
Synthesis of Tricyclic and Tetracyclic Lactone Derivatives of Thieno[2,3-b]pyrazine or Thieno[2,3-b]quinoline: Preliminary Antitumor and Antiparasitic Activity Evaluation


 , ,
, ,  ,
, 
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis of Thieno[2,3-b]pyrazine Derivatives by Pd/Cu-Catalyzed Sonogashira Cross-Coupling Followed by Intramolecular Cyclization to Tricyclic Lactones
2.1.1. Pd/Cu-Catalyzed Sonogashira Cross-Coupling
2.1.2. Tandem One-Pot Reaction: Pd/Cu-Catalyzed Sonogashira Cross-Coupling Followed by 6-Endo-dig Cyclization to Tricyclic Lactones
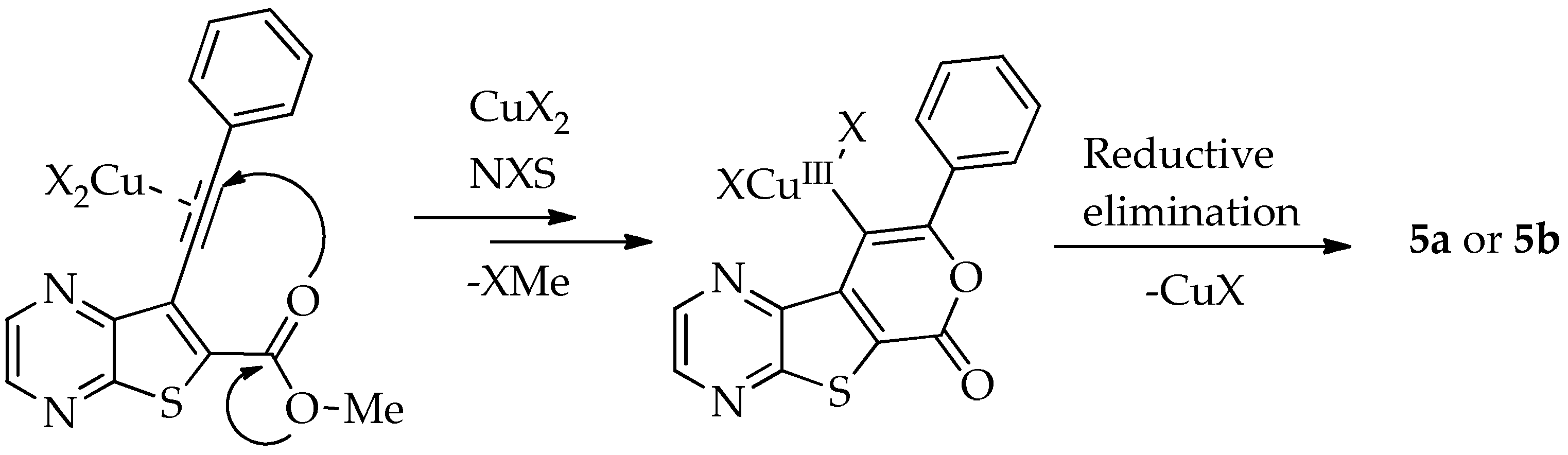
2.1.3. Halocyclization to 9-Halo Tricyclic Lactones
2.2. Synthesis of Tetracyclic Lactones Derived from Thieno[2,3-b]quinoline by Rh(III)-Catalyzed Formal [4+2] Cycloaddition, Triggered by C-H Activation
2.3. Cell Growth Inhibitory Effect of Compounds 2, 3, 5 and 7 on CaCo-2, MCF-7, AGS, HeLa, NCI-H460 Cell Lines and on a Non-Tumor Cell Line (PLP2)
2.4. Cell Growth Inhibitory Effect of Compounds 2c–g, 3a, 5a and 7 on T. brucei and L. infantum Promastigotes and THP-1-Derived Macrophage Cell Line as a Toxicity Model
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Thieno[2,3-b]pyrazine Derivatives by Pd/Cu-Catalyzed Sonogashira Cross-Coupling and Intramolecular Cyclization to Tricyclic Lactones
- 7-bromothieno[2,3-b]pyrazine-6-carboxylic acid (1b): Compound 1a (0.617 g, 2.26 mmol) was dissolved in THF/MeOH/H2O (6:1:1, 60 mL) and LiOH (0.203 g, 8.47 mmol) was added and the mixture was stirred for 3 h at rt, monitored by TLC. After partial removal of the solvents, the mixture was acidified using HClconc till pH = 5 and a precipitate came out. This was filtered under vacuum, washed with H2O and dried in the oven at 50 °C for several hours. Compound 1b was obtained as a white solid (0.530 g, 90%). 1H NMR (400 MHz, DMSO-d6): δ = 8.85 (1 H, d, J = 2.0 Hz, HetAr-H), 8.95 (1 H, d, J = 2.0 Hz, HetAr-H) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C7H479BrN2O2S: 258.9172, found 258.9169. Calculated for C7H481BrN2O2S: 260.9151, found 260.9146.
General Procedure for the Synthesis of Sonogashira Cross-Coupling Products (2a–g)
- Methyl 7-(phenylethynyl)thieno[2,3-b]pyrazine-6-carboxylate (2a) and 8-phenyl-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3a): Compound 1a (0.100 g, 0.366 mmol), phenylacetylene (0.0460 g, 0.0490 mL, 0.403 mmol) heating for 3.5 h. Column chromatography using a solvent gradient from 30 to 50% ether/petroleum ether gave compound 2a as a light yellow solid (0.0700 g, 65%), m.p. 145–147 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.98 (3 H, s, OCH3), 7.48–7.52 (3 H, m, Ar-H), 7.63–7.65 (2 H, m, Ar-H), 8.86 (1 H, d, J = 2.4 Hz, HetAr-H), 8.97 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.3 (OCH3), 81.8 (C), 99.6 (C), 121.5 (C), 121.7 (C), 129.0 (2 × ArCH), 129.8 (4′-CH), 131.7 (2 × Ar-CH), 136.3 (C), 144.3 (HetAr-CH), 144.7 (HetAr-CH), 147.8 (C), 154.3 (C), 161.1 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C16H11N2O2S: 295.0536; found: 295.0540.
- Methyl 7-[(4-aminophenyl)ethynyl]thieno[2,3-b]pyrazine-6-carboxylate (2b) and 8-(4-aminophenyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3b): Compound 1a (0.100 g, 0.366 mmol), 4-ethynylaniline (0.0490 g, 0.403 mmol) heating for 3 h. Column chromatography using a solvent gradient from 20 to 60% ethyl acetate/petroleum ether gave compound 2b as an orange solid (0.0683 g, 60%), m.p. 203–204 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.97 (3 H, s, OCH3), 5.80 (2 H, s, NH2), 6.61 (2 H, d, J = 8.8 Hz, 3′ and 5′-H), 7.31 (2 H, d, J = 8.8 Hz, 2′ and 6′-H), 8.84 (1 H, d, J = 2.4 Hz, HetAr-H), 8.95 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.0 (OCH3), 80.4 (C), 103.3 (C), 107.1 (C), 113.7 (3′ and 5′-CH), 122.9 (C), 133.2 (C), 133.4 (2′ and 6′-CH), 144.0 (HetAr-CH), 144.6 (HetAr-CH), 147.8 (C), 150.6 (C), 154.4 (C), 161.4 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C16H12N3O2S: 310.0645; found: 310.0645.
