Abstract
Hereditary hemochromatosis (HH) is an autosomal recessive disorder characterized by enhanced intestinal absorption of dietary iron. Without therapeutic intervention, iron overload leads to multiple organ damage such as liver cirrhosis, cardiomyopathy, diabetes, arthritis, hypogonadism and skin pigmentation. Most HH patients carry HFE mutant genotypes: homozygosity for p.Cys282Tyr or p.Cys282Tyr/p.His63Asp compound heterozygosity. In addition to HFE gene, mutations in the genes that encode hemojuvelin (HJV), hepcidin (HAMP), transferrin receptor 2 (TFR2) and ferroportin (SLC40A1) have been associated with regulation of iron homeostasis and development of HH. The aim of this review was to identify the main gene mutations involved in the pathogenesis of type 1, 2, 3 and 4 HH and their genetic testing indication. HFE testing for the two main mutations (p.Cys282Tyr and p.His63Asp) should be performed in all patients with primary iron overload and unexplained increased transferrin saturation and/or serum ferritin values. The evaluation of the HJV p.Gly320Val mutation must be the molecular test of choice in suspected patients with juvenile hemochromatosis with less than 30 years and cardiac or endocrine manifestations. In conclusion, HH is an example that genetic testing can, in addition to performing the differential diagnostic with secondary iron overload, lead to more adequate and faster treatment.
1. Introduction
Hereditary hemochromatosis (HH) is an autosomal recessive disorder characterized by enhanced intestinal absorption of dietary iron. Without therapeutic intervention, iron overload leads to multiple organ damage such as liver cirrhosis, cardiomyopathy, diabetes, arthritis, hypogonadism and skin pigmentation. However, iron can be efficiently and safely removed by therapeutic phlebotomy, which is initiated by withdrawing blood at a rate of 500 mL per week until serum ferritin reaches <50 μg/L. The oral iron chelator deferasirox is not registered for genetic iron overload, since conventional phlebotomies have much lower side effects. But recent studies reported that oral chelator could be used in exceptional cases [1–5].
The major mutation that has been associated with disease is the p.Cys282Tyr in the HFE gene that occurs in approximately 80% of HH cases. In addition, a high proportion of the remaining patients are compound heterozygous for the HFE p.Cys282Tyr and the common HFE p.His63Asp alteration [6–8]. In Northern European populations, the HFE p.Cys282Tyr homozygous genotype is particularly common (1 in 200–300 healthy subjects) and the HFE 282Tyr allele frequency is high (5.1 to 8.2%) [9]. In contrast, in countries with racial/ethnic heterogeneity from South America, Asia and Africa a lower prevalence of HH have been observed, and an increased number of patients with primary iron overload do not carry the p.Cys282Tyr/p.Cys282Tyr or p.Cys282Tyr/p.His63Asp genotypes [10,11] (for example, a minor allele frequency of p.Cys282Tyr allele of 2.3% is observed in Brazilian blood donors) [12,13].
In addition to the HFE gene, mutations in the genes that encode hemojuvelin (HJV), hepcidin (HAMP), transferrin receptor 2 (TFR2) and ferroportin (SLC40A1) have been associated with regulation of iron homeostasis and development of HH [9,14,15].
Early diagnostic and initiation of iron-depletion therapy ensure life quality and increase survival times of HH patients. In this scenario, genetic testing applied to HH can, in addition to performing the differential diagnostic with secondary iron overload, lead to more adequate and faster treatment. Thus, the pivotal aim of this review was to identify the main gene mutations involved in the pathogenesis of the type 1, 2, 3 and 4 HH and their genetic testing indications.
2. HH Types, Related Genes and Their Main Mutations
According to OMIM (Online Mendelian Inheritance in Man, www.ncbi.nlm.nih.gov/omin) 5 types of HH have been identified on the basis of clinical, biochemical, and genetic characteristics (Table 1). The classic hemochromatosis is most often caused by a mutation in a gene designated HFE on chromosome 6p21.3. Nonetheless, in minor frequency, there are 4 additional disorders of primary iron overload: juvenile hemochromatosis (JH) or type 2 hemochromatosis, which is divided into 2 forms: type 2A JH, caused by mutations in the HJV gene on chromosome 1q21, and type 2B JH, caused by mutations in the HAMP gene on chromosome 19q13. HH types 3 and 4 are caused by mutations in the TFR2 and SLC40A1 genes on chromosomes 7q22 and 2q32, respectively (Table 1) [16–19].

Table 1.
Characteristics according to HH types.
2.1. HFE
HFE related-HH (OMIM 235200), classified as type 1, is the most frequent form of the disease and the most common autosomal recessive disorder in Northern European populations. HH is characterized by enhanced intestinal absorption of iron leading to multiple organ damage, such as cirrhosis, hepatoma, diabetes mellitus, arthritis, cardiomyopathy, and hypogonadism [20–23].
HFE gene (613609), constituted by 6 exons, encodes a membrane protein that is similar to major histocompatibility class I-like proteins, called HFE.
Most HH patients carry homozygosity for p.Cys282Tyr or p.Cys282Tyr/p.His63Asp compound heterozygous genotypes. Besides the missense mutation at position 282, where cysteine is replaced by tyrosine (p.Cys282Tyr, c.845G>A, rs1800562) and the common substitution of histidine for aspartic acid at position 63 (p.His63Asp, c.187C>G, rs1799945), a third mutation is also commonly assessed: the substitution of cysteine for serine at amino acid position 65 (p.Ser65Cys, c.193A>T, rs1800730). However, recent reports have suggested that rare HFE variants, such as p.Gly43Ala, p.Leu46Trp, p.Val53Met, p.Gly93Arg, p.Ile105Thr, p.Gln127His, p.Asp129Asn, p.Glu168Gln, p.Glu168del, p.Leu183Pro, p.Glu277Lys, p.Gln283Pro, p.Val284Met, p.Arg330Met, and a deletion in the 6p chromosome region containing HFE could also be linked to HH thus contributing to genetic and phenotypic heterogeneity of the disease [6,7,9,15,24,25].
The first proposed pathogenic mechanism for explaining HH was the disruption of a disulfide bond in HFE that is critical for its binding to β2 microglobulin. This complex interacts with transferrin receptor 1, decreasing the affinity with transferrin and consequently modulating iron absorption in enterocytes [26]. However, in recent years, evidences indicating HFE protein as a hepcidin modulator have emerged. The functional loss of HFE in mice and humans has been shown to reduce hepcidin synthesis [27–29] and that HFE loss seems to be associated with blunted signaling responses to BMP6 (bone morphogenetic protein 6), a key regulator of hepcidin, in vitro and in vivo [30,31]. Indeed, HFE related-HH has been associated with reduced hepcidin levels (Figure 1) [24,28,29].
