3. Discussion
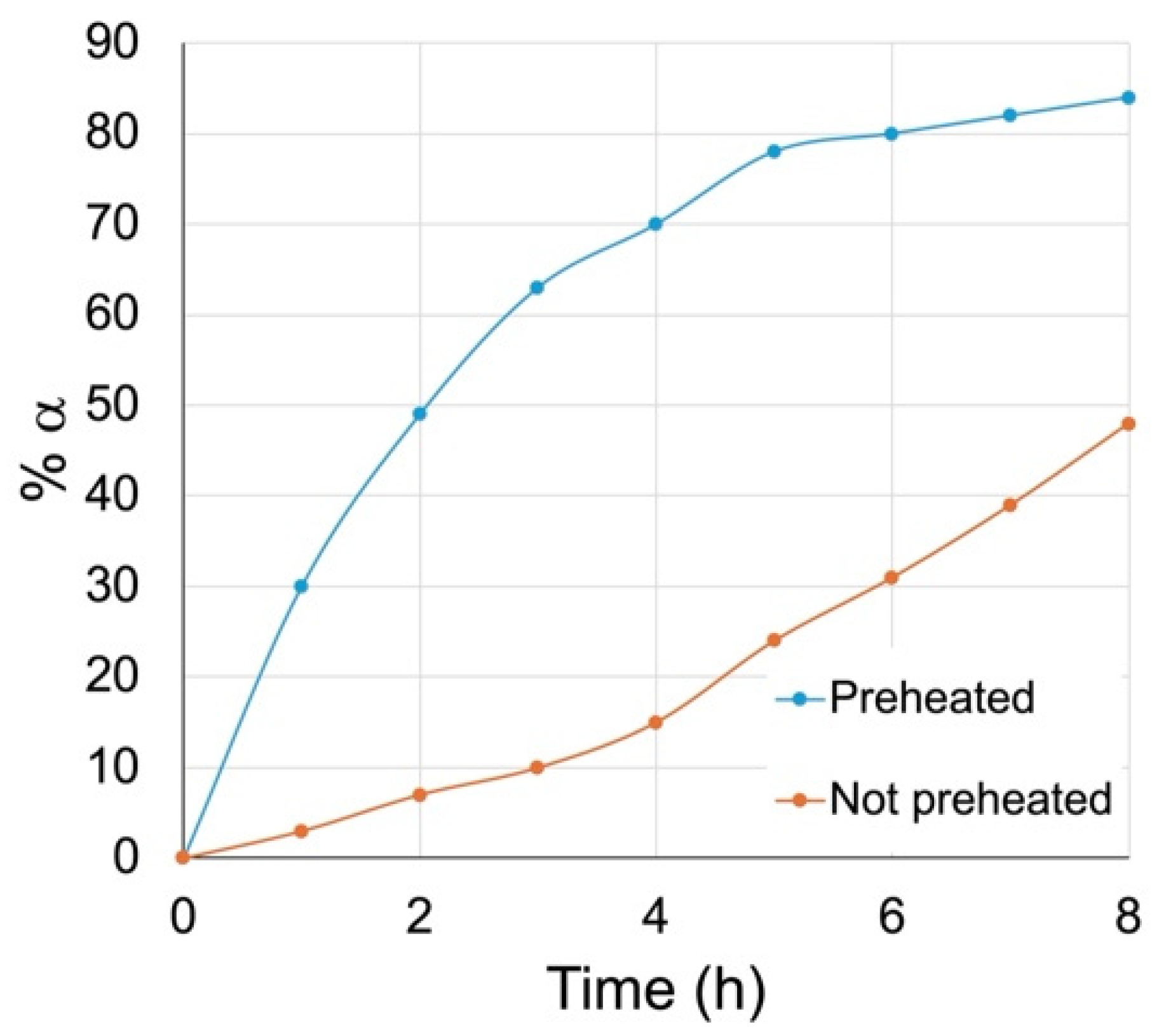
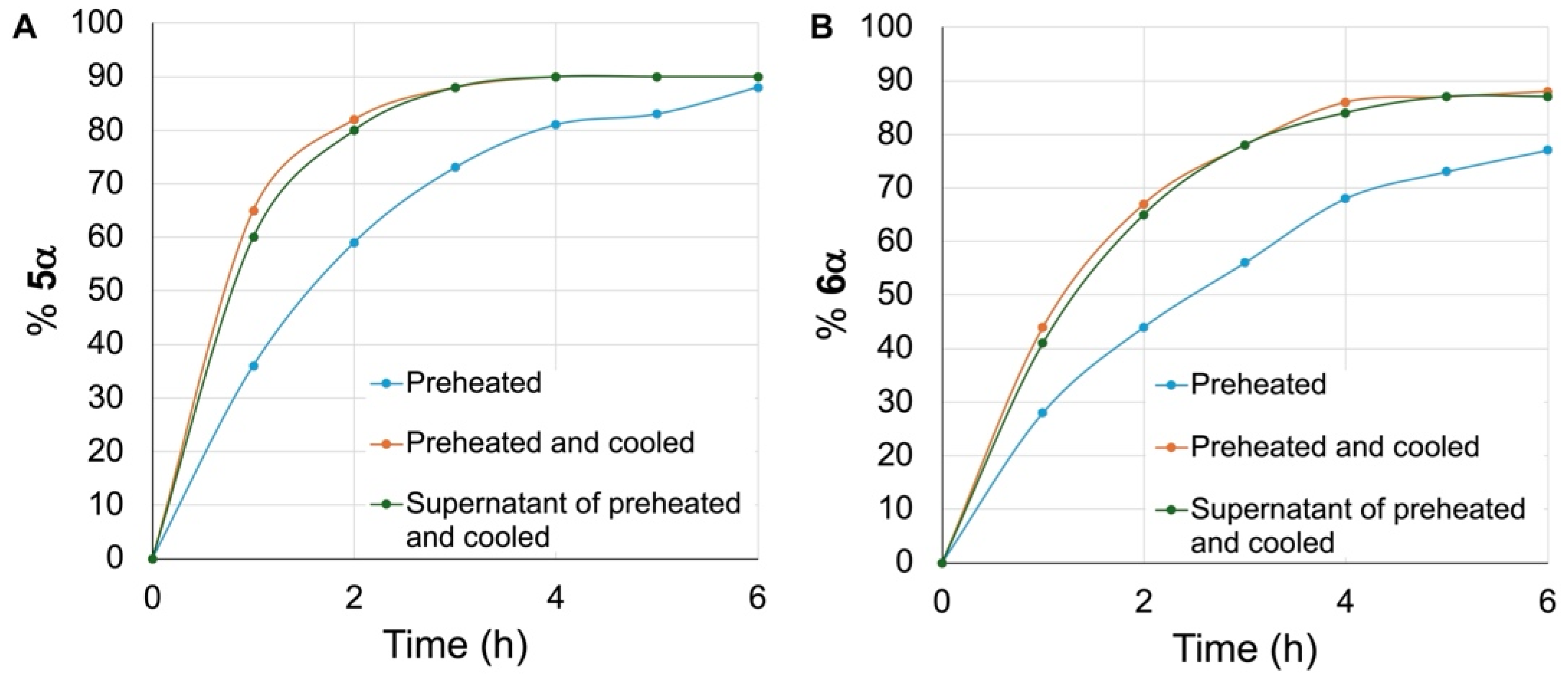
We have discovered simple and cost-effective conditions to promote the anomerization of N-acetylglucosamine β glycosides to their α anomers with up to 90% conversion. These conditions involve dissolving the β glycosides in DBM-DMF (1:2) and placing the solutions at 100 °C for 24 h in a sealed flask. Our study suggests that heating the DBM-DMF mixture at 100 °C leads to the formation of a compound that promotes this anomerization. Upon cooling of a DBM-DMF (1:2) mixture heated at 100 °C for 24 h, we observed the formation of a crystalline solid. We have shown that this hygroscopic solid promoted the anomerization of methyl β glycoside 6β in pure DMF to 4% after 24 h and 7% after 48 h. Since heating the β glycoside in pure DMF did not lead to any anomerization, this result suggests that this solid could have promoted the anomerization. We have shown that the anomerization was faster when the hexyl and methyl β glycosides (5β and 6β) were stirred at 100 °C in the supernatant of a DBM-DMF mixture preheated at 100 °C for 24 h and then cooled to room temperature and decanted before being added to the β glycosides. Interestingly, cooling preheated (24 h, 100 °C) mixtures of DBM-DMF and then adding the β glycosides at room temperature before placing the mixtures at 100 °C further increased the kinetics of anomerization for both hexyl and methyl glycosides 5β and 6β. We propose that the formation of the crystalline solid during the heating/cooling cycle of the mixture prior to the addition of the β glycosides increased the concentration of the active species in solution, thereby enhancing the kinetics of anomerization.
It is known [
23] that DMF and methyl bromide heated to 80 °C in a sealed vessel lead to the formation tetramethylammonium bromide. Indeed, the reaction of
N,
N-disubstituted amides with alkyl halides is a known way to access various quaternary ammonium halide salts [
24]. In these reactions (
Scheme 3A), the amides first degrade to give carbon monoxide and the corresponding dialkylamines (
I), which then undergo two successive nucleophilic displacements by alkyl halides, yielding the quaternary ammonium salts [
24]. It is also known that the reaction of dihalomethane (CH
2I
2 or CH