- Methyl 7-[(4-fluorophenyl)ethynyl]-thieno[2,3-b]pyrazine-6-carboxylate (2c) and 8-(4-fluorophenyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3c): Compound 1a (0.0760 g, 0.278 mmol), 1-ethynyl-4-fluorobenzene (0.0370 g, 0.306 mmol) heating for 4 h. Column chromatography using a solvent gradient from 30 to 50% ether/petroleum ether gave compound 2c as a white solid (0.0473 g, 55%), m.p. 175–176 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.99 (3 H, s, OCH3), 7.32–7.38 (2 H, m, 3′ and 5′-H), 7.68–7.73 (2 H, m, 2′ and 6′-H), 8.88 (1 H, d, J = 2.0 Hz, HetAr-H), 8.98 (1 H, d, J = 2.0 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.3 (OCH3), 81.6 (C), 98.5 (C), 116.4 (d, J = 22.1 Hz, 3′ and 5′-CH), 118.1 (d, J = 3.0 Hz, 1′-C), 121.3 (C), 134.2 (d, J = 9.1 Hz, 2′ and 6′-CH), 136.4 (C), 144.3 (HetAr-CH), 144.7 (HetAr-CH), 147.7 (C), 154.3 (C), 161.0 (C=O), 162.6 (d, J = 248.5 Hz, CF) ppm. 19F NMR (282.85 MHz, DMSO-d6) δ = −104.9 ppm. HRMS (ESI) [M + H]+, m/z Calculated for C16H10FN2O2S: 313.0441; found: 313.0441.
- Methyl 7-[(4-methoxyphenyl)ethynyl]thieno[2,3-b]pyrazine-6-carboxylate (2d): Compound 1a (0.0800 g, 0.290 mmol), 4-ethynylanisole (0.0420 g, 0.0410 mL, 0.320 mmol) heating for 4 h. Column chromatography using a solvent gradient from 10 to 40% ether/petroleum ether gave compound 2d as a yellow solid (0.0710 g, 75%), m.p. 150–152 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.82 (3 H, s, 4′-OCH3), 3.98 (3 H, s, OCH3), 7.04–7.07 (2 H, d, J = 8.8 Hz, 3′ and 5′-H), 7.58–7.60 (2 H, d, J = 8.8 Hz, 2′ and 6′-H), 8.86–8.87 (1 H, d, J = 2.4 Hz, HetAr-H), 8.97 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.2 (OCH3), 55.4 (4′-OCH3), 80.9 (C), 100.3 (C), 113.5 (C), 114.7 (3′ and 5′-CH), 122.0 (C), 133.5 (2′ and 6′-CH), 135.2 (C), 144.2 (HetAr-CH), 144.7 (HetAr-CH), 147.8 (C), 154.3 (C), 160.4 (C), 161.1 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C17H13N2O3S: 325.0641; found: 325.0642.
- Methyl 7-(p-tolylethynyl)thieno[2,3-b]pyrazine-6-carboxylate (2e) and 8-(p-Tolyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3e): Compound 1a (0.100 g, 0.366 mmol), 4-ethynyltoluene (0.0470 g, 0.403 mmol) heating for 4 h. Column chromatography using a solvent gradient from 10 to 20% ether/petroleum ether gave compound 2e as a yellow solid (0.0616 g, 55%) m.p. 146–147 °C. 1H NMR (400 MHz, DMSO-d6): δ = 2.37 (3 H, s, CH3), 3.99 (3 H, s, OCH3), 7.31 (2 H, d, J = 8.0 Hz, 3 ’and 5′-H), 7.53 (2 H, d, J = 8.0 Hz, 2′ and 6′-H), 8.87 (1 H, d, J = 2.0 Hz, HetAr-H), 8.98 (1 H, d, J = 2.0 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 21.2 (CH3), 53.2 (OCH3), 81.4 (C), 100.0 (C), 118.6 (C), 121.7 (C), 129.6 (3′ and 5′-CH), 131.7 (2′ and 6′-CH), 135.9 (C), 139.8 (C), 144.2 (HetAr-CH), 144.7 (HetAr-CH), 147.8 (C), 154.3 (C), 161.1 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C17H13N2O2S: 309.0692; found: 309.0696.
- Methyl 7-(pyridin-2-ylethynyl)thieno[2,3-b]pyrazine-6-carboxylate (2f) and 8-(Pyridin-2-yl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3f): Compound 1a (0.0800 g, 0.290 mmol), 2-ethynylpyridine (0.0340 g, 0.0330 mL, 0.320 mmol) heating for 5 h. Column chromatography using a solvent gradient from 10 to 60% ethyl acetate/petroleum ether gave compound 2f as a white solid (0.0420g, 50%), m.p. 183–185°C. 1H NMR (400 MHz, DMSO-d6): δ = 3.99 (3 H, s, OCH3), 7.48–7.51 (1 H, ddd, J = 7.6, 4.8 and 1.2 Hz, 5′-H), 7.72–7.74 (1 H, dt, J = 7.6, 1.2 and 0.8 Hz, 3′-H), 7.90–7.94 (1 H, app. td, J= 7.6 and 1.6 Hz, 4′-H), 8.67–8.69 (1 H, dq, J = 4.8, 1.6 and 0.8 Hz, 6′-H), 8.89 (1 H, d, J = 2.4 Hz, HetAr-H), 8.99 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.3 (OCH3), 80.4 (C), 98.4 (C), 120.6 (C), 124.3 (5′-CH), 128.0 (3′-CH), 137.0 (4′-CH), 137.7 (C), 141.7 (C), 144.4 (HetAr-CH), 144.8 (HetAr-CH), 147.9 (C), 150.5 (6′-CH), 154.2 (C), 160.9 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C15H10N3O2S: 296.0488; found: 296.0488.
- Methyl 7-(thiophen-3-ylethynyl)thieno[2,3-b]pyrazine-6-carboxylate (2g) and 8-(thiophen-3-yl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3g): Compound 1a (0.100 g, 0.366 mmol), 3-ethynylthiophene (0.0450 g, 0.0410 mL, 0.403 mmol) heating for 3.5 h. Column chromatography using a solvent gradient from 20 to 60% ether/petroleum ether gave compound 2g as a dark orange solid (0.0840 g, 75%), m.p. 158–160 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.98 (3 H, s, OCH3), 7.33 (1 H, dd, J = 5.2 and 1.2 Hz, 4′-H), 7.72 (1 H, dd, J = 5.2 and 2.8 Hz, 5′-H), 8.05 (1 H, dd, J = 2.8 and 1.2 Hz, 2′-H), 8.87 (1 H, d, J = 2.4 Hz, HetAr-H), 8.96 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 53.2 (OCH3), 81.2 (C), 95.2 (C), 120.5 (C), 121.6 (C), 127.5 (5′-CH), 129.6 (4′-CH), 131.7 (2′-CH), 135.9 (C), 144.2 (HetAr-CH), 144.7 (HetAr-CH), 147.8 (C), 154.2 (C), 161.0 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C14H9N2O2S2: 301.0100; found: 301.0096.