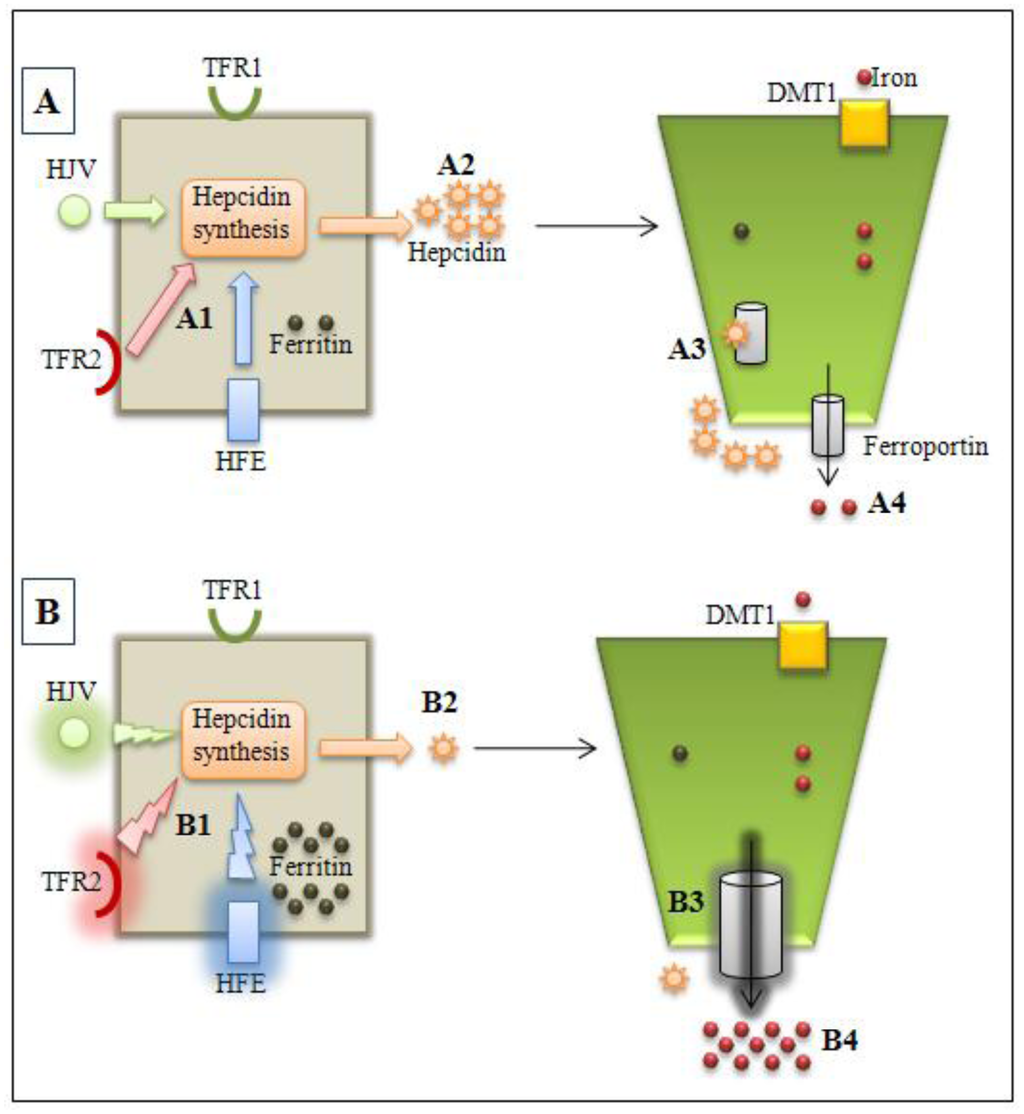
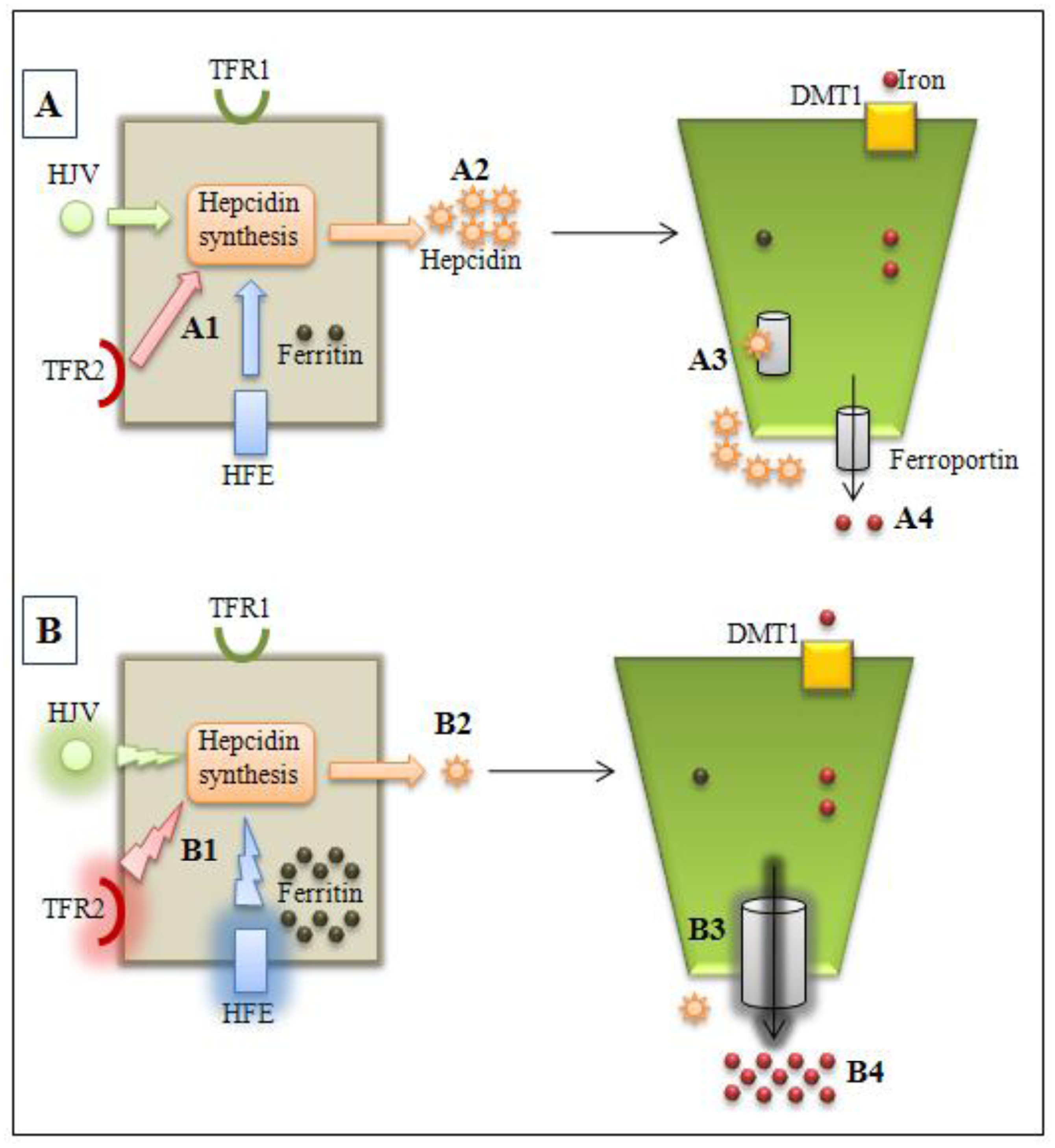
Figure 1.
