Abstract
Four previously undescribed drimane-type sesquiterpenoids, ramaribotrytols A–D (1–4), were isolated from the fruiting bodies of the clavarioid fungus Ramaria botrytoides. Their structures and absolute configurations were elucidated by comprehensive 1D and 2D NMR spectroscopic analyses, high-resolution electrospray ionization mass spectrometry (HRESIMS), and electronic circular dichroism (ECD) calculations. Compounds 1–4 are characterized by oxygenation at C-2, a structural feature that is rare within the drimane sesquiterpene family. These findings expand the chemical diversity of secondary metabolites from the medicinal and edible fungus Ramaria and enrich the structural repertoire of drimane-type sesquiterpenoids derived from higher fungi.
1. Introduction
Higher fungi are recognized as a prolific and distinctive source of structurally diverse natural products, many of which possess unique skeletons and intriguing biological activities [1]. Owing to their complex biosynthetic pathways and specialized enzymatic machinery, fungal secondary metabolites often display unusual oxidation patterns and structural modifications that distinguish them from those of plants and bacteria [2], making higher fungi an important reservoir for the discovery of new chemical entities. Ramaria botrytoides, also known as “broom fungus,” has been used as an edible mushroom since ancient times due to its delicious taste and various nutrients. In Traditional Chinese medicine, this fungus possesses the function of strengthening the body [3], while modern medicine suggests it shows effects on anti-aging and human immunity improvement.
Among fungal-derived sesquiterpenes, drimane-type sesquiterpenes constitute a large class that has been widely reported from higher fungi [4]. Members of this family exhibit a broad range of biological activities, including antimicrobial [5], cytotoxic [6], anti-inflammatory [6], acetylcholinesterase inhibitory effects [7], antifeedant activity [8], and have therefore attracted considerable attention from natural products chemists. Structurally, drimane sesquiterpenes are characterized by a bicyclic decalin core which was reported to arise from the class II type of terpene synthase [9], the haloacid dehalogenase-like (HAD-like) terpene cyclase [10,11].
Drimane scaffolds are frequently decorated with oxygen-containing functional groups introduced during tailoring processes. To date, oxygenation in drimane sesquiterpenoids has most commonly been observed at positions C-3, C-6, C-10, C-11, C-12, C-13, and C-14 [12]. Except for dendocarbin B2 and 2α-hydroxyisodrimeninol from nudibranch Dendrodoris carbunculosa [13]. Oxygenation at the C-2 position is uncommon in drimane sesquiterpenes derived from fungi, indicating a relatively high degree of regioselectivity in the oxidative tailoring of the drimane scaffold. During our ongoing investigation of secondary metabolites from higher fungi, the fruiting bodies of the edible mushroom Ramaria botrytoides yielded drimane sesquiterpenoids 1–4 featuring unprecedented C-2 oxygenation, prompting detailed chemical and spectroscopic studies. Herein, we reported the isolation, structural elucidation, and biological activity of these drimane sesquiterpenes.
