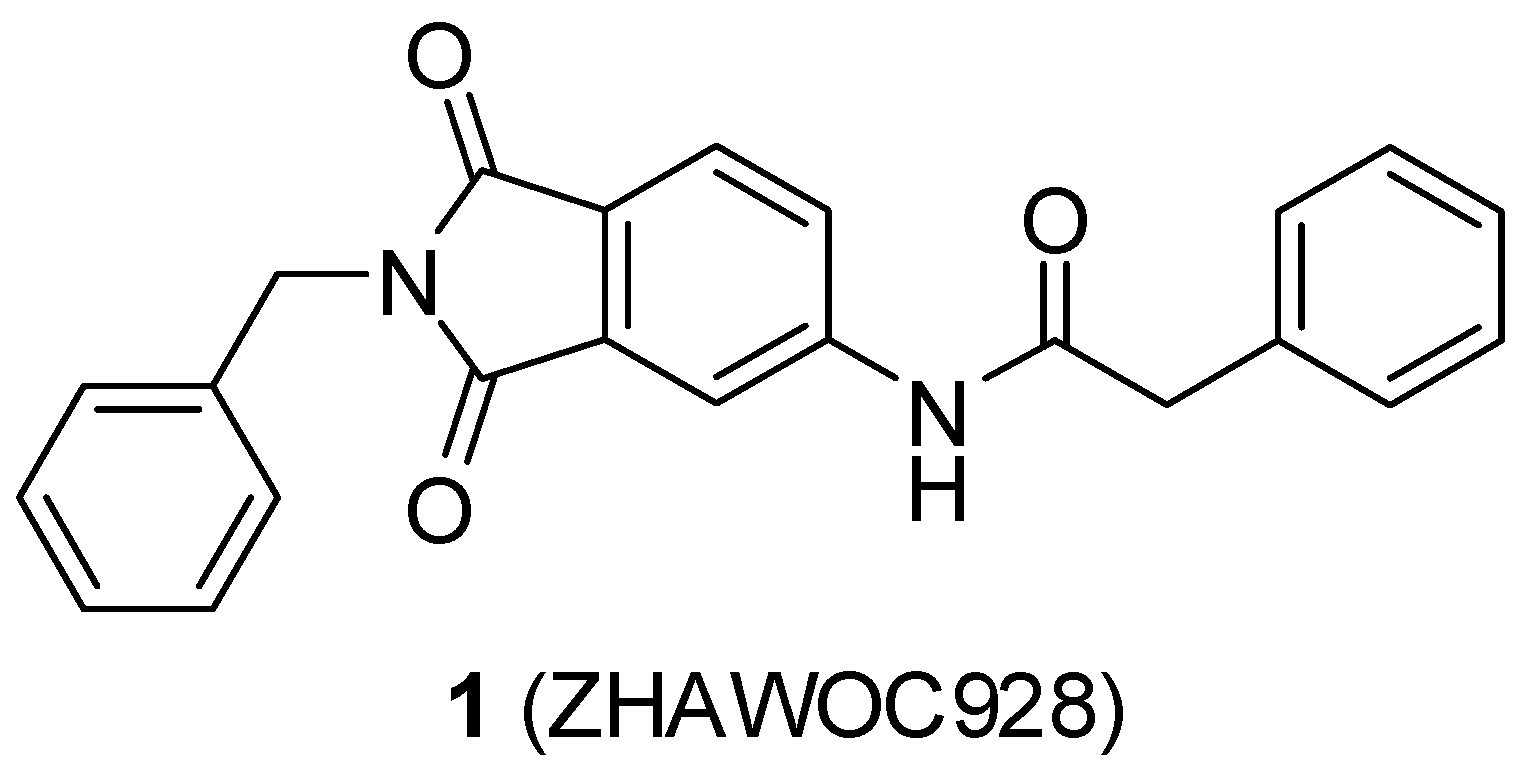

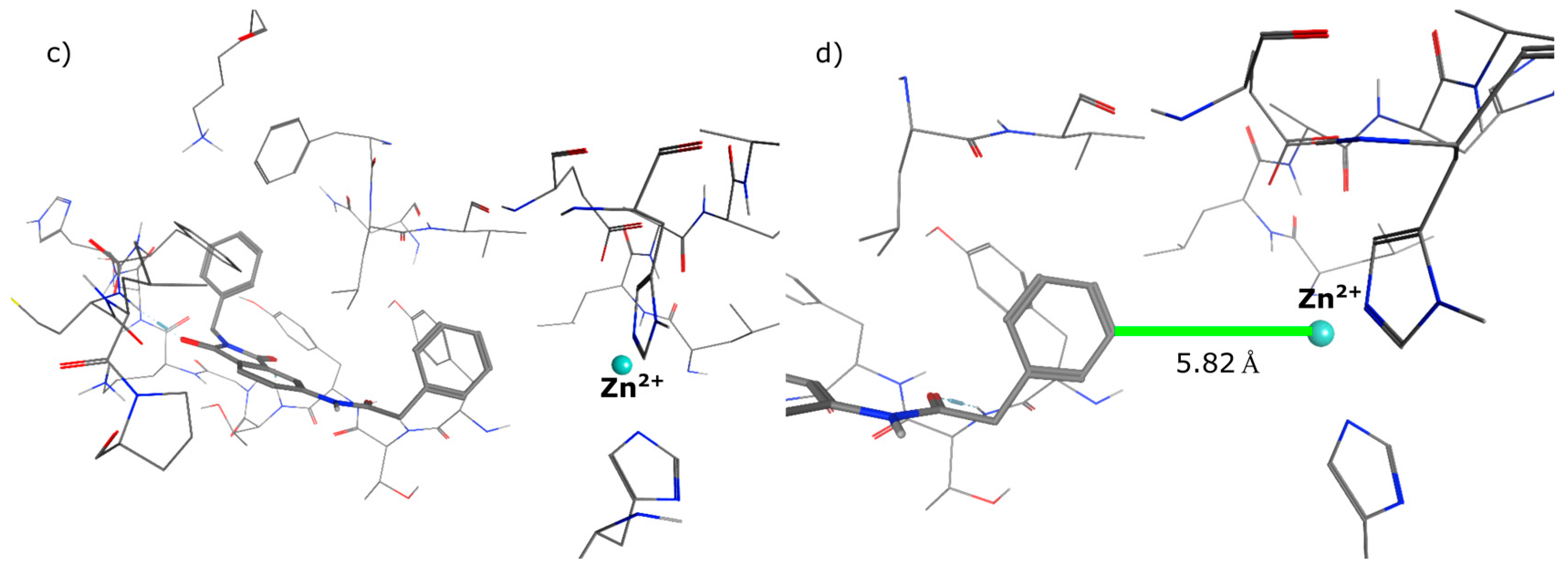