Normal (A) and hemochromatosis (B) conditions. A1: HFE, HJV, and TFR2 modulates hepcidin synthesis by hepatocytes; A2: normal hepcidin levels; A3: hepcidinferroportin interaction with internalization and ferroportin degradation in enterocytes; A4: normal iron absorption. B1: HFE or HJV or TFR2 gene mutations alter hepcidin synthesis modulation; B2: lower hepcidin levels; B3: decreased hepcidin-ferroportin interaction and increased ferroportin activity; B4: iron overload observed in types 1, 2 and 3 hemochromatosis. TFR2: transferrin receptor 2; TFR1: transferrin receptor 1, HFE: HFE protein; HJV: hemojuvelin.
2.2. HJV and HAMP
Juvenile hemochromatosis (JH), also classified as type 2, is a rare autosomal recessive disorder of iron overload that leads to organ damage before the age of 30, and usually causes cardiomyopathy, hypogonadotrophic hypogonadism, liver damages and endocrine dysfunctions. Types 2A (OMIM 602390) and 2B (OMIM 613313) are caused by mutations in HJV and HAMP genes, respectively [32,33].
HJV (608374) gene, constituted by 4 exons, was identified in 2004 and encodes a protein called hemojuvelin [32]. Patients with type 2A JH and knockout mice models demonstrate low hepcidin levels implying that hemojuvelin is involved in the hepcidin synthesis [34]. HAMP gene (606464), constituted by 3 exons, encodes hepcidin, a peptide known as iron hormone. Hepcidin is produced by hepatocytes and it plays a role in iron absorption related to ferroportin degradation of the enterocytes [33,35].
Several HJV mutations have been found in patients: p.Arg54del, p.Cys80Arg, p.Ser85Pro, p.Gly99Arg, p.Gly99Val, p.Leu101Pro, p.Gly116del, p.Cys119Phe, p.Ile222Asn, p.Arg131fs, p.Asp149fs, p.Leu165del, p.Ala168Asp, p.Phe170Ser, p.Asp172Glu, p.Arg176Cys, p.Trp191Cys, p.Asn196Lys, p.Ser205Arg, p.Ile222Asn, p.Lys234del, p.Asp249His, p.Gly250Val, p.Asn269fs, p.Ile281Thr, p.Arg288Trp, p.Cys321Trp, p.Cys321del, p.Arg326del, p.Ser328fs, p.Cys361fs, and p.Arg385del. However, the HJV p.Gly320Val is the most frequent mutation and has been reported in JH patients in several populations around the world [22,32,36–39]. In contrast, mutations in HAMP are a very rare cause of JH: p.Met31fs, p.Met50fs, p.Arg56del, p.Arg59Gly, p.Cys70Arg, p.Gly71Asp, and p.Cys78Thr [22,33,40,41]. In addition, some studies support the concept that digenic inheritance of HFE and HJV or of HFE and HAMP mutations can lead to iron overload or may aggravate the phenotype [37,39,41–44].
For both type 2A and 2B JH, it is well established that the cause of iron overload may be explained by decreases in the synthesis and, consequently depressed hepcidin levels (Figure 1) [34,45]. Cell-surface expression of hemojuvelin was associated with increased expression of hepcidin; likewise, loss of hemojuvelin expression, as in juvenile hemochromatosis, was associated with reduced hepcidin expression [22,46].
HJV seems to play a role in iron absorption and release from cells and has anti-inflammatory properties [47]. An important study revealed that HJV acts as a BMP co-receptor and signals via the SMAD pathway to regulate hepcidin expression [46,48]. A BMP6 dependent signaling pathway has been shown to play a key role in regulation of hepcidin expression [24]. BMPs bind to type I and type II serine threonine kinase receptors, which phosphorylate specific intracellular SMAD proteins (SMAD1,5,8). Phosphorylated SMAD1,5,8 (P-SMAD1,5,8) binds to the common mediator SMAD4, and the SMAD complex translocates to the nucleus to affect transcription of target genes HAMP (encoding hepcidin) is transcriptionally up-regulated by BMPs [46,49–52]. Impaired hepatic signaling through mutations in genes encoding either the ligand BMP6, the BMP coreceptor hemojuvelin or Smad4 leads to low hepcidin levels and iron overload in mice. Collectively, these data show that BMP-SMAD signaling is an important regulatory pathway for hepcidin expression and thus iron metabolism [53–56].
2.3. TFR2
Type 3 HH (OMIM 604250) is an autosomal recessive disease caused by mutations in TFR2 gene and iron overload is similar to HFE related-HH phenotype. TFR2 gene (604720), constituted by 18 exons, encodes transferrin receptor 2 protein (TFR2). TFR2 is involved with uptake of transferrin bound iron by hepatocytes and it is also involved in the hepcidin synthesis [57–60]. One possibility is that it operates in the pathway discussed above for HFE (or in a parallel pathway of its own) facilitating the BMP/SMAD signaling that activates hepcidin expression. Another possibility is that TFR2, which is also able to interact with HFE, forms an iron-sensing complex that modulates hepcidin expression in response to blood levels of diferric transferrin [24,61–63].
This disorder seems to be rare and few TFR2 mutations have been reported: p.His33Asn, p.Glu60del, p.Arg105del, p.Met172Lys, p.Tyr250del, p.Gln317del, p.Arg396del, p.Ala444Thr, p.Arg455Gln, p.Arg481His, p.Leu490Arg, p.Val561del, p.Gln690Pro, and p.Gly792Arg. In both animal models and patients with TFR2 related-HH decreased hepcidin levels were observed (Figure 1) [22,42,57,64–67].
2.4. SLC40A1
Type 4 HH (OMIM 606069) has an autosomal dominant pattern and it is caused by mutations in the SLC40A1 gene. This rare disease can present peculiar clinical features such as high serum ferritin levels plus low or normal transferrin saturation values until the end stage of the disease. It may also be the presence of a mild iron-deficient anemia in the initial stage and a reduced tolerance to therapeutic phlebotomy [45,68,69].