2. Results and Discussion
Compound 1 (Figure 1) was isolated as a colorless oil. Its molecular formula was established as C15H22O4 by (+)-HRESIMS, which showed a protonated molecular ion at m/z 249.14847 [M − H2O + H]+ (calcd. for C15H21O3, 249.14852). The 1H NMR spectroscopic data of 1 (Table 1) displayed two singlet methyl resonances at δH 1.08 (3H, s, H3-13) and δH 1.18 (3H, s, H3-15). Analysis of the 13C NMR (Table 2) and HSQC spectra of 1 revealed 15 carbon signals, including two methyls at δC 28.2 (Me-13) and δC 21.8 (Me-15), six methylenes at δC 44.6 (C-1), δC 45.8 (C-3), δC 19.5 (C-6), δC 26.6 (C-7), δC 72.5 (C-12), and δC 66.0 (C-14), two methines at δC 64.8 (C-2) and δC 53.7 (C-5), two non-protonated carbons at δC 41.2 (C-4), δC 37.2 (C-10), two olefinic carbons at δC 163.1 (C-8) and δC 135.4 (C-9), together with a carbonyl group at δC 175.0 (C-11). The NMR data of 1 were closely related to those of proversilin A [14], a known drimane-type sesquiterpene. The main structural differences were the presence of an additional hydroxyl group at C-2 and the relocation of the carbonyl group from C-12 in proversilin A to C-11 in 1. The 1H-1H COSY (Figure 2) showed correlations of H-1/H-2/H-3 [δH 2.87, δH 1.11 (H-1α/1β)/δH 3.93 (H-2)/δH 0.96, 2.15 (H-3α/3β)], and the chemical shifts in H-2 (δH 3.93, m) and C-2 (δC 64.8) of 1 supported oxygenation at C-2. Key HMBC correlations (Figure 2) from H-12 (δH 4.68, 4.63) to C-11 (δC 175.0), and from H-7 (δH 2.46, 2.34) to C-8 (δC 163.1), C-9 (δC 135.4), and C-12 (δC 72.5), further established the planar structure and confirmed C-12 as a methylene group. The relative configuration of 1 was deduced from ROESY data (Figure 3). The key ROESY correlations of H3-13/H-5, and of H3-15 with H-2 and H-14, indicated an α-oriented hydroxyl group at C-2 and oxygenation at C-14. The absolute configuration of compound 1 was determined to be 2R,4S,5R,10S based on comparison of the experimental circular dichroism (CD) spectrum with the calculated electronic CD (ECD) spectra of tentative stereoisomers (Figure 4). Accordingly, compound 1 was named ramaribotrytol A.
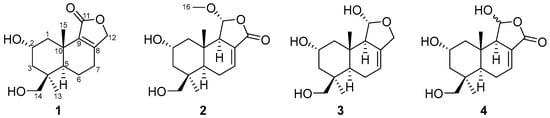
Figure 1.
Structures of compounds 1–4.

Table 1.
1H NMR (600 MHz) data of 1–4 (CD3OD).
Table 2.
13C NMR (150 MHz) data of 1–4 (CD3OD).
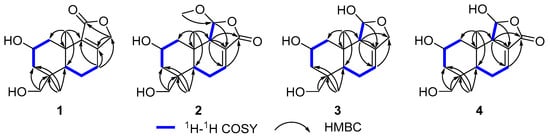
Figure 2.
Key 1H–1H COSY and HMBC correlations of 1–4.
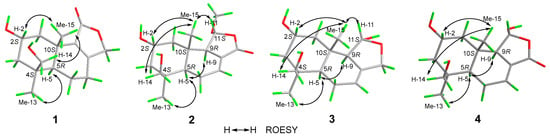
Figure 3.
Key ROESY correlations of 1–4.
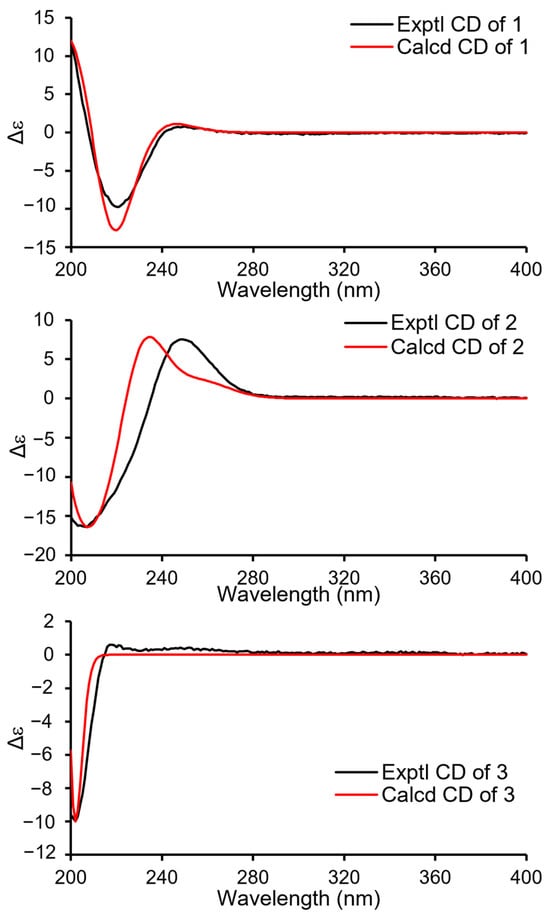
Figure 4.