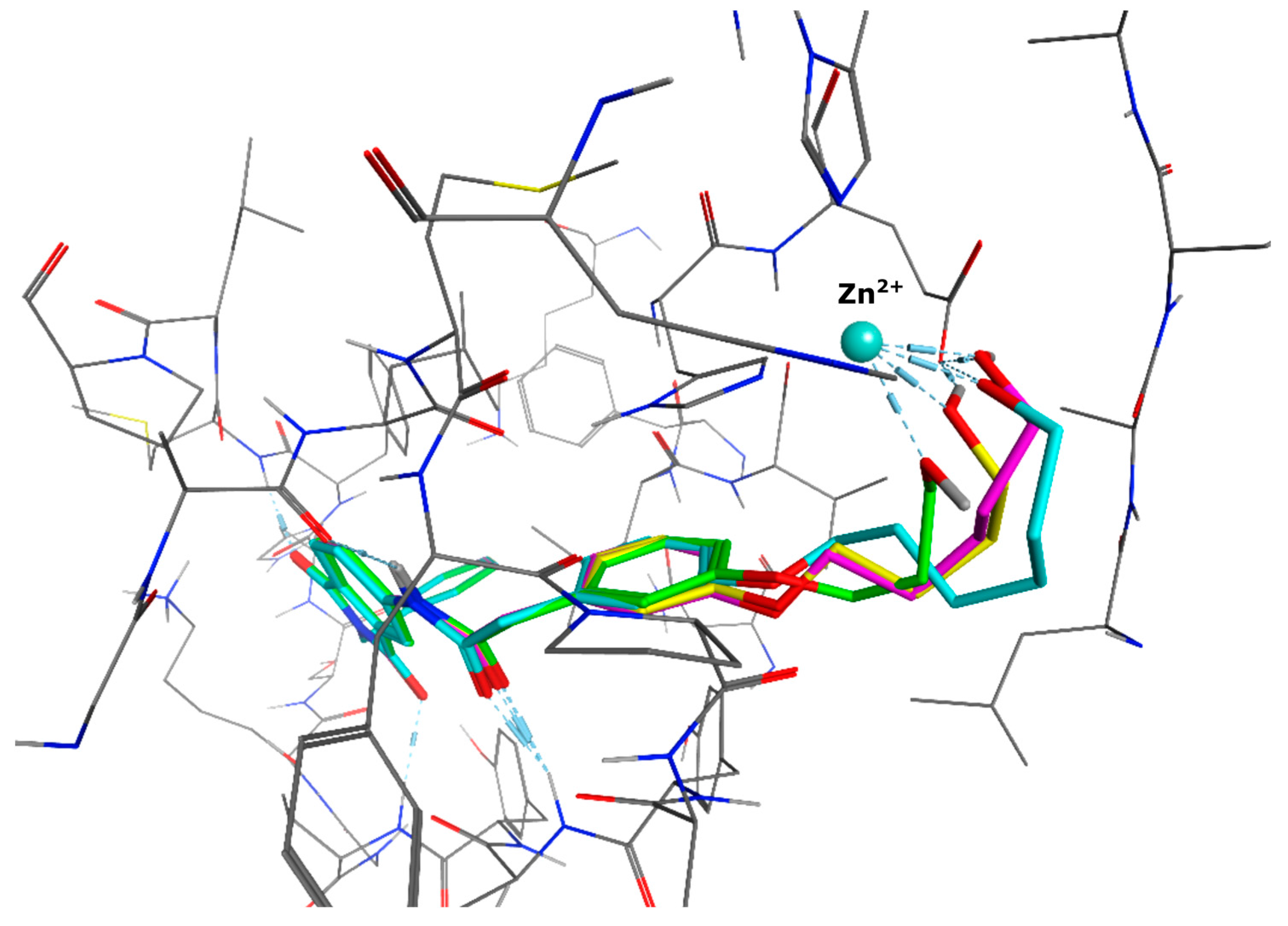