- Methyl 7-[(4-cyanophenyl)ethynyl]thieno[2,3-b]pyrazine-6-carboxylate (2h): Compound 1a (0.050 g, 0.183 mmol), 4-ethynylbenzonitrile (0.0256 g, 0.201 mmol) heating for 4 h. After cooling, H2O was added and a precipitate came out. This was filtered under vacuum, dried in the oven overnight at 50 °C giving a light brown solid, that after some washes with dry ether gave compound 2h as a beige solid (0.0555 g, 95%), m.p. 281–282 °C. 1H NMR (400 MHz, DMSO-d6, 60 °C): δ = 4.00 (3 H, s, OCH3), 7.80 (2 H, d, J = 8.0 Hz, 2′ and 6’-H), 7.92 (2 H, d, J = 8.0 Hz, 3′ and 5’-H), 8.86 (1 H, d, J = 2.0 Hz, HetAr-H), 8.96 (1 H, d, J = 2.0 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 60 °C): δ = 52.9 (OCH3), 84.9 (C), 97.2 (C), 111.6 (C), 117.8 (C), 120.3 (C), 126.2 (C), 132.1 (2′ and 6’-CH), 132.4 (3′ and 5’-CH), 137.4 (C), 144.0 (HetAr-CH), 144.4 (HetAr-CH), 147.4 (C), 154.0 (C), 161.5 (C=O) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C17H10N3O2S: 320.0489; found: 320.0492.
General Procedure for Tricyclic Lactones (3a–g) by One Pot Tandem Sonogashira Coupling and Intramolecular Cyclization
- 8-Phenyl-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3a) and 7-(phenylethynyl)thieno[2,3-b]pyrazine (4a): Compound 1b (0.100 g, 0.386 mmol), phenylacetylene (0.0430 g, 0.0466 mL, 0.425 mmol) and heating for 1 h. Column chromatography using a solvent gradient from 10 to 20% ether/petroleum ether gave compound 3a as a yellow solid (0.0630 g, 59%), m.p. 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.53–7.58 (3 H, m, Ar-H), 7.93 (1 H, s, 9-H), 8.04–8.06 (2 H, m, Ar-H), 8.94 (1 H, d, J = 2.4 Hz, HetAr-H), 9.03 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 96.6 (9-CH), 122.9 (C), 125.5 (2 × Ar-CH), 129.2 (2 × Ar-CH), 130.8 (4′-CH), 130.9 (C), 141.5 (C), 143.8 (C), 143.9 (HetAr-CH), 145.6 (HetAr-CH), 157.5 (C), 157.6 (C), 158.1 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C15H9N2O2S: 281.0379; found: 281.0379.
- 8-(4-Aminophenyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3b): Compound 1b (0.100 g, 0.386 mmol), 4-ethynylaniline (0.0410 g, 0.340 mmol) and heating for 2 h. Column chromatography using a solvent gradient from 20 to 50% ethyl acetate/petroleum ether gave compound 3b as a dark orange solid (0.0570 g, 63%), m.p. 286–287 °C. 1H NMR (400 MHz, DMSO-d6): δ = 5.89 (2 H, s, NH2), 6.66 (2 H, d, J = 8.8 Hz, 3′ and 5′-H), 7.57 (1 H, s, 9-H), 7.72 (2 H, d, J = 8.8 Hz, 2′ and 6′-H), 8.91 (1 H, d, J = 2.4 Hz, HetAr-H), 8.99 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 92.6 (9-CH), 113.7 (3′ and 5′-CH), 117.5 (C), 119.3 (C), 127.1 (2′ and 6′-CH), 142.5 (C), 143.7 (HetAr-CH), 143.9 (C), 145.5 (HetAr-CH), 151.7 (C), 157.9 (C), 158.3 (C), 159.7 (C) ppm. Calculated for C15H10N3O2S: 296.0488; found: 296.0488. HPLC retention time = 4.02 min, 3b purity is 100%.
- 8-(4-Fluorophenyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3c) and 7-[(4-Fluorophenyl)ethynyl]thieno[2,3-b]pyrazine (4c): Compound 1b (0.100 g, 0.386 mmol), 1-ethynyl-4-fluorobenzene (0.0520 g, 0.425 mmol) and heating for 2 h. Column chromatography using a solvent gradient from 10 to 50% ether/petroleum ether gave compound 3c as a yellow solid (0.0580 g, 35%), m.p. 269–271 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.37–7.42 (2 H, m, 3′ and 5′-H), 7.96 (1 H, s, 9-H), 8.12-8.15 (2 H, m, 2′ and 6′-H), 8.95 (1 H, d, J = 2.4 Hz, HetAr-H), 9.04 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 96.7 (9-CH), 116.4 (d, J = 22.1 Hz, 3′ and 5′-CH), 122.9 (C), 127.7 (d, J =3.0 Hz, 1′-C), 128.2 (d, J = 9.1 Hz, 2′ and 6′-CH), 141.6 (C), 143.9 (C), 144.1 (HetAr-CH), 145.8 (HetAr-CH), 156.9 (C), 157.6 (C), 158.3 (C), 163.6 (d, J = 250.5 Hz, CF) ppm. 19F NMR (282.85 MHz, DMSO-d6) δ = −105.5 ppm. HRMS (ESI) [M + H]+, m/z Calculated for C15H8FN2O2S: 299.0285; found: 299.0280. HPLC retention time = 12.31 min, 3c purity is 96%.
- 8-(4-Methoxyphenyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3d) and 7-[(4-methoxyphenyl)ethynyl]thieno[2,3-b]pyrazine (4d): Compound 1b (0.0900 g, 0.347 mmol), 4-ethynylanisole (0.0520 g, 0.382 mmol) and heating for 1 h. Column chromatography using a solvent gradient from 10 to 40% ether/petroleum ether gave compound 3d as a yellow solid (0.0570 g, 53%), m.p. 222–224 °C. 1H NMR (400 MHz, DMSO-d6): δ = 3.83 (3 H, s, OCH3), 7.09 (2 H, d, J = 8.8 Hz, 3′ and 5′-H), 7.80 (1 H, s, 9-H), 7.99 (2 H, d, J = 8.8 Hz, 2′ and 6-H), 8.96 (1 H, d, J = 2.4 Hz, HetAr-H), 9.05 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 55.6 (OCH3), 95.2 (9-CH), 114.8 (3′ and 5′-CH), 121.7 (C), 123.5 (C), 127.4 (2′ and 6′-CH), 142.1 (C), 143.96 (C), 143.98 (HetAr-CH), 145.8 (HetAr-CH), 157.8 (C), 158.1 (C), 158.4 (C), 161.5 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C16H11N2O3S: 311.0485; found: 311.0486. HPLC retention time = 12.68 min, 3d purity is 97%.
- 8-(p-Tolyl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3e) and 7-(p-tolylethynyl)thieno[2,3-b]pyrazine (4e): Compound 1b (0.100 g, 0.386 mmol), 4-ethynyltoluene (0.0490 g, 0.425 mmol) and heating for 2 h. Column chromatography using a solvent gradient from 5 to 40% ether/hexane gave compound 3e as a yellow solid (0.0560 g, 50%), m.p. 241–243 °C. 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (3 H, s, CH3), 7.36 (2 H, d, J = 8.0 Hz, 3′ and 5′-H), 7.78 (1 H, s, 9-H), 7.90 (2 H, d, J = 8.0 Hz, 2′ and 6′-H), 8.90 (1 H, d, J = 2.4 Hz, HetAr-H), 8.99 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 20.5 (CH3), 95.6 (9-CH), 122.1 (C), 125.1 (2′ and 6′-CH), 128.1 (C), 129.4 (3′ and 5′-CH), 140.5 (C), 141.3 (C), 143.45 (HetAr-CH), 143.5 (C), 145.1 (HetAr-CH), 157.1 (C), 157.9 (C), 158.0 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C16H11N2O2S: 295.0536; found: 295.0534. HPLC retention time = 18.5 min, 3e purity is 99%.