ECD calculations of 1–3.
Compound 2 (Figure 1) was isolated as a colorless oil. (+)-HRESIMS analysis of 2 returned a protonated ion peak at m/z 297.16910 [M + H]+ (calcd. for C16H25O5, 297.16965), corresponding to the molecular formula C16H24O5. The 1H NMR spectrum of 2 (Table 1) revealed three methyl resonances at δH 1.04 (3H, s, H-13), δH 0.91 (3H, s, H-15) and δH 3.58 (3H, s, H-16), The 13C NMR (Table 2) and HSQC spectroscopic data of 2 (Table 2) exhibited 16 carbon signals, including three methyls at δC 27.7 (C-13), δC 16.5 (C-15), and δC 58.3 (C-16), four methylenes at δC 49.0 (C-1), δC 45.8 (C-3), δC 25.6 (C-6), and δC 65.5 (C-14), four sp3-hybridized methines at δC 64.3 (C-2), δC 50.8 (C-5), δC 59.1 (C-9), and δC 107.5 (C-11), two quaternary sp3 carbons at δC 40.8 (C-4) and δC 36.4 (C-10), two olefinic carbons at δC 138.1 (C-7) and δC 128.8 (C-8), together with a carbonyl group at δC 169.7 (C-12). The NMR spectroscopic features of 2 closely resembled those of 11α-ethoxycinamolide [15], differing by the presence of hydroxyl groups at C-2 and C-14 and a methoxy group at C-11 in place of an ethoxy substituent. These changes were supported by the 1H–1H COSY correlations of H2-1/H-2/H2-3, and chemical shifts in H2-14 and C-14, together with the HMBC correlations from H2-14 to C-3, C-4, and C-5, and from H3-16 to C-11 (Figure 2). The relative configuration of 2 was established by ROESY analysis. Key correlations of H3-15/H-11, H3-15/H2-14, H3-15/H-2, as well as H3-13/H-5, indicated the 2R*,4S*,5R*,9R*,10S*,11R* configuration of compound 2 (Figure 3). The absolute configuration of compound 2 was subsequently determined as 2R,4S,5R,9R,10S,11R by comparison of the experimental CD spectrum with calculated ECD spectrum (Figure 4). Therefore, compound 2 was named ramaribotrytol B.
Compound 3 (Figure 1) was isolated as a colorless oil. The molecular formula was determined as C15H24O4 based on (+)-HRESIMS, which displayed a protonated molecular ion at m/z 251.16412, [M − H2O + H]+ (calcd. for C15H23O3, 251.16417). The 1D NMR data of compound 3 (Table 1 and Table 2) showed high similarities to those of compound 2. The main difference between these two compounds is that the resonances for carbonyl group (C-12: δC 169.7) and methoxy (δC 58.3) in 2 were absent in 3, but with the presence of an oxygenated methylene (C-12: δC 69.3) in 3. These structural changes were confirmed by the HMBC correlations from H2-12 to C-7, C-8, C-9, and C-11 (Figure 2). Analysis of the ROESY spectrum (Figure 3) indicated that 3 shares the same relative configuration as 2, and its absolute configuration was determined as 2R,4S,5R,9R,10S,11R by comparison of the experimental CD spectrum with calculated ECD spectrum (Figure 4). Therefore, compound 3 was named ramaribotrytol C.