2Br
2) with trimethylamine (
Scheme 3B, [
25]) or
N,
N,
N′,
N′-tetramethylmethylenediamine (TMMD) (
Scheme 3C, [
26]) produces dimethymethyleniminium salts (
II and
III).
We observed constant gas evolution while heating the sealed DBM-DMF reaction mixtures, which strongly suggested that DMF dissociated into dimethylamine and carbon monoxide. The
1H NMR (
d6-DMSO) spectrum of the dried solid showed four products (
Supplementary Material). The major compound was characterized by two signals of a 3-to-1 relative integration at 3.65 ppm and 8.18 ppm, correlating in the HSQC to methyl and methylene carbons at 48 and 168 ppm, respectively. These signals are in agreement with those reported for the iminium iodide salt (
II,
Scheme 3) described by Schreiber et al. [
25] at 3.67 and 8.18 ppm, respectively. Further examination of the
1H NMR spectrum of the solid also revealed two signals of a 3-to-1 relative integration that, depending on the sample, appeared as doublets or broad singlets at 2.66 ppm and 4.44 ppm, both correlating in the COSY spectrum to a broad NH signal at 9.5 ppm. In the HSQC, these signals correlated to methyl and methylene carbons at 38 and 79 ppm, respectively. We propose that these signals correspond respectively to the two methyl and the hydroxymethyl substituents in hydroxymethyl(dimethyl)ammonium bromide (
Scheme 4,
VI), which was likely formed by the reaction of the iminium salt (
III) with water contained in the NMR solvent. Finally, NMR of the solid in
d6-DMSO also showed two unrelated signals: a doublet at 2.77 ppm and a triplet at 2.54 ppm, which correlated in the COSY spectrum to broad NH signals at 9.6 and 8.4 ppm, respectively. In the HSQC, these signals correlated to methyl carbons at 44 and 34 ppm, respectively. We propose that these signals correspond to dimethylamine (
Scheme 4,
IV) and dimethylammonium bromide, respectively.
X-ray crystallography of the crystalline solid confirmed that it was indeed the dimethylmethyleniminium bromide salt
III (
Figure 5). Crystallographic data and experimental details are given in the
Supplementary Material. The structure obtained was deposited at the Cambridge Crystallographic Data Centre (CCDC) under the number 2425977 and is in complete agreement with that previously reported by Clark et al. [
26] for the same compound.