SLC40A1 (604353) gene, constituted by 8 exons, encodes a membrane transporter called ferroportin that modulates iron efflux [70]. SLC40A1 mutations, such as p.His32Arg, p.Tyr64Asn, p.Val72Asp, p.Ala77Asp, p.Gly80Val, p.Arg88Thr, p.Asn144His, p.Asp157Gly, p.Asp157Asn, p.Val162del, p.Asn174Ile, p.Arg178Gly, p.Ile180Thr, p.Asp181Val, p.Gln182His, p.Asn185Asp, p.Gln248His, p.Gly267Asp, p.Gly323Val, p.Cys326Ser, p.Cys326Tyr, p.Gly330del, p.Ser338Arg, p.Arg489Ser, p.Gly490Asp, and p.Gly490Val were associated with type 4 HH. Two hypotheses have been proposed to account for this disease: the trapping of iron in macrophages that are unable to export iron and the failure to be degraded by interaction with hepcidin [22,42,69–74].
3. Biochemical Assays for Body Iron Store Analysis
The most common biochemical assays performed in laboratorial routine for iron overload analysis are serum iron, TIBC (total iron binding capacity), transferrin saturation (TS, which is a ratio between serum iron and TIBC expressed as percentage), and serum ferritin. Serum ferritin is a highly sensitive test for iron overload in HH, but it has low specificity, being also elevated in inflammatory process, diabetes, alcohol consumption, and liver damage.
Usually, TS values can be a helpful tool as a marker of iron overload. Some studies reported that TS values are usually higher than 50% in females and 60% in males with iron overload caused by genetic alterations [16,75–77]. In addition, a scale has been proposed by the Haute Autorité de Santé as clinical recommendations on the HH management: stage 0: without biochemical and clinical abnormalities; stage 1: increased TS (>45%), normal serum ferritin, and no clinical symptoms; stage 2: increased TS, increased serum ferritin (>200 μg/L in females and >300 μg/L in males), but no clinical symptoms; stage 3: abnormal biochemical values and initial clinical symptoms (fatigue, arthritis, impotence, skin hyperpigmentation); and stage 4: abnormal biochemical values, and clinical symptoms manifesting organ damage (cirrhosis, diabetes, hypogonadism, or cardiomyopathy) [76,78].
In this context, patients with suspect iron overload should primarily be evaluated through fasting TS and serum ferritin. HFE mutations molecular assay should be performed only in those with increased biochemical values [79].
4. Genetic Testing and Methodology
4.1. Genetic Testing
HFE testing for the two main mutations (p.Cys282Tyr and p.His63Asp) should be performed in all patients with unexplained increased TS and/or serum ferritin values (Figure 2). In these cases, the molecular diagnostic of HFE related-HH is usually associated with the presence of the p.Cys282Tyr homozygosity and p.Cys282Tyr/p.His63Asp compound heterozygous genotypes. However, p.His63Asp homozygous and p.His63Asp/p.Ser65Cys compound heterozygous genotypes have been associated with HH phenotype [3,15,23].
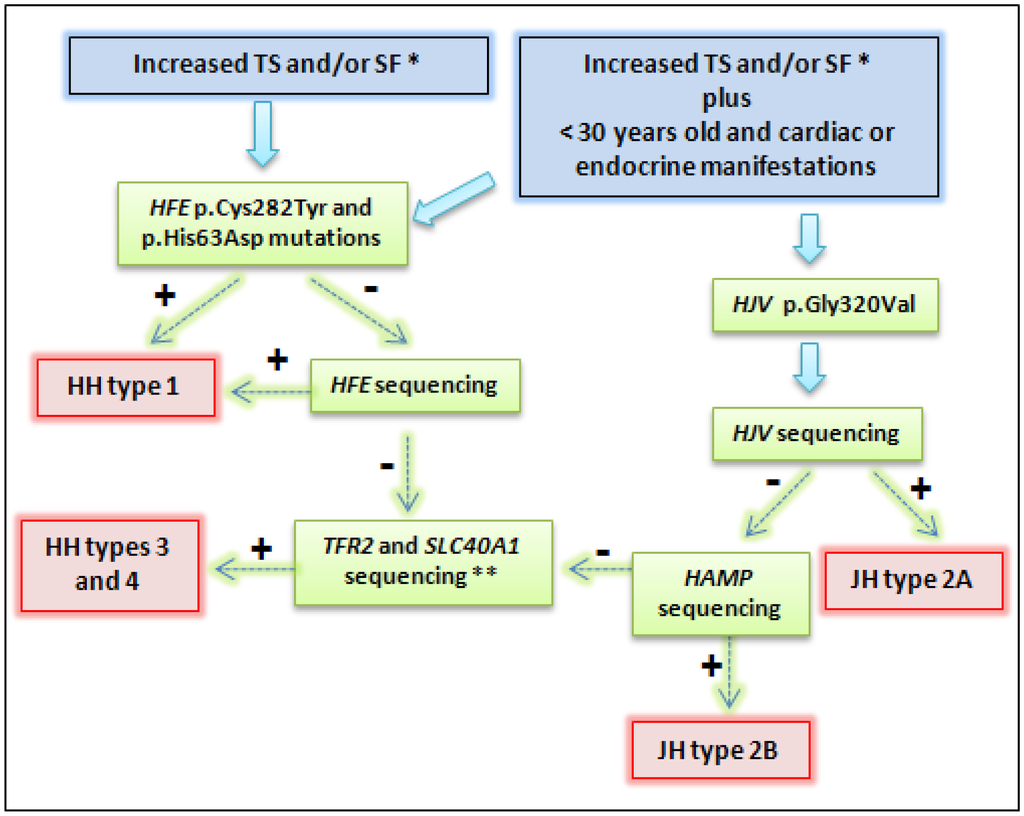
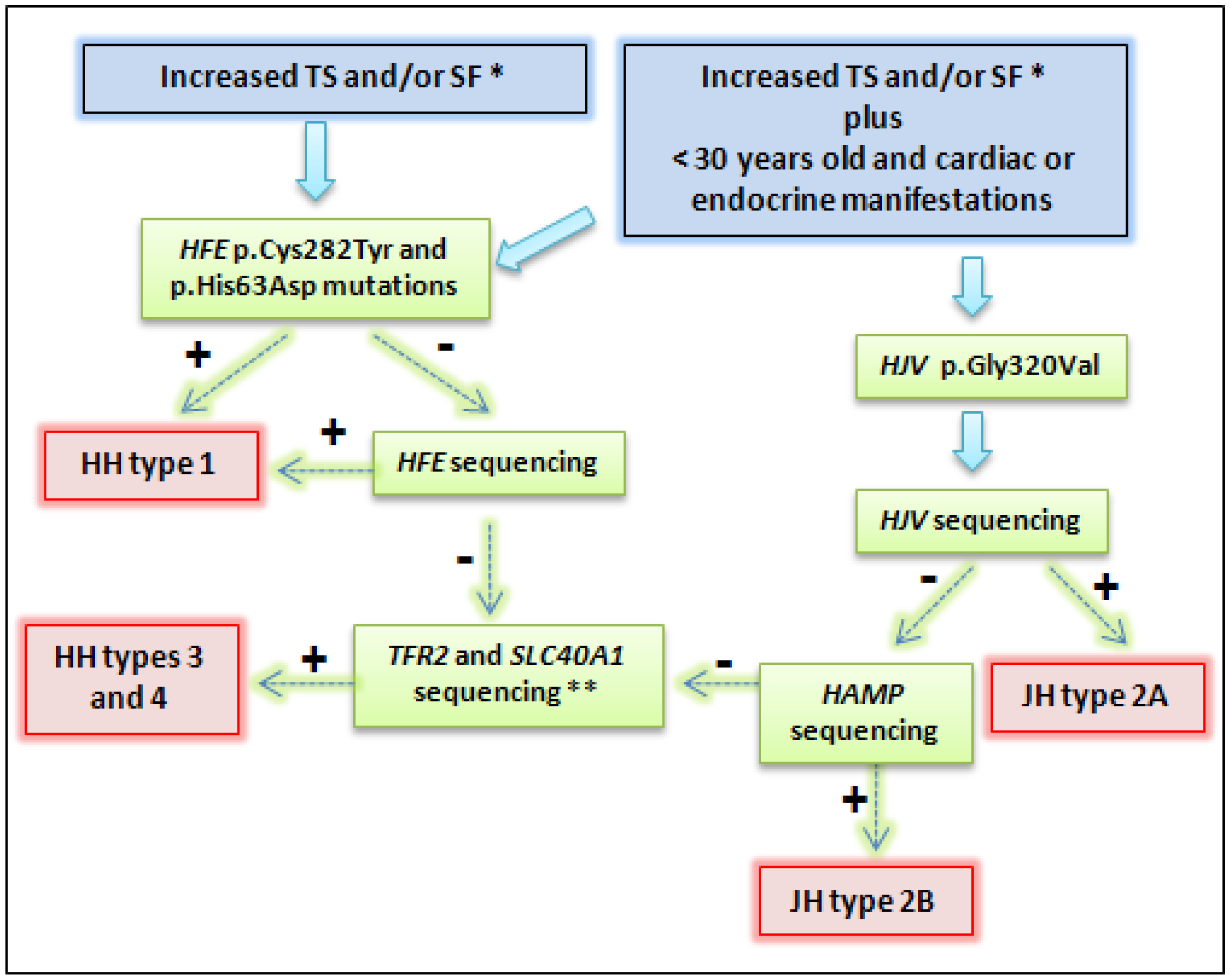
Figure 2.
Representation of diagnostic strategy for patients suspected hereditary hemochromatosis (HH). * Recommendations report TS > 45%, SF > 200 μg/L in females and > 300 μg/L in males; or in advanced stages: TS > 50% in females and TS > 60% in males, in the absence of secondary causes [79,80]. ** Some patients with primary iron overload may not present mutation during this genetic approach. Very rare mutations in other genes can be involved [15,81]. Abbreviations: TS: transferrin saturation; SF: serum ferritin; JH: juveline hemochromatosis. + means positive result, and − means negative result.