Compound 4 (Figure 1) was isolated as a colorless oil. (+)-HRESIMS analysis of 4 returned a protonated ion peak at m/z 283.15387 [M + H]+ (calcd. for C15H23O5, 283.15400), which was consistent with the molecular formula C15H22O5. The 1D NMR spectroscopic data of 4 (Table 1 and Table 2) closely resembled those of compound 2, except for the absence of resonances attributable to H-11 and C-11. Notably, the methoxy signal observed in 2 was absent in 4, suggesting the formation of a hemiacetal functionality at C-11 in 4. This change was consistent with the molecular formula and with the known propensity of hemiacetals to undergo epimerization. A similar phenomenon has been found in the related case of dendocarbin A [15]. The ROESY spectroscopic analysis indicated that compound 4 shares the same relative configuration as 2 at all stereogenic centers (Figure 3) except C-11, which existed as an epimeric mixture. On this basis, compound 4 was identified as a C-11 epimeric hemiacetal and was named ramaribotrytol D.
The isolated compounds were evaluated for antibacterial activity against Escherichia coli ATCC 25922, Staphylococcus aureus subsp. aureus ATCC 29213, Salmonella enterica subsp. enterica ATCC 14028, and Pseudomonas aeruginosa ATCC 27853. At a concentration of 200 μM, compounds 1–4 exhibited no significant inhibitory activity against any of the tested bacterial strains. In addition, these compounds showed no cytotoxic activity toward the human cancer cell lines examined.
3. Experimental Section
3.1. General Experimental Procedures
Measurements of optical rotations were carried out with an Autopol IV-T digital polarimeter (Rudolph, Hackettstown, NJ, USA). UV and CD spectra were obtained on a Hitachi UH5300 spectrophotometer (Hitachi, Tokyo, Japan) and a Chirascan Circular Dichroism Spectrometer (Applied Photophysics Limited, Leatherhead, Surrey, UK), respectively. NMR data, including both 1D and 2D spectra, were recorded on a Bruker Avance III 600 MHz spectrometer (Bruker Corporation, Karlsruhe, Germany). High-resolution mass spectrometry (HRESIMS) measurements were performed using a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Preparative high-performance liquid chromatography (prep-HPLC) was conducted on an Agilent 1260 Infinity II system, equipped with a Zorbax SB-C18 column (particle size: 5 μm, dimensions: 9.4 mm × 150 mm or 7 μm, 21.1 mm × 250 mm) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA). For column chromatography (CC), silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China) and Sephadex LH-20 (GE Healthcare, Upplasa, Sweden) were employed.
3.2. Fungal Material
The fruiting bodies of mushroom Ramaria botrytoides were bought from Mushuihua wild mushroom trading market, Kunming, Yunnan Province, China in July 2022. The fruiting bodies of the Ramaria botrytoides (10 kg) were washed three times with sterilized water and then chopped into small pieces. A small portion of sample was then freeze-dried using liquid nitrogen and ground into powder. Genomic DNA was extracted using a fungal genomic DNA rapid extraction kit (Shanghai Sangon Biotech, Shanghai, China). PCR amplification with the primers ITS1 and ITS4 was performed using the fungal genomic DNA as template, followed by Sanger sequencing. The sequence data has been submitted to the GenBank (Accession No. PX890821.1). The sequencing results were compared against the NCBI database, confirming the identification of the fungus as R. botrytoides.
3.3. Extraction and Isolation
The crushed fruiting bodies of R. botrytoides were soaked in chloroform/methanol (v/v 1:1) mixed solvent (40 L) for one week, followed by centrifugation to separate the fungal biomass and the liquid extract. The soaking and centrifugation steps were repeated once. The liquid layer was then concentrated into a brown viscous crude extract using a rotary evaporator at 45 °C under reduced pressure. The crude extract was completely resuspended in water and extracted twice with an equal volume of ethyl acetate. Then the ethyl acetate layer was collected and evaporated at 45 °C to dry. A total of 8.161 g of crude extract from the ethyl acetate layer was obtained.
The total crude extract was preliminarily separated by normal-phase silica gel column chromatography (column size: 6 cm i.d. × 50 cm length; silica gel bed height: 15 cm). The stationary phase consisted of 200–300 mesh silica gel, and the mobile phase was a gradient of petroleum ether/acetone mixed solvents (petroleum ether/acetone: v/v, 20:1, 10:1, 1:1, 500 mL each gradient) followed by methanol (500 mL), yielding fractions A–D.