Concentration of the supernatant and
1H NMR (
d6-DMSO) analysis (
Supplementary Material) of the residue showed the presence of what we assume to be dimethylamine (doublet at 2.77 ppm) and dimethylammonium bromide (triplet at 2.54 ppm) as major compounds. We also observed the presence of what we proposed to be hydroxymethyl(dimethyl)ammonium bromide (
VI), resulting from the hydration of the dimethylmethyleniminium bromide salt by water contained in DMSO and characterized by the two doublets mentioned above (2.66 ppm and 4.44 ppm).
We propose (
Scheme 4) that the main reaction taking place between DMF and DBM results from the nucleophilic displacement of bromide in dibromomethane by the dimethylamine
IV formed after decomposition of the DMF. This reaction leads to the bromomethyl(dimethyl)ammonium bromide intermediate
V, which promptly loses HBr to give dimethylmethyleniminium bromide
III. Thus, we conclude that anomerization could be promoted either by the HBr released during the formation of the dimethylmethyleniminium bromide
III or by the highly reactive dimethylmethyleniminium bromide salt
III itself. Without attempting to optimize the reaction, we observed up to 7% anomerization when adding dry dimethylmethyleniminium bromide
III salt to a solution of the β glycoside
6β in pure DMF and heating the solution at 100 °C for 48 h. Since stirring the β glycoside
6β in pure DMF at 100 °C did not lead to any anomerization, this initial result strongly suggests that the highly reactive Mannich salt
III is indeed capable of promoting anomerization. Further experiments are ongoing to confirm this hypothesis and clarify if the HBr formed is involved in the anomerization.
It has been known since the late 1920s that Lewis acids can promote the anomerization of β glucosides, as Pacsu [
27,
28,
29] described the anomerization of methyl, hexyl, and cyclohexyl tetraacetyl β-
d-glucopyranosides using SnCl
4 or TiCl
4. Subsequently, Ikemoto et al. [
30] described the anomerization of various acylated β glycosides using FeCl
3 at room temperature and reported that acetylated and benzoylated β glycosides yielded the best results. Interestingly when attempting the anomerization of peracetylated β methyl
N-acetylglucosamine glycoside
6β, they observed the formation of the oxazoline in a 32% yield, which suggested an exocyclic cleavage of the glycosidic bond. Expanding on these early results, the Murphy group studied the TiCl
4- and SnCl
4-promoted anomerization of various β glycosides [
31,
32,
33,
34]. These studies confirmed the early work by Pacsu that showed that TiCl
4 was more efficient at promoting anomerization than SnCl
4 [
32]. The Murphy group discovered that β glucuronic acids were more easily anomerized than glycosides, 6-deoxy glycosides, or xylopyranosides and that fuco-, xylo-, arabino-, gluco-, and galactopyranosides, including β glycosides of
N-acetylglucosamine, could be anomerized in these conditions [
31,
32,
34]. Furthermore, they also studied the impact of the O-2 protecting group, showing that 2-
O-acetylated β glycosides were less reactive toward anomerization than their 2-
O-benzoylated analogues [
33]. Interestingly, Crich and Vinod made the serendipitous discovery that a β methyl glycoside of 2-
N-acetyl-2-
N,3-
O-oxazolidinone of glucosamine underwent anomerization to the α anomer when treated with NaCNBH
3 and HCl in diethyl ether [
35]. The Oscarson and Ito groups also independently reported the anomerization of
N-acetyl or
N-benzyl 2,3-
trans-oxazolidinones of glucosamine promoted by AgOTf or BF
3•Et
2O, respectively [
36,
37]. Crich [
35] suggested that the anomerization of these 2,3-oxazolidinones was assisted by the strain created by the 2,3-
trans-fused ring, which facilitated a O5-C1
endo cleavage, eventually leading to the more stable α anomer. Indeed, subsequent calculations by the Ito group showed that the
N-benzyl-2,3-
trans-oxazolidinone group gave lower energies for the transition state and
endo O5-C1 cleavage than pyranosides not carrying this 2,3-
trans-fused ring [
38]. More recently, Frem et al. [
39] also reported anomerization of the n-butyl β glycoside of peracetylated Glc
NAc promoted by Cu(OTf)
2 or triflic acid at 130 °C in 1,2-dichloroethane. In the latter case, they observed considerable degradation of the glycoside. They also reported transglycosylation of the n-butyl β glycoside to the methyl β glycoside when adding excess methanol to the reaction mixture. Interestingly, they observed limited anomerization of the β methyl glycoside, only reaching an α/β ratio of 1/6 in this reaction.
Glycosidic bonds can be cleaved either via an exocyclic C1-O1 or endocyclic O5-C1 cleavage (
Scheme 5 (a and b)).
It is generally accepted that when submitted to solvolysis in protic conditions, α glycosides undergo exocyclic C1-O1 cleavage assisted by the stereoelectronic effect of the ring oxygen O-5 [
40,
41,
42]. In contrast, the path of cleavage for β glycosides in protic acid has been shown to occur through both
exo (C1-O1) and
endo (O5-C1) cleavage [
40,
41,
42]. Furthermore, the ring opening of β glycosides under Lewis acid catalysis such as Me
2BBr [
43], AlMe
3 [
44], and AlMe
3/MeTiCl
3 [
45] was shown to proceed mostly through
endo cleavage. In his studies, Murphy [
33] proposed that anomerization of β glycosides promoted by TiCl
4 or SnCl
4 occurred via an endocyclic O5-C1 cleavage induced by the chelation of the O5 and O6 oxygens by the metal catalyst, as shown in
Scheme 6.
We do not have unquestionable evidence that the anomerizations described here follow endocyclic opening at the O5-C1 bond. However, the fact that we do not observe either hemiacetal formation (even when adding traces of water to our reactions) or the formation of oxazoline as reported by Ikemoto et al. [
30] strongly suggests that the glycosidic C1-O1 bond is not cleaved during the anomerization. Thus, our results suggest that the anomerizations follow an endocyclic mechanism.