- 8-(Pyridin-2-yl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3f): Compound 1b (0.100 g, 0.386 mmol), 2-ethynylpyridine (0.0450 g, 0.0440 mL, 0.425 mmol) and heating for 1 h. Column chromatography using a solvent gradient from 5 to 30% ethyl acetate/petroleum ether gave compound 3f as a yellow solid (0.040 g, 50%), m.p. 238–239 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.53–7.56 (1 H, m, 5′-H), 8.02–8.03 (2 H, m, 3′ and 4′-H), 8.13 (1 H, s, 9-H), 8.74–8.76 (1 H, m, 6′-H), 8.95 (1 H, d, J = 2.4 Hz, HetAr-H), 9.05 (1 H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 97.6 (9-CH), 120.1 (3′ or 4′-CH), 124.6 (C), 125.4 (5′-CH), 138.0 (3′ or 4′-CH), 141.0 (C), 143.9 (C), 144.3 (HetAr-CH), 145.8 (HetAr-CH), 148.1 (C), 150.3 (6′-CH), 156.5 (C), 157.2 (C), 158.2 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C14H8N3O2S: 282.0332; found: 282.0333.
- 8-(Thiophen-3-yl)-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (3g) and 7-(thiophen-3-ylethynyl)thieno[2,3-b]pyrazine (4g): Compound 1b (0.0900 g, 0.347 mmol), 3-ethynylthiophene (0.0430 g, 0.0390 mL, 0.382 mmol) and heating for 1 h. Column chromatography using a solvent gradient from 20 to 40% ethyl ether/petroleum ether gave compound 3g as a yellow solid (0.0600 g, 54%), m.p. 247–249 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.73–7.75 (1 H, dd, J = 5.2 and 2.8 Hz, 5′-H), 7.81 (1 H, dd, J = 5.2 and 1.2 Hz, 4′-H), 7.84 (1 H, s, 9-H), 8.27 (1 H, dd, J = 2.8 and 1.2 Hz, 2′-H), 8.93 (1 H, d, J = 2.4 Hz, HetAr-H), 9.02 (1H, d, J = 2.4 Hz, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 96.3 (9-CH), 122.2 (C), 125.2 (4′-CH), 126.1 (2′-CH), 128.4 (5′-CH), 133.4 (C), 141.8 (C), 143.88 (C), 143.91 (HetAr-CH), 145.7 (HetAr-CH), 154.6 (C), 157.5 (C), 158.2 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C14H9N2O2S2: 301.0100; found: 301.0096.
General Procedure for the 9-HaloTricyclic Lactones (5a and 5b)
- 9-Chloro-8-phenyl-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (5a): Compound 2a (0.0500 g, 0.170 mmol), NCS (0.0450 g, 0.340 mmol) and CuCl2 (0.0580 g, 0.425 mmol), gave compound 5a as a yellow solid (0.0324 g, 60%), m.p. 313–314 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.57–7.59 (3 H, m, Ar-H), 7.82–7.84 (2 H, m, Ar-H), 8.98 (1 H, broad s, HetAr-H), 9.08 (1 H, broad s, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 108.2 (C), 125.2 (C), 128.3 (2 × Ar-CH), 129.0 (2 × Ar-CH), 130.2 (C), 130.4 (4′-CH), 136.6 (C), 143.6 (HetAr-CH), 143.8 (C), 144.8 (HetAr-CH), 153.5 (C), 156.5 (C), 157.5 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C15H835ClN2O2S: 314.9989; found: 314.9989.
- 9-Bromo-8-phenyl-6H-pyrano[4′,3′:4,5]thieno[2,3-b]pyrazin-6-one (5b): Compound 2a (0.0560 g, 0.190 mmol), NBS (0.0680 g, 0.381 mmol) and CuBr2 (0.107 g, 0.478 mmol), gave compound 5b as a yellow solid (0.0340 g, 50%), m.p. 313–315 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.56 (3 H, m, Ar-H), 7.77 (2 H, m, Ar-H), 8.95 (1 H, broad s, HetAr-H), 9.06 (1 H, broad s, HetAr-H) ppm. 13C NMR (100.6 MHz, DMSO-d6): δ = 94.4 (C), 125.3 (C), 127.9 (2 × Ar-CH), 129.2 (2 × Ar-CH), 130.1 (4′-CH), 131.5 (C), 136.9 (C), 142.9 (HetAr-CH), 143.8 (C), 144.6 (HetAr-CH), 154.5 (C), 156.6 (C), 157.5 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C15H879BrN2O2S: 358.9484; found: 358.9484. m/z Calculated for C15H881BrN2O2S: 360.9464; found: 360.9464.
3.1.2. Synthesis of Tetracyclic Lactones Derived from Thieno[2,3-b]quinoline by Rh(III)-Catalyzed Formal [4+2] Cycloaddition, Triggered by C-H Activation
General Procedure for the C-H Activation/Cycloaddition (7a–f)
- 3,4-Diphenyl-1H-pyrano[4′,3′:4,5]thieno[2,3-b]quinolin-1-one (7a): Compound 6 (0.0500 g, 0.218 mmol) and diphenylacetylene (0.0400 g, 0.218 mmol). Column chromatography using a solvent gradient from 5 to 40% ether/petroleum ether gave compound 7a as a yellow solid (0.0500 g, 56%), m.p. 304–305 °C. 1H NMR (400 MHz, DMSO-d6, 80 °C): δ = 7.25 (1 H, s, 5-H), 7.30–7.35 (3 H, m, Ar-H), 7.44–7.47 (2 H, m, Ar-H), 7.50–7.66 (7 H, m, Ar-H), 7.87–7.91 (1 H, m, Ar-H), 8.07–8.10 (1 H, broad d, J = 8.4 Hz, Ar-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 80 °C): δ = 115.8 (C) 122.0 (C), 124.0 (C), 126.2 (CH), 127.0 (C), 127.3 (CH), 127.7 (2 × CH), 128.6 (2 × CH), 128.7 (2 × CH), 129.0 (2 × CH), 129.1 (CH), 130.6 (2 × CH), 131.7 (CH), 131.8 (C), 133.0 (C), 134.2 (5-CH), 141.2 (C), 147.3 (C), 154.3 (C), 157.1 (C), 162.2 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C26H16NO2S: 406.0896; found: 406.0897.