Fraction D was subjected to normal-phase silica gel column chromatography (column size: 3 cm i.d. × 30 cm length; silica gel bed height: 15 cm) with a gradient elution using a petroleum ether/acetone (v/v, 20:1 to 1:1, 250 mL each gradient). The petroleum ether/acetone v/v = 1:1 eluted fraction was subjected to preparative HPLC (acetonitrile-H2O: 5–25%, 0–25 min, 4 mL/min) to afford compounds 1 (3.2 mg), 2 (3.9 mg), 3 (2.7 mg) and 4 (2.1 mg).
3.4. Characterization Data
3.4.1. Ramaribotrytol A (1)
Colorless oil; C15H22O4; [α]25.0D +32.0 (c 0.50, MeOH); UV (MeOH) λmax (log ε) 220 (3.78) nm; 1H NMR (600 MHz, CD3OD) data: Table 1 (Figure S1, Supplementary Materials), 13C NMR (150 MHz, CD3OD) data: Table 2 (Figure S2, Supplementary Materials); HRESIMS m/z 249.14847 [M − H2O + H]+ (calcd. for C15H21O3, 249.14852) (Figure S7, Supplementary Materials).
3.4.2. Ramaribotrytol B (2)
Colorless oil; C16H24O5; [α]25.0D −75.3 (c 0.50, MeOH); UV (MeOH) λmax (log ε) 220 (3.81) nm; 1H NMR (600 MHz, CD3OD) data: Table 1 (Figure S8, Supplementary Materials), 13C NMR (150 MHz, CD3OD) data: Table 2 (Figure S9, Supplementary Materials); HRESIMS m/z 297.16910 [M + H]+ (calcd. for C16H25O5, 297.16965) (Figure S14, Supplementary Materials).
3.4.3. Ramaribotrytol C (3)
Colorless oil; C15H24O4; [α]25.0D −41.1 (c 0.50, MeOH); UV (MeOH) λmax (log ε) 210 (6.35) nm; 1H NMR (600 MHz, CD3OD) data: Table 1 (Figure S15, Supplementary Materials), 13C NMR (150 MHz, CD3OD) data: Table 2 (Figure S16, Supplementary Materials); HRESIMS m/z 251.16412 [M − H2O + H]+ (calcd. for C15H23O3, 251.16417) (Figure S21, Supplementary Materials).
3.4.4. Ramaribotrytol D (4)
Colorless oil; C15H22O5; [α]25.0D −107.8 (c 0.50, MeOH); UV (MeOH) λmax (log ε) 225 (3.85) nm; 1H NMR (600 MHz, CD3OD) data: Table 1 (Figure S22, Supplementary Materials), 13C NMR (150 MHz, CD3OD) data: Table 2 (Figure S23, Supplementary Materials); HRESIMS m/z 283.15387 [M + H]+ (calcd. for C15H23O5, 283.15400) (Figure S28, Supplementary Materials).
3.5. ECD Calculation
The Gaussian 16 program was employed for computational tasks [16]. Conformers with a distribution above 1%, generated using the MMFF94s force field, were selected for further optimization at DFT/B3LYP/6-31G(d) level. Time-dependent DFT (TD-DFT) calculations of the ECD spectra were performed at the B3LYP/6-31G(d,p) level in MeOH, utilizing the IEFPCM model. ECD curves were processed by Boltzmann weighing of the selected conformers, and the results were plotted using SpecDis 1.71 [17] and a custom Microsoft Office Excel sheet.