In the absence of the mentioned HFE genotype combinations, other HH types could be considered. When there is genetic iron overload in a patient with less than 30 years and cardiac or endocrine manifestations, JH diagnostic is suggestive (Figure 2). Thus, the evaluation of the p.Gly320Val mutation in the HJV gene must be the molecular test of choice. According to several studies, this procedure would confirm the majority of JH cases [5,29]. Early diagnosis is paramount. If result is negative, sequencing should be performed to evaluate the HJV and HAMP genes (Figure 2). Our group reported a case with both clinical and molecular diagnostic of JH, and the use of deferasirox therapy adjunct to venesections during the initial treatment presented significant improvements as cardiomyopathy and liver disease were prevented, and endocrine functions were normalized [5].
Mutations in the TFR2 and SLC40A1 genes are rare compared with HFE mutations and they have also been reported in children, adolescents, and adults. These genes should be sequenced after negative results for other genes (Figure 2). Nowadays, the costs for sequencing have come down, especially if it evaluates the number of bases per dollar of the next generation sequencings. However, for the most part of clinical practice around the world, screening of HFE, HJV, HAMP, TFR2 and SLC40A1 through direct sequencing is not widely available. This approach is usually reserved for scientific studies and for very specific cases such as patients who are not responsive to treatment and had more severe complications due to iron overload. In addition and for the most part of cases, the treatment is not dependent on molecular diagnosis. Our group reported that direct sequencing does not significantly improve the diagnostic throughput as when it was compared to the sole HFE main mutations testing [15,19,79,80].
4.2. Methodology
Genotyping of HFE p.Cys282Tyr and p.His63Asp mutations is one of the most requested molecular assays in the laboratorial routine. The analysis of single nucleotide polymorphisms, for example HFE p.Cys282Tyr, HFE p.His63Asp, and HJV p.Gly320Val, can be performed by several available methods for genotyping (Figure 2), such as restriction fragment length polymorphisms (RFLP), allele-specific amplification analysis real time-polymerase, denaturing HPLC, sequencing strategies, TaqMan assay, multiplex amplification followed by reverse hybridization [79,82–84]. A high-resolutionmelting (HRM) assay was developed by our group for genotyping HFE p.Cys282Tyr and p.His63Asp mutations in a unique procedure being capable of ensuring the result in approximately 112 minutes and, with cost-effectiveness especially in a large-scale demand, compared to methods cited above. The advantages of genotyping with this procedure were the non-dependence on gel electrophoresis and on mutagenic reagents for visualization of fragments, and the reduction of the chances for contamination due to sample preparation compared to RFLP and sequencing strategies. There are disadvantages for the HRM method: interference from another genetic variant that may be present in the amplicon leading to misdiagnosis by altering the curve pattern of the target-mutation. The amplicon for the HFE p.Cys282Tyr mutation may present the following known genetic variants: p.Thr281Thr, p.Gln283Pro, p.Val284Met; and the amplicon for the HFE p.His63Asp mutation may present the p.Ser65Cys known genetic variants. Moreover, non-described mutations may also be present [83,85,86].
5. Conclusions
Advances in the understanding of HH have been obtained over the years: association of the HFE p.Cys282Tyr as the main mutation involved, genetic markers for juvenile hemochromatosis and several pathogenic mutations associated with non-HFE HH, hepcidin as an iron hormone, new techniques for the laboratorial evaluation, and increased knowledge about HH management. Nonetheless, there are still unclear points to be explored in the HH context: the exact role of the HFE protein, molecular pathways of the hepcidin synthesis, the identification of non-genetic factors that affect penetrance, more robust functional prediction tools, and protein functionality assays more informative and easier for the study of identified genetic alterations.