- 3,4-Dibutyl-1H-pyrano[4′,3′:4,5]thieno[2,3-b]quinolin-1-one (7b): Compound 6 (0.0500 g, 0.218 mmol) and 5-decyne (0.0300 g, 0.0390 mL, 0.218 mmol). Column chromatography using a solvent gradient from 5 to 20% ether/petroleum ether afforded compound 7b as a yellow solid (0.0680 g, 85%), m.p.172–173 °C. 1H NMR (400 MHz, DMSO-d6, 80 °C): δ = 0.92–0.99 (6 H, m, 2 × CH3), 1.41–1.43 (2 H, m, CH2), 1.59–1.67 (6 H, m, 3 × CH2), 2.72–2.76 (2 H, t, J = 7.6 Hz, CH2), 3.05–3.09 (2 H, m, CH2), 7.71–7.75 (1 H, m, Ar-H), 7.94–7.99 (1 H, m, Ar-H), 8.11–8.13 (1H, broad d, J = 8.4 Hz, Ar-H), 8.31–8.33 (1 H, broad d, J = 8.4 Hz, Ar-H), 9.26 (1 H, s, 5-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 80 °C): δ = 13.7 (CH3), 13.9 (CH3), 21.7 (CH2), 21.8 (CH2), 25.8 (CH2), 29.5 (CH2) 29.8 (CH2), 31.5 (CH2), 113.6 (C), 121.8 (C), 125.1 (C), 126.5 (CH), 127.0 (C), 127.5 (CH), 130.0 (CH), 132.2 (CH), 135.7 (5-CH), 141.6 (C), 147.4 (C), 158.0 (C), 158.4 (C), 162.3 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C22H24NO2S: 366.1522; found: 366.1526.
- 3,4-Bis(4-bromophenyl)-1H-pyrano[4′,3′:4,5]thieno[2,3-b]quinolin-1-one (7c): Compound 6 (0.0500 g, 0.218 mmol) and bis(4-bromophenyl)acetylene (0.0730 g, 0.218 mmol). Column chromatography using a solvent gradient from 5 to 20% ethyl acetate/petroleum ether gave compound 7c as a yellow solid (0.0920 g, 75%), m.p. 307–308 °C. 1H NMR (400 MHz, DMSO-d6, 80 °C): δ = 7.36 (2 H, d, J = 8.4 Hz, Ar-H), 7.40 (1 H, s, 5-H), 7.51 (2 H, d, J = 8.4 Hz, Ar-H), 7.56 (2 H, d, J = 8.4 Hz, Ar-H), 7.61–7.62 (2 H, m, Ar-H), 7.79 (2 H, d, J = 8.4 Hz, Ar-H), 7.88–7.92 (1 H, m, Ar-H), 8.08 (1 H, broad d, J = 8.8 Hz, Ar-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 80 °C): δ = 115.0 (C), 122.4 (C), 122.9 (C), 123.9 (C), 126.3 (CH), 126.7 (C), 127.2 (CH), 128.9 (CH), 130.7 (2 × CH), 130.8 (C), 130.9 (2 × CH), 131.8 (CH), 132.0 (C), 132.1 (2 × CH), 132.7 (2 × CH), 134.0 (5-CH), 140.5 (C), 147.3 (C), 153.4 (C), 156.8 (C), 162.0 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C26H1479Br2NO2S: 561.9106; found: 561.9105; m/z Calculated for C26H1479Br81BrNO2S: 563.9086; found: 563.9085; m/z Calculated for C26H1481Br2NO2S: 565.9066; found: 565.9064.
- 3,4-Bis(4-methoxyphenyl)-1H-pyrano[4′,3′:4,5]thieno[2,3-b]quinolin-1-one (7d): Compound 6 (0.0500 g, 0.218 mmol) and 1,2-bis(4-methoxyphenyl)ethyne (0.0520 g, 0.218 mmol). Column chromatography using a solvent gradient from 5 to 15% ethyl acetate/petroleum ether gave compound 7d as a yellow solid (0.0620 g, 61%), m.p. 304–306 °C.1H NMR (400 MHz, DMSO-d6, 100 °C): δ = 3.77 (3 H, s, OCH3), 3.93 (3 H, s, OCH3), 6.88 (2 H, d, J = 8.8 Hz, Ar-H), 7.16 (2 H, d, J = 8.8 Hz, Ar-H), 7.38–7.45 (5 H, m, 5-H and 4 × Ar-H), 7.57–7.62 (2 H, m, Ar-H), 7.86–7.90 (1 H, m, Ar-H), 8.08 (1 H, broad d, J = 8.6 Hz, Ar-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 100 °C): δ = 54.8 (OCH3), 55.1 (OCH3), 113.3 (2 × CH), 114.6 (C), 114.7 (2 × CH), 121.0 (C), 123.9 (C), 124.2 (C), 125.2 (C), 126.0 (CH), 127.1 (C), 127.14 (CH), 128.7 (CH), 130.0 (2 × CH), 131.4 (CH), 131.7 (2 × CH), 134.2 (5-CH), 141.8 (C), 147.2 (C), 154.4 (C), 157.0 (C), 159.7 (C), 159.8 (C), 162.2 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C28H20NO4S: 466.1107; found: 466.1107.
- 3,4-Bis(4-(trifluoromethyl)phenyl)-1H-pyrano[4’,3’:4,5]thieno[2,3-b]quinolin-1-one (7e): Compound 6 (0.0400 g, 0.175 mmol) and 1,2-bis(4-(trifluoromethyl)phenyl)ethyne (0.0550 g, 0.175 mmol). Column chromatography using a solvent gradient from 5 to 30% ether/petroleum ether gave compound 7e as a yellow solid (0.0470 g, 50%), m.p. 276–278 °C. 1H NMR (400 MHz, DMSO-d6, 80 °C): δ = 7.23 (1 H, s, 5-H), 7.50 (1 H, broad d, J = 8.4 Hz, Ar-H), 7.59–7.71 (5 H, m, Ar-H), 7.81 (2 H, d, J = 8.0 Hz, Ar-H), 7.83–7.93 (1 H, m, Ar-H), 9.94 (2 H, d, J = 8.0 Hz, Ar-H) 8.09 (1 H, broad d, J = 8.4 Hz, Ar-H) ppm. 13C NMR (100.6 MHz, DMSO-d6, 80 °C): δ = 115.6 (C), 122.3 (C), 123.1 (C) 123.4 (q, J = 271.2 Hz, CF3), 123.7 (q, J = 271.2 Hz, CF3), 124.6 (q, J = 3.0 Hz, 2 × CH), 125.8 (q, J = 3.0Hz, 2 × CH), 126.3 (CH), 126.6 (C), 127.3 (CH), 128.6 (CH), 129.6 (q, J = 32.2 Hz, CCF3), 129.7 (2 × CH), 129.9 (q, J = 32.2 Hz, CCF3), 131.8 (2 × CH), 131.9 (CH), 133.9 (5-CH), 135.4 (C), 137.0 (C), 140.1 (C), 147.4 (C) 152.9 (C), 156.7 (C), 162.0 (C) ppm. 19F NMR (282.85 MHz, DMSO-d6) δ = -57.4 (s), −57.0 (s) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C28H14F6NO2S: 542.0644; found: 542.0643. HPLC retention time = 7.9 min, 7e purity is 96%.