3.6. Biological Activity Assay
3.6.1. Antibacterial Assays
The bacterial strains used in this study, Escherichia coli ATCC 25922, Staphylococcus aureus subsp. aureus ATCC 29213, Salmonella enterica subsp. enterica ATCC14028 and Pseudomonas aeruginosa ATCC27853, were obtained from the China General Microbiological Culture Collection Center (CGMCC), Beijing, China. Both strains were cultured overnight in Mueller–Hinton broth (Oxoid, Thermo Fisher Scientific) at 37 °C with shaking at 200 rpm. Following incubation, each culture was diluted 40-fold into fresh MHB broth and incubated for an additional 2–3 h under the same conditions. Mid-log phase cultures were adjusted to a concentration of 5 × 105 CFU/mL using McFarland Standards and used as the seeding solution for subsequent antibacterial assays. A volume of 100 μL of this seeding solution was added to each well of compound-containing 96-well plates, resulting in a final compound concentration of 200 μM. The plates were then covered and incubated at 37 °C for 24 h. Inhibitory rates were calculated using the formula [(1 − OD625sample)/OD625negative control] × 100%. Penicillin G sodium and ceftazidime were served as positive controls, with inhibitory rates exceeding 99% [4]. Initially, compounds were screened for antibacterial activity against two bacterial strains at a concentration of 200 μM. If an inhibitory rate greater than 50% was observed, additional concentrations of the compounds were tested to assess their antibacterial efficacy.
3.6.2. Cytotoxicity Assays
A549 human lung carcinoma cells (Conservation Genetics CAS Kunming Cell Bank, Yunnan, China) were cultured in high-glucose DMEM containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin, and incubated at 37 °C in a 5% CO2 atmosphere. Viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which measures the reduction in MTT to purple formazan by NAD(P)H-dependent cellular oxidoreductases, indicative of viable cells. For adherent cells, 100 μL of the cell suspension was seeded into 96-well plates and incubated for 12 h to allow attachment. The test compounds in DMSO were then added to each well, achieving a final concentration of 40 μM. Suspended cells were seeded at 1 × 105 cells/mL before compound addition. Cisplatin was used as a positive control. After 48 h of incubation, 100 μg of MTT was introduced into each well, and the cells were incubated for an additional 4 h. The culture medium was then removed, and the formazan product was solubilized using 100 μL of 20% SDS-50% DMF. Absorbance was measured at 595 nm using a microplate reader (Tecan Spark 10M, Männedorf, Switzerland). All assays were performed in triplicate, and the IC50 values were determined using a published method [6].
4. Conclusions
In conclusion, four previously undescribed drimane-type sesquiterpenoids (1–4) were isolated and characterized from the fruiting bodies of the wild mushroom Ramaria botrytoides, a chemically underexplored edible fungus. Compounds 1–4 represent highly oxidized drimane sesquiterpenes featuring a rare hydroxyl substitution at C-2, suggesting the involvement of unique oxidative enzymes in the post-biosynthetic modification of drimane scaffold in R. botrytoides. Compounds 1–4 were evaluated for antibacterial activity and cytotoxicity but did not exhibit significant biological effects. Overall, this study expands the chemical diversity of secondary metabolites from the medicinal and edible fungus R. botrytoides and enriches the structural repertoire of sesquiterpenoids derived from higher fungi. In addition, the identification of these metabolites provides preliminary chemical evidence supporting the future safety evaluation of R. botrytoides as an edible mushroom.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules31040645/s1, Figures S1–S32: HRESIMS, 1D and 2D NMR, and CD data of compounds 1–4; Tables S1–S16: ECD calculation details for compounds 1–3.
Author Contributions
H.-P.C. and X.L. designed the experiment; G.-K.P. and F.S. performed the isolation and identification of all the compounds, and also wrote this manuscript; X.L. made the funding acquisition; J.-K.L., H.-P.C. and X.L. provided comments and suggestions on structural elucidation and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant number 22307148), the Fundamental Research Funds for the Central Universities, South-Central Minzu University (grant numbers CZQ24022).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data in this research were presented in the manuscript and Supplementary Materials.