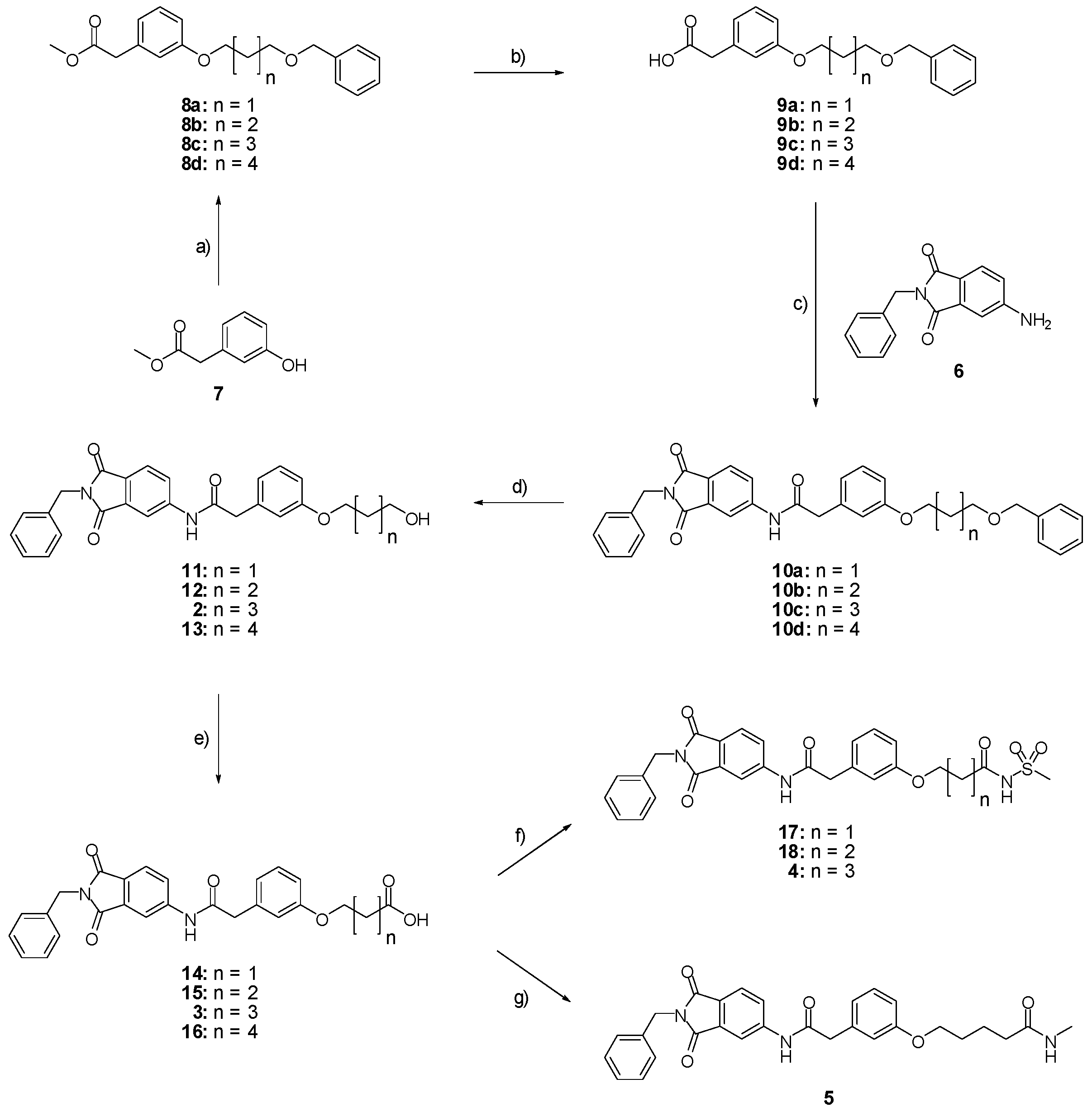

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