- 4-Methyl-3-phenyl-1H-pyrano[4’,3’:4,5]thieno[2,3-b]quinolin-1-one (7f): Compound 6 (0.0500 g, 0.218 mmol) and 1-phenyl-1-propyne (0.0250 g, 0.0270 mL, 0.218 mmol). Column chromatography using a solvent gradient from 5 to 10% ethyl acetate/petroleum ether gave compound 7f as a yellow solid (0.0520 g, 70%), m.p. 301–302 °C. 1H NMR (400 MHz, DMSO-d6, 80 °C): δ = 2.72 (3 H, s, CH3), 7.55–7.61 (3 H, m, Ar-H), 7.66–7.73 (3 H, m, Ar-H), 7.93–7.97 (1 H, m, Ar-H), 8.12 (1 H, broad d, J = 8.8 Hz, Ar-H), 8.32–8.35 (1 H, broad, J = 8.8 Hz, Ar-H), 9.52 (1 H, s, 5-H) ppm. The regioisomer 7f was assigned using nOe. NMR (100.6 MHz, DMSO-d6, 80 °C): δ = 15.0 (CH3), 110.1 (C), 122.2 (C), 124.9 (C), 126.0 (CH), 127.2 (CH), 127.6 (C), 128.2 (2 × CH), 129.1 (2 × CH); 129.5 (CH), 129.6 (CH), 131.7 (CH), 131.9 (C), 135.7 (5-CH), 142.2 (C), 147.3 (C), 154.3 (C), 157.3 (C), 162.2 (C) ppm. HRMS (ESI) [M + H]+, m/z Calculated for C21H14NO2S: 344.0740; found: 344.0740.
3.2. Biological Activities
3.2.1. In Vitro Antitumor Evaluation (SRB Assay)
3.2.2. In Vitro Anti-Parasitic Evaluation
Parasite Cultures
Cell Cultures
In Vitro Evaluation of Anti-T. brucei Activity
Evaluation of Activity Against L. infantum MHOM/MA/67/ITMAP-263 Promastigotes
Cytotoxicity in THP-1 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Folmer, B.J.B.; Man, A.P.A.; Gernette, E.S.; Azevedo, R.C.R.G.; Ibrahim, H. Thieno[2,3-b]pyrazine Compounds as B-RAF Inhibitors. WO 2011147764, A1, 1 December 2011. [Google Scholar]
- Gong, Y.D.; Kwak, S.; Lee, E.S. Substituted Thieno[3,2-b]Pyrazines for Inhibiting Cancer Cell Proliferation and Inducing Cancer Cell Apoptosis. WO 2016093554, A1, 16 June 2016. [Google Scholar]
- Lim, J.; Altman, M.D.; Gibeau, C.R. Thienopyrazine Inhibitors of IRAK4 Activity. WO 2016144849, A1, 15 September 2016. [Google Scholar]
- Guerin, D.J.; Bair, K.W.; Caravella, J.A.; Ioannidis, S.; Lancia, D.R., Jr.; Li, H.; Mischke, S.; Ng, P.Y.; Richard, D.; Sciller, S.E.R.; et al. Thienopyrazine Carboxamides as Ubiquitin-Specific Proteases Inhibitors. WO 2017139779, A1, 17 August 2017. [Google Scholar]
- Teja, C.; Nawaz Khan, F.R. Recent Advances in the Synthesis of Thienoquinolines (Quinoline-fused heterocycle). Asian. J. Org. Chem. 2020, 9, 1889–1900. [Google Scholar] [CrossRef]
- Rechfeld, F.; Gruber, P.; Kirchmair, J.; Boehler, M.; Hauser, N.; Hechenberger, G.; Garczarczyk, D.; Lapa, G.B.; Preobrazhenskaya, M.N.; Goekjian, P.; et al. Thienoquinolines as novel disruptors of the PKCε/RACK2 protein-protein interaction. J. Med. Chem. 2014, 57, 3235–3246. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.; Nikam, M.; Asrondkar, A.; Bobade, A.; Gill, C. Synthesis, Antioxidant, and Anti-Inflammatory Evaluation of Novel Thiophene-Fused Quinoline Based β-Diketones and Derivatives. J. Heterocycl. Chem. 2017, 54, 1415–1422. [Google Scholar] [CrossRef]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Ramadan, M.; Abdelrahman, M.H.; Abbas Bukhari, S.N.; Ali, T.F.S.; Abuo-Rahma, G.E.D.A. Discovery of novel thienoquinoline-2-carboxamide chalcone derivatives as antiproliferative EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. 2019, 27, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Larock, R.C. Synthesis of isocoumarins and α-pyrones via electrophilic cyclization. J. Org. Chem. 2003, 68, 5936–5942. [Google Scholar] [CrossRef]
- Jithunsa, M.; Ueda, M.; Miyata, O. Copper(II)chloride-mediated cyclization reaction of N-alkoxy- ortho-alkynylbenzamides. Org. Lett. 2011, 13, 518–521. [Google Scholar] [CrossRef]
- Calhelha, R.C.; Vale-Silva, L.A.; Pinto, E.; São-José Nascimento, M.; Queiroz, M.J.R.P. Synthesis of Novel 3-(Aryl)Benzothieno[2,3-c]Pyran-1-ones from Sonogashira Products and Intramolecular Cyclization: Antitumoral Activity Evaluation. Eur. J. Med. Chem. 2009, 44, 1893–1899. [Google Scholar] [CrossRef]
- Rodrigues, J.M.; Cendón, B.; Gulías, M.; Mascareñas, J.L.; Queiroz, M.J.R.P. Rhodium(III)-Catalyzed Formal Cycloaddition between Thienopyridine/Thienopyrazine Carboxylic Acids and Alkynes, Triggered by C−H Activation. Eur. J. Org. Chem. 2021, 2021, 3234–3240. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Kanwal, I.; Mujahid, A.; Rasool, N.; Rizwan, K.; Malik, A.; Ahmad, G.; Shah, S.A.A.; Rashid, U.; Nasir, N.M. Palladium and Copper Catalyzed Sonogashira cross Coupling an Excellent Methodology for C-C Bond Formation over 17 Years: A review. Catalysts 2020, 10, 443. [Google Scholar] [CrossRef]
- Yogeshwaran, V.; Dhayanithi, S.; Vinoth Kumar, P.; Mohana Roopan, S. 6-Endo-Dig Cyclization: Flexible Enforce to Develop Synthetic Route in Organic Syntheses. Results Chem. 2023, 6, 101131. [Google Scholar] [CrossRef]
- Rodrigues, J.M.; Calhelha, R.C.; Nogueira, A.; Ferreira, I.C.F.R.; Barros, L.; Queiroz, M.J.R.P. Synthesis of novel methyl 7-[(Hetero)arylamino]thieno[2,3-b]pyrazine-6-carboxylates and antitumor activity evaluation: Effects in human tumor cells growth, cell cycle analysis, apoptosis and toxicity in non-tumor cells. Molecules 2021, 26, 4823. [Google Scholar] [CrossRef] [PubMed]