As we have described above, while all β glycosides
1,
2,
5, and
6 reached a 9/1 α/β ratio after 24 h of reaction, the β methyl glycoside
6β was slower to anomerize that the β hexyl glycoside
5β (
Figure 2 and
Figure 4), suggesting that the size of the glycoside impacts the reaction. These results are in agreement with the results reported by Pacsu [
28], who observed faster anomerization of the peracetylated β hexyl glucopyranoside than that of its methyl glucoside analog under TiCl
4 activation.
Further experiments are ongoing in our laboratory to confirm whether dimethylmethyleniminium bromide III, HBr, or the combination of both promotes anomerization. Furthermore, we are currently investigating the scope of this reaction and its applicability to other glycosides, especially those that do not carry an acetamido group at C-2. Preliminary results have shown that the presence of a more electron-withdrawing N-Troc, N-trichloroacetyl (N-TCA), or O-Ac had a significant impact on the degree of anomerization. These results will be reported in due course.
4. Materials and Methods
4.1. General Experimental
All chemicals were purchased from Fisher Scientific (Ottawa, ON, Canada) or Sigma Aldrich (Oakville, ON, Canada) and were used without purification. Anhydrous dichloromethane was obtained by distillation from phosphorus pentoxide. 1H NMR and 13C NMR spectra were recorded at 298 K on Bruker Avance-400 or 600 spectrometers (Bruker Biospin, Boston, MA, USA) for solutions in CDCl3 (calibrated at δC 77.0 ppm, using residual CHCl3 at δH 7.24 ppm) or d6-DMSO (calibrated at 13C δC 39.5 ppm, using residual CHD2SOCD3 at δH 2.50 ppm). All chemical shifts are reported in parts per million (ppm). All coupling constants (J) are reported in hertz (Hz) and were obtained from analysis of 1H NMR spectra. Structural assignments were made with additional information from gCOSY, gHSQC. 1D TOCSY experiments were acquired with mixing times of 150 ms and pre-scan delays of 2 s. Multiplicities are abbreviated as singlet (s), broad singlet (bs), doublet (d), doublet of a doublet (dd), triplet (t), and multiplet (m). Organic solutions were dried over Na2SO4 and concentrated under reduced pressure. Thin layer chromatography (TLC) was performed on aluminum plates coated with silica gel and charred with 20% H2SO4 in EtOH. Compounds were purified by flash column chromatography using Silica Gel 60 (230–400 mesh). Mass spectra were obtained under electrospray ionization (ESI) on a Thermo Scientific Orbitrap Velos Pro spectrometer equipped with a TOF analyzer at the Queen’s Mass Spectrometry Facility (Kingston, ON, Canada). The reported masses are for 35Cl isotope of chlorine and the 79Br isotope for bromine. Optical rotations for new compounds were measured on a Autopol® IV automatic polarimeter by Rudolph Research Analytical (Hackettstown, NJ, USA).
4.2. General Method for the Anomerization Reactions
Glycosides
1,
2,
5β, and
6β (10–100 mg) were dissolved in a 1:2 volumetric ratio of DBM-DMF at concentrations of 0.04 M or 0.4 M. For glycosides
1 and
2, 10 equiv of NaBr was added in all reactions except for that described in
Table 1, entry 3. The reaction mixtures were sealed and placed in an oil bath preheated to the desired temperature (70–100 °C), and the reactions were left to proceed for 24 h. The solutions were diluted in 5 mL of DCM, washed with water (2 × 5 mL), and the water phases were re-extracted with DCM (3 × 5 mL). The combined organic layers were dried on Na
2SO
4, concentrated under reduced pressure at 40 °C to yield tan syrups or amorphous solids. The products were analyzed by TLC (hexanes:EtOAc) and NMR.
4.3. 8-Chlorooctyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (2)
Known [
17] 2-acetamido-3,4,6-tri-
O-acetyl-2-deoxy-β-
d-glucopyranosyl chloride (Horton’s chloride, 4 g, 11 mmol) was dissolved in anhyd DCM (40 mL) under N
2. Molecular sieves (4 Å, 5 g) were added along with 8-chloro-1-octanol (2.8 mL, 16 mmol, 1.5 equiv), and the reaction was stirred for 1 h. HgCl
2 (3.86 g, 14 mmol, 1.3 equiv) was added to the reaction, which was left to stir for 16 h at rt. The mixture was diluted with DCM (50 mL), filtered over Celite
®, and the filtrate was washed with satd aq NaHCO
3 (2 × 50 mL) and water (3 × 50 mL), and the water phases were re-extracted with DCM (3 × 20 mL). The combined organic layers were dried over Na
2SO
4 and concentrated under reduced pressure. The pure 8-chlorooctyl glycoside
2 was precipitated in cold 1:1 Et
2O–toluene and filtered off as a white solid (3.6 g, 67%). Chromatography of the mother liquor (EtOAc-hexanes, 8:2) gave additional pure 8-chlorooctyl glycoside
2 (0.32 g, 6%). [α]
D −8.80 (
c 0.5, MeOH).
1H NMR (CDCl
3, 400 MHz)
δH 5.42 (d, 1H,
J = 8.7 Hz, NH), 5.28 (dd, 1H,
J = 9.3, 10.6 Hz, H-3), 5.04 (t, 1H,
J = 9.7 Hz, H-4), 4.66 (d, 1H,
J = 8.3 Hz, H-1), 4.24 (dd, 1H,
J = 4.8, 11.9 Hz, H-6a), 4.11 (dd, 1H,
J = 2.5, 12.2 Hz, H-6b), 3.86–3.75 (m, 2H, H-2, OCH
HCH
2), 3.69–3.65 (m, 1H, H-5), 3.51 (t, 2H,
J = 6.7 Hz, CH
2Cl), 3.47–3.41 (m, 1H, OC
HHCH
2), 2.06, 2.01, 2.00, 1.92 (4s, 12H, 4 × COCH
3), 1.73 (m, 2H,
J = 7.8 Hz, C
H2CH
2Cl), 1.56–1.50 (m, 2H, OCH
2C
H2), 1.41–1.35 (m, 2H, C
H2CH
2CH
2Cl), 1.29–1.25 (m, 6H, CH
2(C
H2)
3CH
2).