HFE testing for the two main mutations (p.Cys282Tyr and p.His63Asp) should be performed in all suspected patients with primary iron overload and unexplained increased TS and/or serum ferritin values. The evaluation of the HJV p.Gly320Val mutation must be the molecular test of choice in suspected patients with juvenile hemochromatosis.
In conclusion, hereditary hemochromatosis is an example that genetic testing can, in addition to performing the differential diagnostic with secondary iron overload, lead to more adequate and faster treatment.
Acknowledgments
PCJL Santos is recipient of a fellowship from FAPESP, Proc. 2010-17465-8, Brazil.
- Conflict of InterestThe authors declare no conflict of interest.
References
- Alexander, J.; Kowdley, K.V. HFE-associated hereditary hemochromatosis. Genet. Med 2009, 11, 307–313. [Google Scholar]
- Bacon, B.R. Hemochromatosis: Diagnosis and management. Gastroenterology 2001, 120, 718–725. [Google Scholar]
- Moyer, T.P.; Highsmith, W.E.; Smyrk, T.C.; Gross, J.B., Jr. Hereditary hemochromatosis: Laboratory evaluation. Clin. Chim. Acta 2011, 412, 1485–1492. [Google Scholar]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1779. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Pereira, A.C.; Chiattone, C.S.; Krieger, J.E.; Guerra-Shinohara, E.M. HJV hemochromatosis, iron overload, and hypogonadism in a Brazilian man: Treatment with phlebotomy and deferasirox.
- Dupradeau, F.Y.; Pissard, S.; Coulhon, M.P.; Cadet, E.; Foulon, K.; Fourcade, C.; Goossens, M.; Case, D.A.; Rochette, J. An unusual case of hemochromatosis due to a new compound heterozygosity in HFE (p.[Gly43Asp;His63Asp]+[Cys282Tyr]): Structural implications with respect to binding with transferrin receptor 1. Hum. Mutat 2008, 29. [Google Scholar] [CrossRef]
- Mendes, A.I.; Ferro, A.; Martins, R.; Picanco, I.; Gomes, S.; Cerqueira, R.; Correia, M.; Nunes, A.R.; Esteves, J.; Fleming, R.; Faustino, P. Non-classical hereditary hemochromatosis in Portugal: Novel mutations identified in iron metabolism-related genes. Ann. Hematol 2009, 88, 229–234. [Google Scholar]
- Pietrangelo, A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut 2006, 55, 564–568. [Google Scholar]
- Swinkels, D.W.; Janssen, M.C.; Bergmans, J.; Marx, J.J. Hereditary hemochromatosis: Genetic complexity and new diagnostic approaches. Clin. Chem 2006, 52, 950–968. [Google Scholar]
- Merryweather-Clarke, A.T.; Pointon, J.J.; Shearman, J.D.; Robson, K.J. Global prevalence of putative haemochromatosis mutations. J. Med. Genet 1997, 34, 275–278. [Google Scholar]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol 2009, 122, 78–86. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Terada, C.T.; Rostelato, S.; Gonzales, I.; Hirata, R.D.; Hirata, M.H.; Chiattone, C.S.; Guerra-Shinohara, E.M. HFE gene mutations and iron status of Brazilian blood donors. Braz. J. Med. Biol. Res 2010, 43, 107–114. [Google Scholar] [Green Version]
- Terada, C.T.; Santos, P.C.; Cancado, R.D.; Rostelato, S.; Lopreato, F.R.; Chiattone, C.S.; Guerra-Shinohara, E.M. Iron deficiency and frequency of HFE C282Y gene mutation in Brazilian blood donors. Transfus. Med 2009, 19, 245–251. [Google Scholar]
- Lok, C.Y.; Merryweather-Clarke, A.T.; Viprakasit, V.; Chinthammitr, Y.; Srichairatanakool, S.; Limwongse, C.; Oleesky, D.; Robins, A.J.; Hudson, J.; Wai, P.; et al. Iron overload in the Asian community. Blood 2009, 114, 20–25. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Pereira, A.C.; Schettert, I.T.; Soares, R.A.; Pagliusi, R.A.; Hirata, R.D.; Hirata, M.H.; Teixeira, A.C.; Figueiredo, M.S.; et al. Hereditary hemochromatosis: Mutations in genes involved in iron homeostasis in Brazilian patients. Blood Cells Mol. Dis 2010, 46, 302–307. [Google Scholar]
- Kaplan, J.; Ward, D.M.; de Domenico, I. The molecular basis of iron overload disorders and iron-linked anemias. Int. J. Hematol 2011, 93, 14–20. [Google Scholar]
- Lee, P.L.; Beutler, E. Regulation of hepcidin and iron-overload disease. Annu. Rev. Pathol 2009, 4, 489–515. [Google Scholar]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, e1–e2. [Google Scholar]
- Pietrangelo, A. Hepcidin in human iron disorders: Therapeutic implications. J. Hepatol 2011, 54, 173–181. [Google Scholar]
- Adams, P.C. Nonexpressing homozygotes for C282Y hemochromatosis: Minority or majority of cases? Mol. Genet. Metab 2000, 71, 81–86. [Google Scholar]
- Leone, P.E.; Gimenez, P.; Collantes, J.C.; Paz-y-Mino, C. Analysis of HFE gene mutations (C282Y, H63D, and S65C) in the Ecuadorian population. Ann. Hematol 2005, 84, 103–105. [Google Scholar]
- Potekhina, E.S.; Lavrov, A.V.; Samokhodskaya, L.M.; Efimenko, A.Y.; Balatskiy, A.V.; Baev, A.A.; Litvinova, M.M.; Nikitina, L.A.; Shipulin, G.A.; Bochkov, N.P.; et al. Unique genetic profile of hereditary hemochromatosis in Russians: High frequency of C282Y mutation in population, but not in patients. Blood Cells Mol. Dis 2005, 35, 182–188. [Google Scholar]
- Santos, P.C.; Pereira, A.C.; Cancado, R.D.; Schettert, I.T.; Sobreira, T.J.; Oliveira, P.S.; Hirata, R.D.; Hirata, M.H.; Figueiredo, M.S.; Chiattone, C.S.; et al. HFE gene mutations in patients with primary iron overload: Is there a significant improvement in molecular diagnosis yield with HFE sequencing? Blood Cells Mol. Dis 2010, 45, 302–307. [Google Scholar]
- Thakur, V.; Guptan, R.C.; Hashmi, A.Z.; Sakhuja, P.; Malhotra, V.; Sarin, S.K. Absence of hemochromatosis associated Cys282Tyr HFE gene mutation and low frequency of hemochromatosis phenotype in nonalcoholic chronic liver disease patients in India. J. Gastroenterol. Hepatol 2004, 19, 86–90. [Google Scholar]
- Pelucchi, S.; Mariani, R.; Bertola, F.; Arosio, C.; Piperno, A. Homozygous deletion of HFE: The Sardinian hemochromatosis. Blood 2009, 113. [Google Scholar] [CrossRef]
- Bennett, M.J.; Lebron, J.A.; Bjorkman, P.J. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature 2000, 403, 46–53. [Google Scholar]