- Duplais, C.; Forman, A.J.; Baker, B.A.; Lipshutz, B.H. UC Pd: A New form of Pd/C for Sonogashira Couplings. Chem. Eur. J. 2010, 16, 3366–3371. [Google Scholar] [CrossRef]
- Campos, J.F.; Berteina-Raboin, S. Eucalyptol, an All-Purpose Product. Catalysts 2022, 12, 48. [Google Scholar] [CrossRef]
- Gilmore, K.; Mohamed, R.K.; Alabugin, I.V. The Baldwin Rules: Revised and Extended. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 487–514. [Google Scholar] [CrossRef]
- Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Regiocontrolled Intramolecular Cyclizations of Carboxylic Acids to Carbon−Carbon Triple Bonds Promoted by Acid or Base Catalyst. Org. Lett. 2006, 8, 5517–5520. [Google Scholar] [CrossRef]
- Gilmore, K.; Alabugin, I.V. Cyclizations of Alkynes: Revisiting Baldwins Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. [Google Scholar] [CrossRef]
- Song, G.; Wang, F.; Li, X. C-C, C-O and C-N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef]
- Roudesly, F.; Oble, J.; Poli, G. Metal-Catalyzed C-H Activation/Functionalization: The Fundamentals. J. Mol. Catal. A Chem. 2017, 426, 275–296. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C-H Functionalisation Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)-H Bond Functionalization Assisted by Removable Directing Groups. Acc. Chem. Res. 2019, 131, 8390–8416. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, M.P.; Gooßen, L.J. Carboxylic Acids as Directing Groups for C−H Bond Functionalization. Chem. Eur. J. 2016, 22, 18654–18677. [Google Scholar] [CrossRef] [PubMed]
- Font, M.; Quibell, J.M.; Perry, G.J.P.; Larrosa, I. The Use of Carboxylic Acids as Traceless Directing Groups for Regioselective C-H Bond Functionalisation. Chem. Commun. 2017, 53, 5584–5597. [Google Scholar] [CrossRef]
- Gulías, M.; Mascareñas, J.L. Metal-Catalyzed Annulations through Activation and Cleavage of C-H Bonds. Angew. Chem. Int. Ed. Engl. 2016, 128, 11164–11184. [Google Scholar] [CrossRef]
- Yang, Y.; Li, K.; Cheng, Y.; Wan, D.; Li, M.; You, J. Rhodium-catalyzed annulation of arenes with alkynes through weak chelation-assisted C-H activation. Chem. Commun. 2016, 52, 2872–2884. [Google Scholar] [CrossRef]
- Long, S.; Resende, D.I.S.P.; Kijjoa, A.; Silva, A.M.S.; Pina, A.; Fernández-Marcelo, T.; Helena Vasconcelos, M.; Sousa, E.; Pinto, M.M.M. Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products. Mar. Drugs 2018, 16, 261. [Google Scholar] [CrossRef]
- Long, S.; Duarte, D.; Carvalho, C.; Oliveira, R.; Santarém, N.; Palmeira, A.; Resende, D.I.S.P.; Silva, A.M.S.; Moreira, R.; Kijjoa, A.; et al. Indole-Containing Pyrazino[2,1-b]quinazoline-3,6-diones Active against Plasmodium and Trypanosomatids. ACS Med. Chem. Lett. 2022, 13, 225–235. [Google Scholar] [CrossRef]
- Abreu, R.M.V.; Ferreira, I.C.F.R.; Calhelha, R.C.; Lima, R.T.; Vasconcelos, M.H.; Adega, F.; Chaves, R.; Queiroz, M.J.R.P. Anti-Hepatocellular Carcinoma Activity Using Human HepG2 Cells and Hepatotoxicity of 6-Substituted Methyl 3-Aminothieno[3,2-b]Pyridine-2- Carboxylate Derivatives: In Vitro Evaluation, Cell Cycle Analysis and QSAR Studies. Eur. J. Med. Chem. 2011, 46, 5800–5806. [Google Scholar] [CrossRef]
- Santarém, N.; Cunha, J.; Silvestre, R.; Silva, C.; Moreira, D.; Ouellette, M.; Cordeiro-Da-Silva, A. The Impact of Distinct Culture Media in Leishmania infantum Biology and Infectivity. Parasitology 2014, 141, 192–205. [Google Scholar] [CrossRef]
- Bowling, T.; Mercer, L.; Don, R.; Jacobs, R.; Nare, B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis. Int. J. Parasitol. Drugs Resist. 2012, 2, 262–270. [Google Scholar] [CrossRef]
- Lopes, A.; Teixeira, S.; Santarém, N.; Greco, A.; Pagliaro, A.; Keminer, O.; Gul, S.; Cordeiro-Da-Silva, A.; Carvalho, M.A. SAR Study of 4,8-Disubstituted Pyrimido[5,4-d]Pyrimidines Exhibiting Antitrypanosomal and Antileishmanial Activity. ACS Med. Chem. Lett. 2024, 15, 1541–1548. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Alkyne | Catalysts | Ligand | Base | Solvent | T/Time | 2a Yield (%) |
| 1 | 1.1 equiv | PdCl2(PPh3)2 5 mol% CuI 5 mol% | _ | Et3N (3 equiv.) | DMF | 100 °C/3.5 h, under Ar | 65% + tricyclic lactone 3a (12%) |
| 2 | 2.0 equiv | Pd/C 5 mol% | XPhos | K2CO3 (3 equiv.) | Eucalyptol | 100 °C/5 h | 30% + 60% of a compound ESI [M + H]+ m/z 397 |
 | ||
|---|---|---|
| Entry | Sonogashira Products | Tricyclic Lactones |
| 1 | 2a, 65% |  |
| 2 |  |  |
| 3 |  |  |
| 4 |  | N.O. |
| 5 |  |  |
| 6 |  |  |
| 7 |  |  |
| 8 |  | N.O. |
 | ||
|---|---|---|
| Entry | Tricyclic Lactones | Decarboxylation Product |
| 1 |  |  |
| 2 |  | N.O. |
| 3 |  |  |
| 4 |  |  |
| 5 |  |  |
| 6 |  | N.O. |
| 7 |  |  |
| Compounds 3 | δ of 9-H Signal (ppm) |
|---|---|
| 3a | 7.93 |
| 3b | 7.57 |
| 3c | 7.96 |
| 3d | 7.80 |
| 3e | 7.78 |
| 3f | 8.13 |
| 3g | 7.84 |
 | |||
|---|---|---|---|