13C NMR (CDCl
3, 100 MHz)
δC 170.9, 170.7, 170.0, 169.4 (C=O), 100.7 (C-1), 72.3 (C-4), 71.8 (C-5), 69.8 (O
CH
2CH
2), 68.7 (C-3), 62.2 (C-6), 54.9 (C-2), 45.1 (CH
2Cl), 32.6 (
CH
2CH
2Cl), 29.4, 29.1, 28.8, 26.8, 25.7 (CH
2(
CH
2)
5CH
2), 23.4, 20.8, 20.7, 20.6 (CO
CH
3). HRESIMS
m/z: [M + H]
+ calcd for C
22H
37ClNO
9 494.2157; found 494.2151.
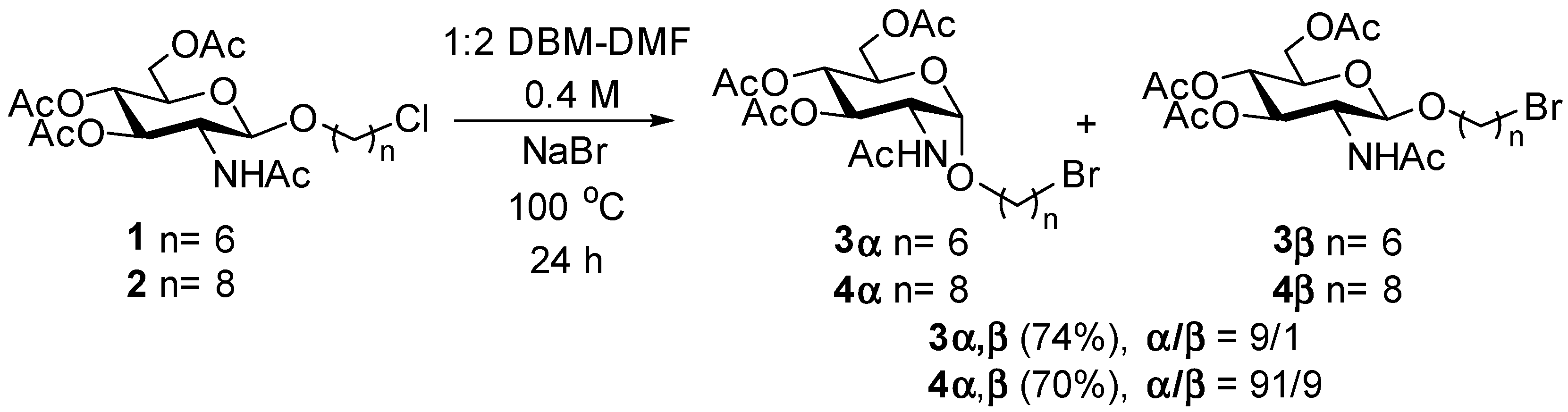
4.4. 6-Bromohexyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (3α) and 6-Bromohexyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (3β)
Known [
16] 6-chlorohexyl 2-acetamido-3,4,6-tri-
O-acetyl-2-deoxy-β-
d-glucopyranoside
1 (
Supplementary Material, 100 mg, 0.215 mmol) was reacted in DBM-DMF (1:2) at a 0.4 M concentration with 10 equiv of NaBr in a sealed flask as described above in the general method (100 °C, 24 h). Work-up as described above gave a brown syrup (82 mg, 74%). The α/β (9:1) ratio was determined by NMR. Purification of the residue with chromatography (EtOAc-hexanes, 7:3) gave analytical samples of pure
3α and
3β.
Analytical data for compound 3α: [α]D +73.1 (c 1.0, MeOH). 1H NMR (CDCl3, 400 MHz) δH 5.64 (d, 1H, J = 9.4 Hz, NH), 5.18 (dd, 1H, J = 9.3, 10.5 Hz, H-3), 5.09 (t, 1H, J = 9.6 Hz, H-4), 4.81 (d, 1H, J = 3.7 Hz, H-1), 4.31 (m, 1H, H-2), 4.21 (dd, J = 4.8, 11.9 Hz, H-6a), 4.07 (dd, 1H, J = 2.5, 12.2 Hz, H-6b), 3.93–3.89 (m, 1H, H-5), 3.69–3.63 (m, 1H, OCHHCH2), 3.44–3.38 (m, 3H, CH2Br, OCHHCH2), 2.08, 2.01, 2.00, 1.92 (4s, 12H, 4 × COCH3), 1.86 (m, 2H, CH2CH2Br), 1.65–1.58 (m, 2H, OCH2CH2), 1.51–1.43 (m, 2H, CH2CH2CH2Br), 1.40–1.33 (m, 2H, OCH2CH2CH2). 13C NMR (CDCl3, 100 MHz) δC 171.4, 170.7, 169.8, 169.3 (C=O), 97.1 (C-1), 71.4 (C-4), 68.2 (OCH2), 68.1 (C-5), 67.7 (C-3), 62.0 (C-6), 51.9 (C-2), 33.7 (CH2Br), 32.5, 29.1, 27.8, 25.3 (CH2(CH2)4CH2Br), 23.2, 20.7, 20.7, 20.6 (COCH3). HRESIMS m/z: [M + H]+ calcd for C20H33BrNO9 510.1339; found 510.1323.