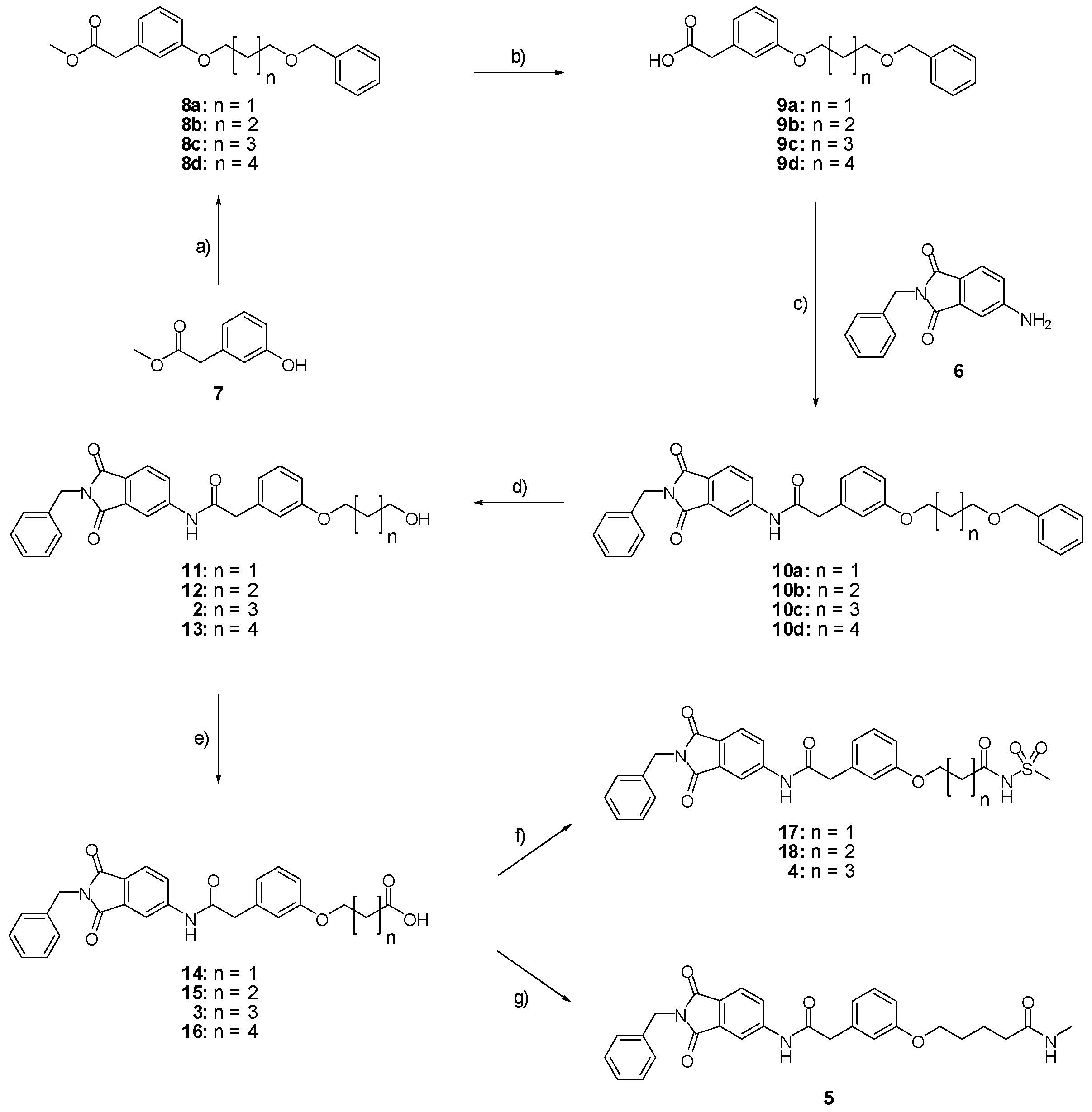{kind=link}