| Entry | Halogenated Tricyclic Lactones | Entry | Halogenated Tricyclic Lactones |
| 1 |  | 2 |  |
 | |||
|---|---|---|---|
| Entry | Tetracyclic Lactones | Entry | Tetracyclic Lactones |
| 1 | 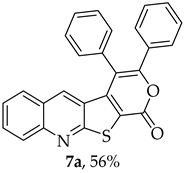 | 4 | 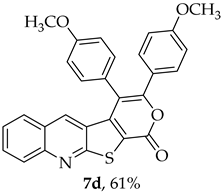 |
| 2 |  | 5 |  |
| 3 |  | 6 |  |
| Tetracyclic Lactones | 5-H Signal (ppm) | 5-CH Signal (ppm) |
|---|---|---|
| 7a | 7.25 | 134.2 |
| 7b | 9.26 | 135.7 |
| 7c | 7.50 | 134.0 |
| 7d | 7.40 | 134.2 |
| 7e | 7.23 | 133.9 |
| 7f | 9.52 | 135.7 |
| GI50 (µM) 1 | ||||||
|---|---|---|---|---|---|---|
| Caco-2 | MCF-7 | AGS | HeLa | NCI-H460 | PLP2 | |
| 2a | 70.03 ± 3.78 | 94.82 ± 6.43 | 86.78 ± 4.99 | 68.67 ± 3.05 | 91.45 ± 9.12 | >125 |
| 2b | 59.24 ± 1.76 | 49.73 ± 2.41 | 51.35 ± 2.87 | 47.92 ± 3.58 | 48.56 ± 4.31 | >125 |
| 2c | 52.11 ± 0.89 | 51.76 ± 1.07 | 70.73 ± 4.72 | 48.32 ± 1.66 | 67.21 ± 3.05 | >125 |
| 2d | 79.52 ± 3.58 | 67.89 ± 2.47 | 59.26 ± 1.97 | 62.33 ± 2.98 | 82.47 ± 5.35 | >125 |
| 2e | 92.97 ± 7.23 | 95.8 ± 8.08 | 89.17 ± 5.33 | 76.9 ± 6.55 | 101.67 ± 8.13 | >125 |
| 2f | 3.72 ± 2.42 | 4.08 ± 0.27 | 9.25 ± 0.61 | 5.44 ± 4.31 | 9.33 ± 4.21 | 57.87 ± 4.31 |
| 2g | 30.73 ± 2.35 | 59.96 ± 3.19 | 46.05 ± 1.77 | 36.38 ± 2.85 | 62.13 ± 3.05 | 90.48 ± 6.47 |
| 3a | 79.93 ± 3.56 | 77.52 ± 1.23 | 56.39 ± 3.76 | 49.73 ± 2.69 | 36.52 ± 0.69 | >125 |
| 3b | 67.49 ± 4.19 | 75.69 ± 6.48 | 74.28 ± 5.96 | 58.07 ± 4.29 | 82.74 ± 7.41 | >125 |
| 3c | 44.05 ± 0.93 | 56.1 ± 3.56 | 51.54 ± 3.29 | 48.38 ± 3.19 | 72.4 ± 6.24 | >125 |
| 3d | 90.48 ± 6.47 | 58.6 ± 1.86 | 56.2 ± 3.02 | 50.28 ± 4.79 | 45.33 ± 3.27 | >125 |
| 3e | 28.81 ± 1.85 | 47.55 ± 4.5 | 43.79 ± 1.76 | 38.21 ± 3.29 | 29.27 ± 1.47 | 90.48 ± 6.47 |
| 3f | 58.76 ± 2.44 | 59.27 ± 2.45 | 42.65 ± 3.96 | 58.76 ± 2.44 | 61.25 ± 4.17 | >125 |
| 3g | 52.54 ± 1.31 | 48.63 ± 2.15 | 45.33 ± 0.89 | 38.98 ± 1.78 | 44.01 ± 3.28 | >125 |
| 5a | 41.71 ± 1.02 | 52.59 ± 3.18 | 36.34 ± 1.47 | 35.78 ± 2.08 | 51.54 ± 4.18 | 66.81 ± 3.58 |
| 5b | 18.33 ± 0.92 | 45.14 ± 1.74 | 32.78 ± 1.56 | 29.06 ± 2.81 | 48.34 ± 1.08 | 89.76 ± 1.79 |
| 7a | 75.04 ± 4.49 | 97.87 ± 6.31 | 93.49 ± 5.37 | 78.92 ± 6.06 | 93.88 ± 3.18 | >125 |
| 7b | 91.08 ± 2.46 | 100.78 ± 7.33 | 99.76 ± 4.94 | 82.79 ± 8.02 | 109.28 ± 9.16 | >125 |
| 7c | 87.76 ± 8.01 | 90.34 ± 4.16 | 103.6 ± 9.12 | 77.13 ± 1.12 | 108.41 ± 5.44 | >125 |
| 7d | 90.01 ± 3.29 | 91.23 ± 8.71 | 88.57 ± 5.08 | 75.18 ± 6.72 | 97.81 ± 6.14 | >125 |
| 7f | 87.02 ± 3.51 | 76.44 ± 2.58 | 83.44 ± 6.27 | 71.52 ± 2.36 | 92.08 ± 3.89 | >125 |
| T.brucei | L. infantum | CC50 Interval (μM) in THP-1-Derived Macrophages | |||
|---|---|---|---|---|---|
| % of Activity ± ST.DEV (Single Dose 20 μM) | IC50 (μM) | % of Activity ± ST.DEV (Single Dose 20 μM) | IC50 (μM) | ||
| 2c | no activity * | — | no activity * | -- | >100 |
| 2d | no activity * | -- | no activity * | -- | >100 |
| 2e | 26 ± 7 | -- | no activity * | -- | >100 |
| 2f | 98 ± 5 | 6.43 | 57 ± 6 | 16 | 100 > CC50 > 25 |
| 2g | no activity * | -- | no activity * | -- | >100 |
| 3a | 33 ± 5 | -- | 30 ± 9 | -- | 100 > CC50 > 50 |
| 5a | 74 ± 11 | 10.80 | 32 ± 8 | -- | 100 > CC50 > 50 |
| 7a | 31 ± 7 | -- | no activity * | -- | <12.5 |
| 7b | no activity * | -- | 51 ± 9 | 19 | 50 > CC50 > 12.5 |
| 7c | 28 ± 7 | -- | 41 ± 6 | -- | <12.5 |
| 7d | no activity * | -- | no activity * | -- | <12.5 |
| 7e | no activity * | -- | no activity * | -- | 50 > CC50 > 25 |
| 7f | 27 ± 3 | -- | 34 ± 8 | -- | 50 > CC50 > 12.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, M.F.; Ribeiro, F.; Borges, A.; Calhelha, R.C.; Santarém, N.; Cordeiro-da-Silva, A.; Queiroz, M.-J.R.P. Synthesis of Tricyclic and Tetracyclic Lactone Derivatives of Thieno[2,3-b]pyrazine or Thieno[2,3-b]quinoline: Preliminary Antitumor and Antiparasitic Activity Evaluation. Molecules 2025, 30, 1999. https://doi.org/10.3390/molecules30091999
Martins MF, Ribeiro F, Borges A, Calhelha RC, Santarém N, Cordeiro-da-Silva A, Queiroz M-JRP. Synthesis of Tricyclic and Tetracyclic Lactone Derivatives of Thieno[2,3-b]pyrazine or Thieno[2,3-b]quinoline: Preliminary Antitumor and Antiparasitic Activity Evaluation. Molecules. 2025; 30(9):1999. https://doi.org/10.3390/molecules30091999
Chicago/Turabian StyleMartins, Maria F., Francisco Ribeiro, Ana Borges, Ricardo C. Calhelha, Nuno Santarém, Anabela Cordeiro-da-Silva, and Maria-João R. P. Queiroz. 2025. "Synthesis of Tricyclic and Tetracyclic Lactone Derivatives of Thieno[2,3-b]pyrazine or Thieno[2,3-b]quinoline: Preliminary Antitumor and Antiparasitic Activity Evaluation" Molecules 30, no. 9: 1999. https://doi.org/10.3390/molecules30091999
APA StyleMartins, M. F., Ribeiro, F., Borges, A., Calhelha, R. C., Santarém, N., Cordeiro-da-Silva, A., & Queiroz, M.-J. R. P. (2025). Synthesis of Tricyclic and Tetracyclic Lactone Derivatives of Thieno[2,3-b]pyrazine or Thieno[2,3-b]quinoline: Preliminary Antitumor and Antiparasitic Activity Evaluation. Molecules, 30(9), 1999. https://doi.org/10.3390/molecules30091999