Analytical data for compound 3β: [α]D −11.1 (c 0.54, MeOH). 1H NMR (CDCl3, 400 MHz) δH 5.44 (d, 1H, J = 8.9 Hz, NH), 5.27 (dd, 1H, J = 9.3, 10.6 Hz, H-3), 5.04 (t, 1H, J = 9.6 Hz, H-4), 4.65 (d, 1H, J = 8.3 Hz, H-1), 4.23 (dd, 1H, J = 4.8, 11.9 Hz, H-6a), 4.10 (dd, 1H, J = 2.5, 12.2 Hz, H-6b), 3.87–3.75 (m, 2H, H-2, OCHHCH2), 3.69–3.65 (m, 1H, H-5), 3.48–3.42 (m, 1H, OCHHCH2), 3.38 (t, 2H, J = 6.7 Hz, CH2Br), 2.09, 2.03, 2.02, 1.93 (4s, 12H, 4 × COCH3), 1.82 (m, 2H, CH2CH2Br), 1.63–1.52 (m, 2H, OCH2CH2), 1.47–1.28 (m, 4H, CH2(CH2)2CH2). 13C NMR (CDCl3, 100 MHz) δC 170.9, 170.7, 170.1, 169.4 (C=O), 100.7 (C-1), 72.3 (C-4), 71.8 (C-5), 69.6 (OCH2), 68.6 (C-3), 62.1 (C-6), 54.9 (C-2), 33.8 (CH2Br), 32.6, 29.2, 27.7 25.0 (CH2(CH2)4CH2Br), 23.4, 20.7, 20.7, 20.6 (COCH3). HRESIMS m/z: [M + H]+ calcd for C20H33BrNO9 510.1339; found 510.1339.
4.5. 8-Bromooctyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (4α) and 8-Bromooctyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (4β)
The 8-chlorooctyl glycoside 2 (105 mg, 0.213 mmol) was reacted in DBM-DMF (1:2) at a 0.4 M concentration with 10 equiv of NaBr in a sealed flask as described above in the general method (100 °C, 24 h). Work-up as described above gave a brown syrup (80 mg, 70%. The α/β (91:9) ratio was determined by NMR. Analytical samples of 4α and 4β were obtained by chromatography (EtOAc/hexanes, 7:3).
Analytical data for compound 4α: [α]D +49.0 (c 0.69, MeOH). 1H NMR (CDCl3, 400 MHz) δH 5.64 (d, 1H, J = 8.7 Hz, NH), 5.18 (dd, 1H, J = 9.3, 10.5 Hz, H-3), 5.09 (t, 1H, J = 9.6, 10.5 Hz, H-4), 4.80 (d, 1H, J = 3.7 Hz, H-1), 4.34–4.28 (m, 1H, H-2), 4.21 (dd, J = 4.6, 12.3 Hz, H-6a), 4.07 (dd, 1H, J = 2.5, 12.3 Hz, H-6b), 3.93–3.89 (m, 1H, H-5), 3.68–3.62 (m, 2H, H-2, OCHHCH2), 3.43–3.37 (m, 3H, OCHHCH2, CH2Br), 2.07, 2.01, 2.00, 1.93 (4s, 12H, 4 × COCH3), 1.84 (m, 2H, CH2CH2Br), 1.61–1.55 (m, 2H, OCH2CH2), 1.46–1.39 (m, 2H, CH2CH2CH2Br), 1.37–1.28 (m, 6H, CH2(CH2)3CH2). 13C NMR (CDCl3, 100 MHz) δC 171.4, 170.7, 169.8, 169.3 (C=O), 97.1 (C-1), 71.4 (C-4), 68.5 (OCH2), 68.1 (C-5), 67.7 (C-3), 62.0 (C-6), 51.9 (C-2), 33.9 (CH2Br), 32.7 (CH2CH2Br), 29.2, 29.1, 28.6, 28.0, 26.0 (CH2(CH2)5CH2CH2Br), 23.2, 20.7, 20.7, 20.6 (COCH3). HRESIMS m/z: [M + H]+ calcd for C22H37BrNO9 538.1652; found 538.1646.
Analytical data for compound 4β: [α]D -12.1 (c 0.69, MeOH). 1H NMR (CDCl3, 400 MHz) δH 5.42 (d, 1H, J = 8.7Hz, NH), 5.28 (dd, 1H, J = 9.3, 10.6 Hz, H-3), 5.04 (t, 1H, J = 9.7 Hz, H-4), 4.66 (d, 1H, J = 8.3 Hz, H-1), 4.24 (dd, 1H, J = 4.8, 12.2 Hz, H-6a), 4.11 (dd, 1H, J = 2.5, 12.2 Hz, H-6b), 3.86–3.75 (m, 2H, H-2, OCHHCH2), 3.69–3.65 (m, 1H, H-5), 3.47–3.41 (m, 1H, OCHHCH2), 3.38 (t, 2H, J = 6.8 Hz, CH2Br), 2.06, 2.01, 2.00, 1.92 (4s, 12H, 4 × COCH3), 1.82 (m, 2H, CH2CH2Br), 1.57–1.50 (m, 2H, OCH2CH2), 1.43–1.35 (m, 2H, CH2CH2CH2Br), 1.31–1.23 (m, 6H, CH2(CH2)3CH2). 13C NMR (CDCl3, 100 MHz) δC 170.9, 170.7, 170.0, 169.4 (C=O), 100.7 (C-1), 72.3 (C-4), 71.8 (C-5), 69.8 (OCH2), 68.7(C-3), 62.2 (C-6), 54.9 (C-2), 34.0 (CH2Br), 32.7 (CH2CH2Br), 29.4, 29.1, 28.7, 28.0, 25.7 (CH2(CH2)5CH2CH2Br), 23.4, 20.8, 20.7, 20.6 (COCH3). HRESIMS m/z: [M + H]+ calcd for C22H37BrNO9 538.1652; found 538.1652.