- Ahmad, K.A.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Britton, R.S.; Bacon, B.R.; Sly, W.S.; Fleming, R.E. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol. Dis 2002, 29, 361–366. [Google Scholar]
- Bridle, K.R.; Frazer, D.M.; Wilkins, S.J.; Dixon, J.L.; Purdie, D.M.; Crawford, D.H.; Subramaniam, V.N.; Powell, L.W.; Anderson, G.J.; Ramm, G.A. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003, 361, 669–673. [Google Scholar]
- Gehrke, S.G.; Pietrangelo, A.; Kascak, M.; Braner, A.; Eisold, M.; Kulaksiz, H.; Herrmann, T.; Hebling, U.; Bents, K.; Gugler, R.; et al. HJV gene mutations in European patients with juvenile hemochromatosis. Clin. Genet 2005, 67, 425–428. [Google Scholar]
- Corradini, E.; Garuti, C.; Montosi, G.; Ventura, P.; Andriopoulos, B., Jr; Lin, H.Y.; Pietrangelo, A.; Babitt, J.L. Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis. Gastroenterology 2009, 137, 1489–1497. [Google Scholar]
- Kautz, L.; Meynard, D.; Besson-Fournier, C.; Darnaud, V.; Al Saati, T.; Coppin, H.; Roth, M.P. BMP/Smad signaling is not enhanced in Hfe-deficient mice despite increased Bmp6 expression. Blood 2009, 114, 2515–2520. [Google Scholar]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.; Franchini, P.L.; Dube, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet 2004, 36, 77–82. [Google Scholar]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet 2003, 33, 21–22. [Google Scholar]
- Lin, L.; Goldberg, Y.P.; Ganz, T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood 2005, 106, 2884–2889. [Google Scholar]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar]
- Aguilar-Martinez, P.; Lok, C.Y.; Cunat, S.; Cadet, E.; Robson, K.; Rochette, J. Juvenile hemochromatosis caused by a novel combination of hemojuvelin G320V/R176C mutations in a 5-year old girl. Haematologica 2007, 92, 421–422. [Google Scholar]
- De Lima Santos, P.C.; Pereira, A.C.; Cancado, R.D.; Schettert, I.T.; Hirata, R.D.; Hirata, M.H.; Figueiredo, M.S.; Chiattone, C.S.; Krieger, J.E.; Guerra-Shinohara, E.M. Hemojuvelin and hepcidin genes sequencing in Brazilian patients with primary iron overload. Genet. Test Mol. Biomark 2010, 14, 803–806. [Google Scholar]
- Lanzara, C.; Roetto, A.; Daraio, F.; Rivard, S.; Ficarella, R.; Simard, H.; Cox, T.M.; Cazzola, M.; Piperno, A.; Gimenez-Roqueplo, A.P.; et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood 2004, 103, 4317–4321. [Google Scholar]
- Lee, P.L.; Beutler, E.; Rao, S.V.; Barton, J.C. Genetic abnormalities and juvenile hemochromatosis: Mutations of the HJV gene encoding hemojuvelin. Blood 2004, 103, 4669–4671. [Google Scholar]
- Jacolot, S.; Le Gac, G.; Scotet, V.; Quere, I.; Mura, C.; Ferec, C. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood 2004, 103, 2835–2840. [Google Scholar]
- Porto, G.; Roetto, A.; Daraio, F.; Pinto, J.P.; Almeida, S.; Bacelar, C.; Nemeth, E.; Ganz, T.; Camaschella, C. A Portuguese patient homozygous for the -25G>A mutation of the HAMP promoter shows evidence of steady-state transcription but fails to up-regulate hepcidin levels by iron. Blood 2005, 106, 2922–2923. [Google Scholar]
- Biasiotto, G.; Roetto, A.; Daraio, F.; Polotti, A.; Gerardi, G.M.; Girelli, D.; Cremonesi, L.; Arosio, P.; Camaschella, C. Identification of new mutations of hepcidin and hemojuvelin in patients with HFE C282Y allele. Blood Cells Mol. Dis 2004, 33, 338–343. [Google Scholar]
- Altes, A.; Bach, V.; Ruiz, A.; Esteve, A.; Felez, J.; Remacha, A.F.; Sarda, M.P.; Baiget, M. Mutations in HAMP and HJV genes and their impact on expression of clinical hemochromatosis in a cohort of 100 Spanish patients homozygous for the C282Y mutation of HFE gene. Ann. Hematol 2009, 88, 951–955. [Google Scholar]
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; McHugh, P.J.; Chapman, R.W.; Pointon, J.J.; Wimhurst, V.L.; et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet 2003, 12, 2241–2247. [Google Scholar]
- Camaschella, C.; Poggiali, E. Rare types of genetic hemochromatosis. Acta Haematol 2009, 122, 140–145. [Google Scholar]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet 2006, 38, 531–539. [Google Scholar]
- Papanikolaou, G.; Tzilianos, M.; Christakis, J.I.; Bogdanos, D.; Tsimirika, K.; MacFarlane, J.; Goldberg, Y.P.; Sakellaropoulos, N.; Ganz, T.; Nemeth, E. Hepcidin in iron overload disorders. Blood 2005, 105, 4103–4105. [Google Scholar]
- Malyszko, J. Hemojuvelin: The hepcidin story continues. Kidney Blood Press. Res 2009, 32, 71–76. [Google Scholar]
- Andriopoulos, B., Jr; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; Babitt, J.L. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar]
- Babitt, J.L.; Huang, F.W.; Xia, Y.; Sidis, Y.; Andrews, N.C.; Lin, H.Y. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J. Clin. Invest 2007, 117, 1933–1939. [Google Scholar]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. (Berl.) 2009, 87, 471–480. [Google Scholar]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet 2009, 41, 478–481. [Google Scholar]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron regulation of hepcidin despite attenuated smad1,5,8 signaling in mice without transferrin receptor 2 or hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar]
- Huang, F.W.; Pinkus, J.L.; Pinkus, G.S.; Fleming, M.D.; Andrews, N.C. A mouse model of juvenile hemochromatosis. J. Clin. Invest 2005, 115, 2187–2191. [Google Scholar]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Invest 2005, 115, 2180–2186. [Google Scholar]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; Deng, C.X. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005, 2, 399–409. [Google Scholar]
- Camaschella, C.; Roetto, A.; Cali, A.; de Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet 2000, 25, 14–15. [Google Scholar]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem 1999, 274, 20826–208232. [Google Scholar]