Acknowledgments
The authors thank the Bioactivity Screening Center, Kunming Institute of Botany, Chinese Academy of Sciences for screening the bioactivity of the compounds.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, H.P.; Liu, J.K. Secondary Metabolites from Higher Fungi. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Cham, Switzerland, 2018; Volume 106, pp. 1–201. [Google Scholar] [CrossRef]
- Gressler, M.; Löhr, N.A.; Schäfer, T.; Lawrinowitz, S.; Seibold, P.S.; Hoffmeister, D. Mind the mushroom: Natural product biosynthetic genes and enzymes of Basidiomycota. Nat. Prod. Rep. 2021, 38, 702–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Han, Y.-J.; Zhang, M.-H.; Zhang, K.-R.; Ng, T.B.; Liu, F. Purification and characterization of a novel ubiquitin-like antitumour protein with hemagglutinating and deoxyribonuclease activities from the edible mushroom Ramaria botrytis. AMB Exp. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Yang, Q.; Xu, H.; Dong, L. Drimane-type sesquiterpenoids from fungi. Chin. J. Nat. Med. 2022, 20, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-Z.; Wei, J.; Su, F.; Chen, H.-P.; Liu, J.-K. Matsulongifolins A–J: (−)-longifolane-type sesquiterpenoids from the mushroom Tricholoma matsutake (S. Ito & Imai) Singer. Phytochemistry 2025, 235, 114483. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, Z.-X.; Peng, G.-K.; Li, X.; Chen, H.-P.; Liu, J.-K. Vibralactone Derivatives Isolated from Co-cultures of the Basidiomycetes Stereum hirsutum and Boreostereum vibrans. Nat. Prod. Bioprospect. 2025, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-Y.; Gao, Y.; Yao, J.-N.; Zhou, L.; Chen, H.-P.; Liu, J.-K. Penisimplicins A and B: Novel Polyketide–Peptide Hybrid Alkaloids from the Fungus Penicillium simplicissimum JXCC5. Molecules 2024, 29, 613. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, H.-P.; Zhou, L.; Fan, J.; Awakawa, T.; Mori, T.; Ushimaru, R.; Abe, I.; Liu, J.-K. Cordycicadins A−D, Antifeedant Polyketides from the Entomopathogenic Fungus Cordyceps cicadae JXCH1. Org. Lett. 2022, 24, 8627–8632. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Du, W.; Zhang, X.; Lin, X.; Li, F.-R.; Yang, Q.; Wang, H.; Rudolf, J.D.; Zhang, B.; Dong, L.-B. Discovery, Structure, and Mechanism of a Class II Sesquiterpene Cyclase. J. Am. Chem. Soc. 2022, 144, 22067–22074. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hoefgen, S.; Valiante, V. Biosynthesis of Fungal Drimane-Type Sesquiterpene Esters. Angew. Chem. Int. Ed. 2021, 60, 23763–23770. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Chen, C.-T.; Lee, C.-F.; Huang, R.-J.; Chen, K.-L.; Lu, Y.-C.; Liang, S.-Y.; Pham, M.-T.; Rao, Y.K.; Wu, S.-H.; et al. The Biosynthetic Gene Cluster of Mushroom-Derived Antrocin Encodes Two Dual-Functional Haloacid Dehalogenase-like Terpene Cyclases. Angew. Chem. Int. Ed. 2023, 62, e202215566. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Valiante, V. Chemical Diversity and Biosynthesis of Drimane-Type Sesquiterpenes in the Fungal Kingdom. ChemBioChem 2022, 23, e202200173. [Google Scholar] [CrossRef] [PubMed]
- Sakio, Y.; Hirano, Y.J.; Hayashi, M.; Komiyama, K.; Ishibashi, M. Dendocarbins A-N, New Drimane Sesquiterpenes from the Nudibranch Dendrodoris carbunculosa. J. Nat. Prod. 2001, 64, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, R.; Cao, F.; Wang, J.; Hu, Z.; Zhang, Y. Proversilins A−E, Drimane-Type Sesquiterpenoids from the Endophytic Aspergillus versicolor. J. Nat. Prod. 2020, 83, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, Y.; Ludwiczuk, A.; Harinantenaina, L.; Toyota, M.; Nishiki, M.; Bardon, A.; Nii, K. Distribution of Drimane Sesquiterpenoids and Tocopherols in Liverworts, Ferns and Higher Plants: Polygonaceae, Canellaceae and Winteraceae Species. Nat. Prod. Commun. 2012, 7, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.