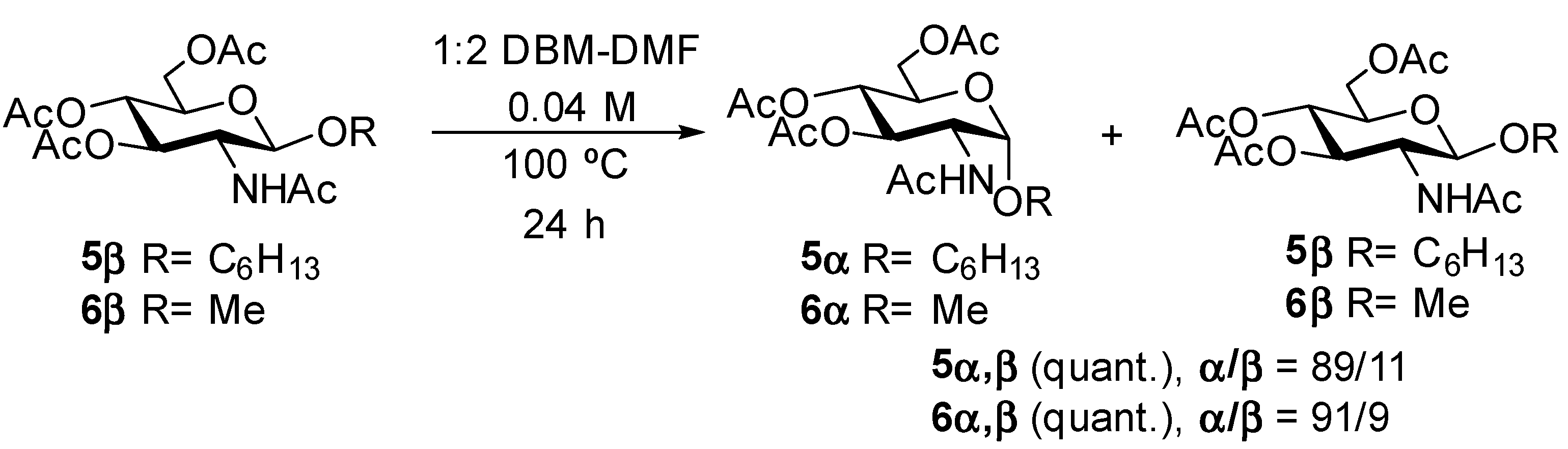
4.6. Hexyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (5α) and Hexyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (5β)
The known [
19] hexyl glycoside
5β (
Supplementary Material, 12 mg, 0.027 mmol) was reacted in DBM-DMF (1:2) at a 0.04 M concentration in a sealed flask as described above in the general method (100 °C, 24 h). Work-up as described above gave a brown syrup (12 mg, quant). The α/β (89:11) ratio was determined by NMR.
NMR data for known major compound
5α are in agreement with those previously reported [
21] and were assigned in the mixture using 1D TOCSY (irradiation of H-1) and HSQC experiments (
Supplementary Material):
1H NMR (CDCl
3, 400 MHz) δ
H 5.63 (d, 1H,
J = 9.5 Hz, NH), 5.19 (dd, 1H,
J = 9.6, 10.6 Hz, H-3), 5.09 (t, 1H,
J = 9.6 Hz, H-4), 4.80 (d, 1H,
J = 3.7 Hz, H-1), 4.31 (m, 1H, H-2), 4.21 (dd, 1H,
J = 4.8, 12.3 Hz, H-6a), 4.07 (dd, 1H,
J = 2.5, 12.1 Hz, H-6b), 3.91 (m, 1H, H-5), 3.68–3.63 (m, 1H, OCH
HCH
2), 3.43–3.38 (m, 1H, OC
HHCH
2), 2.07, 2.01, 2.00, 1.93 (4 s, 12H, 4 × COCH
3), 1.59 (m, 2H, OCH
2C
H2), 1.36–1.21 (m, 6H, CH
2(C
H2)
3CH
3), 0.88 (m, 3H, (CH
2)
5C
H3).
13C NMR (CDCl
3, 100 MHz) δ
C 171.4, 170.7, 169.8, 169.3 (C=O), 97.1 (C-1), 71.5 (C-4), 68.7 (O
CH
2CH
2), 68.6 (C-5), 67.7 (C-3) 62.1 (C-6), 51.9 (C-2), 31.5 (OCH
2CH
2), 29.2, 25.8 (CH
2(
CH
2)
2CH
2), 23.3 (COCH
3), 22.6 (
CH
2CH
3), 20.7, 20.7, 20.6 (CO
CH
3), 14.0 (CH
2CH
3).
NMR data for known minor compound
5β are in agreement with those previously reported [
19]:
1H NMR (CDCl
3, 400 MHz) δ
H 5.46 (d, 1H,
J = 8.7 Hz, NH), 5.29 (dd, 1H,
J = 9.4, 10.5 Hz, H-3), 5.04 (t, 1H,
J = 9.6 Hz, H-4), 4.66 (d, 1H,
J = 8.3 Hz, H-1), 4.23 (dd, 1H,
J = 4.7, 12.2 Hz, H-6a), 4.10 (dd, 1H,
J = 2.4, 12.2 Hz, H-6b), 3.96–3.75 (m, 2H, H-2, OCH
HCH
2), 3.69–3.65 (m, 1H, H-5), 3.48–3.41 (m, 1H, OC
HHCH
2), 2.10, 2.02, 2.01, 1.92 (4 s, 12H, 4 × COCH
3), 1.55 (m, 2H, OCH
2C
H2), 1.31–1.20 (m, 6H, CH
2(C
H2)
3CH
3), 0.85 (m, 3H, (CH
2)
5C
H3).
13C NMR (CDCl
3, 100 MHz) δ
C 170.9, 170.7, 170.1, 169.4 (C=O), 100.7 (C-1), 72.3 (C-4), 71.7 (C-5), 70.0 (O
CH
2CH
2), 68.7 (C-3) 62.2 (C-6), 54.9 (C-2), 31.5 (OCH
2CH
2), 29.4, 25.5, (CH
2(
CH
2)
2CH
2), 23.3 (CO
CH
3), 22.6 (
CH
2CH
3), 20.7, 20.7, 20.6 (CO
CH
3), 14.0 (CH
2CH
3).
4.7. Methyl 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (6α) and Methyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranoside (6β)
The known [
20] methyl glycoside
6β (
Supplementary Material, 10 mg, 0.028 mmol) was reacted in DBM-DMF (1:2) at a 0.04 M concentration in a sealed flask as described above in the general method (100 °C, 24 h). Work-up as described above gave a brown syrup (10 mg, quant.). The α/β (91:9) ratio was determined by NMR.