- Nemeth, E.; Roetto, A.; Garozzo, G.; Ganz, T.; Camaschella, C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005, 105, 1803–1806. [Google Scholar]
- Wallace, D.F.; Summerville, L.; Lusby, P.E.; Subramaniam, V.N. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut 2005, 54, 980–986. [Google Scholar]
- Chen, J.; Chloupkova, M.; Gao, J.; Chapman-Arvedson, T.L.; Enns, C.A. HFE modulates transferrin receptor 2 levels in hepatoma cells via interactions that differ from transferrin receptor 1-HFE interactions. J. Biol. Chem 2007, 282, 36862–36870. [Google Scholar]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 2008, 7, 205–214. [Google Scholar]
- Waheed, A.; Britton, R.S.; Grubb, J.H.; Sly, W.S.; Fleming, R.E. HFE association with transferrin receptor 2 increases cellular uptake of transferrin-bound iron. Arch. Biochem. Biophys 2008, 474, 193–197. [Google Scholar]
- Fleming, R.E.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Koeffler, H.P.; Kawabata, H.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658. [Google Scholar]
- Koyama, C.; Wakusawa, S.; Hayashi, H.; Ueno, T.; Suzuki, R.; Yano, M.; Saito, H.; Okazaki, T. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: Hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern. Med 2005, 44, 990–993. [Google Scholar]
- Majore, S.; Milano, F.; Binni, F.; Stuppia, L.; Cerrone, A.; Tafuri, A.; de Bernardo, C.; Palka, G.; Grammatico, P. Homozygous p.M172K mutation of the TFR2 gene in an Italian family with type 3 hereditary hemochromatosis and early onset iron overload. Haematologica 2006, 91, e92–e93. [Google Scholar]
- Mattman, A.; Huntsman, D.; Lockitch, G.; Langlois, S.; Buskard, N.; Ralston, D.; Butterfield, Y.; Rodrigues, P.; Jones, S.; Porto, G.; et al. Transferrin receptor 2 (TfR2) and HFE mutational analysis in non-C282Y iron overload: Identification of a novel TfR2 mutation. Blood 2002, 100, 1075–1077. [Google Scholar]
- Mayr, R.; Janecke, A.R.; Schranz, M.; Griffiths, W.J.; Vogel, W.; Pietrangelo, A.; Zoller, H. Ferroportin disease: A systematic meta-analysis of clinical and molecular findings. J. Hepatol 2010, 53, 941–949. [Google Scholar]
- Schimanski, L.M.; Drakesmith, H.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; Robson, K.J.; et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 2005, 105, 4096–4102. [Google Scholar]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem 2000, 275, 19906–19912. [Google Scholar]
- Beutler, E. Hemochromatosis: Genetics and pathophysiology. Annu. Rev. Med 2006, 57, 331–347. [Google Scholar]
- Cremonesi, L.; Forni, G.L.; Soriani, N.; Lamagna, M.; Fermo, I.; Daraio, F.; Galli, A.; Pietra, D.; Malcovati, L.; Ferrari, M.; et al. Genetic and clinical heterogeneity of ferroportin disease. Br. J. Haematol 2005, 131, 663–670. [Google Scholar]
- Hetet, G.; Devaux, I.; Soufir, N.; Grandchamp, B.; Beaumont, C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood 2003, 102, 1904–1910. [Google Scholar]
- Kasvosve, I.; Gomo, Z.A.; Nathoo, K.J.; Matibe, P.; Mudenge, B.; Loyevsky, M.; Gordeuk, V.R. Effect of ferroportin Q248H polymorphism on iron status in African children. Am. J. Clin. Nutr 2005, 82, 1102–1106. [Google Scholar]
- Aguilar-Martinez, P.; Schved, J.F.; Brissot, P. The evaluation of hyperferritinemia: An updated strategy based on advances in detecting genetic abnormalities. Am. J. Gastroenterol 2005, 100, 1185–1194. [Google Scholar]
- Brissot, P.; Troadec, M.B.; Bardou-Jacquet, E.; Le Lan, C.; Jouanolle, A.M.; Deugnier, Y.; Loreal, O. Current approach to hemochromatosis. Blood Rev 2008, 22, 195–210. [Google Scholar]
- Le Gac, G.; Dupradeau, F.Y.; Mura, C.; Jacolot, S.; Scotet, V.; Esnault, G.; Mercier, A.Y.; Rochette, J.; Ferec, C. Phenotypic expression of the C282Y/Q283P compound heterozygosity in HFE and molecular modeling of the Q283P mutation effect. Blood Cells Mol. Dis 2003, 30, 231–237. [Google Scholar]
- Brissot, P.; Troadec, M.B.; Loreal, O. The clinical relevance of new insights in iron transport and metabolism. Curr. Hematol. Rep 2004, 3, 107–115. [Google Scholar]
- EASL clinical practice guidelines for HFE hemochromatosis. J. Hepatol. 2010, 53, 3–22.
- Brissot, P.; de Bels, F. Current approaches to the management of hemochromatosis. Hematol. Am. Soc. Hematol. Educ. Program 2006, 36–41. [Google Scholar]
- Van Bokhoven, M.A.; van Deursen, C.T.; Swinkels, D.W. Diagnosis and management of hereditary haemochromatosis. BMJ 2011, 342. [Google Scholar] [CrossRef]
- de Juan Jimenez, I.; Cardenosa, E.E.; Suela, S.P.; Gonzalez, E.B.; Trejo, D.S.; Lluch, O.F.; Gilabert, P.B. Advantage of high-resolution melting curve analysis over conformation-sensitive gel electrophoresis for mutational screening of BRCA1 and BRCA2 genes. Clin. Chim. Acta 2011, 412, 578–582. [Google Scholar]
- Santos, P.C.; Soares, R.A.; Krieger, J.E.; Guerra-Shinohara, E.M.; Pereira, A.C. Genotyping of the hemochromatosis HFE p.H63D and p.C282Y mutations by high-resolution melting with the Rotor-Gene 6000((R)) instrument. Clin. Chem. Lab. Med 2011, 49, 1633–1636. [Google Scholar]
- Tag, C.G.; Gressner, A.M.; Weiskirchen, R. An unusual melting curve profile in LightCycler multiplex genotyping of the hemochromatosis H63D/C282Y gene mutations. Clin. Biochem 2001, 34, 511–515. [Google Scholar]
- Castiglioni, E.; Soriani, N.; Girelli, D.; Camaschella, C.; Spiga, I.; Della Porta, M.G.; Ferrari, M.; Cremonesi, L. High resolution melting for the identification of mutations in the iron responsive element of the ferritin light chain gene. Clin. Chem. Lab. Med 2011, 48, 1415–1418. [Google Scholar]
- Lin, J.T.; Hsiao, K.J.; Chen, C.Y.; Wu, C.C.; Lin, S.J.; Chou, Y.Y.; Shiesh, S.C. High resolution melting analysis for the detection of SLC25A13 gene mutations in Taiwan. Clin. Chim. Acta 2011, 412, 460–465. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).