NMR data for known major compound
6α are in agreement with those previously reported [
22] and were assigned using 1D TOCSY (irradiation of H-1) and HSQC experiments (
Supplementary Material):
1H NMR (CDCl
3, 400 MHz) δ
H 5.69 (d, 1H,
J = 9.7 Hz, NH), 5.19 (dd, 1H,
J = 9.6, 10.6 Hz, H-3), 5.09 (t, 1H,
J = 9.8 Hz, H-4), 4.70 (d, 1H,
J = 3.6 Hz, H-1), 4.32 (m, 1H, H-2), 4.21 (dd, 1H,
J = 3.7, 9.7 Hz, H-6a), 4.08 (dd, 1H,
J = 3.6, 9.6 Hz, H-6b), 3.89 (m, 1H, H-5), 3.38 (s, 3H, OCH
3), 2.07, 2.00, 1.99, 1.93 (4 s, 12H, 4 × COCH
3).
13C NMR (CDCl
3, 100 MHz) δ
C 171.4, 170.7, 169.9, 169.3 (C=O), 98.3 (C-1), 71.3 (C-4), 68.1 (C-5), 67.6 (C-3), 62.0 (C-6), 55.4 (CH
3), 54.6 (C-2), 23.2, 22.7, 20.7, 20.6 (CO
CH
3).
NMR data for known minor compound
6β are in agreement with those previously reported [
20]:
1H NMR (CDCl
3, 400 MHz) δ
H 5.45 (d, 1H,
J = 8.9 Hz, NH), 5.25 (dd, 1H,
J = 9.3, 10.4 Hz, H-3), 5.06 (t, 1H,
J = 9.8 Hz, H-4), 4.56 (d, 1H,
J = 8.3 Hz, H-1), 4.26 (dd, 1H,
J = 4.7, 12.3 Hz, H-6a), 4.13 (dd, 1H,
J = 2.5, 12.3 Hz, H-6b), 3.84 (m, 1H, H-2), 3.68 (m, 1H, H-5), 3.48 (s, 3H, OCH
3), 2.08, 2.01, 2.00, 1.94 (4 s, 12H, 4 × COCH
3).
13C NMR (CDCl
3, 100 MHz) δ
C 171.0, 170.7, 170.2, 169.4 (C=O), 101.6 (C-1), 72.4 (C-4), 71.8 (C-5), 68.5 (C-3), 62.1 (C-6), 56.8 (
CH
3), 54.6 (C-2), 23.4, 20.7, 20.7, 20.6 (CO
CH
3).
5. Conclusions
We have described the synthesis of a new 8-chlorooctyl β Glc
NAc glycoside. We have established that the chlorine atom in this compound as well as in the 6-chlorohexyl analog can be swiftly displaced by bromide in a mixture of DBM and DMF at 90–100 °C with or without the addition of sodium bromide. However, these reactions must be carefully monitored to maximize the nucleophilic displacement while limiting anomerization of the glycosidic bond. At 100 °C, the reaction is best stopped after ~2 h. The bromide in these glycosides should then be easily displaced with
N-hydroxyphthalimide in mild conditions [
14], leading, after deprotection, to the corresponding aminooxy functional group needed for conjugation to PS A1 [
10,
11,
12,
13] or other carrier molecules.
In the course of this work, we discovered that these mild conditions led to the anomerization of the β glycosidic bond in Glc
NAc to the corresponding α anomer. At 100 °C, the α/β was 9/1 after 24 h of reaction in a mixture (1:2) of DBM and DMF. While we initially assumed that anomerization happened preferentially from the bromoalkyl glycosides, we established that the alkyl glycosides also anomerized in these conditions. We discovered that applying a heating–cooling cycle to the solvent mixtures prior to adding the β glycosides and placing the reaction at 100 °C increased the kinetics of anomerization. Upon cooling the preheated DBM-DMF mixtures, we observed the formation of a crystalline solid, which was characterized by NMR spectroscopy and X-ray crystallography to be the known [
25,
26] Mannich salt dimethylmethyleniminium bromide. Although we only observed limited anomerization (4–7%) when the solid was reacted with the β glycoside
6β in pure DMF at 100 °C, we propose that this highly reactive carbocation could play the role of a Lewis acid in promoting anomerization in these reactions. However, it is also possible that the HBr formed together with the dimethylmethyleniminium bromide plays a role in the anomerization. Further experiments are ongoing in our laboratory to clarify the mechanism of this reaction. Our studies strongly suggest that anomerization of these Glc
NAc β glycosides occurs via an
endo O5-C1 cleavage, followed by bond rotation and cyclization to the more stable α anomer. We have also shown that the nature of the aglycon impacted the rate of anomerization, with hexyl β glycosides of Glc
NAc anomerizing at a faster rate than the analogous methyl β glycoside. The conditions discovered and described here are cost-effective and simple when compared to using heavy-metal-containing Lewis acids such as TiCl
4 or SnCl
4 [
27,
28,
29,
31,
32,
33,
34] to promote the anomerization of β glycosides. Furthermore, these conditions are mild and do not lead to the degradation of the glycoside like the use of a strong protic acid such as TfOH does [
39]. In the present work, we focused our investigation on the anomerization of β glycosides of peracetylated
N-acetylated glucosamine. Following this serendipitous discovery, additional experiments are ongoing to optimize the reaction conditions and further maximize the anomerization. Furthermore, we are currently investigating the scope of this reaction and its applicability to other glycosides, including disaccharides. Preliminary results suggest that the presence of a more electron-withdrawing
N-Troc,
N-TCA, or
O-Ac at C-2 of a β-
d-glucopyranoside has a significant impact on the degree of anomerization. Finally, further mechanistic studies are also ongoing to clarify the involvement of the Mannich salt dimethylmethyleniminium bromide and HBr in the anomerization; our results will be reported in due course.


 and
and 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}