
N-Alkylamino Stilbene Compounds as Amyloid β Inhibitors for Alzheimer’s Disease Research

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Design and Synthesis of Compounds
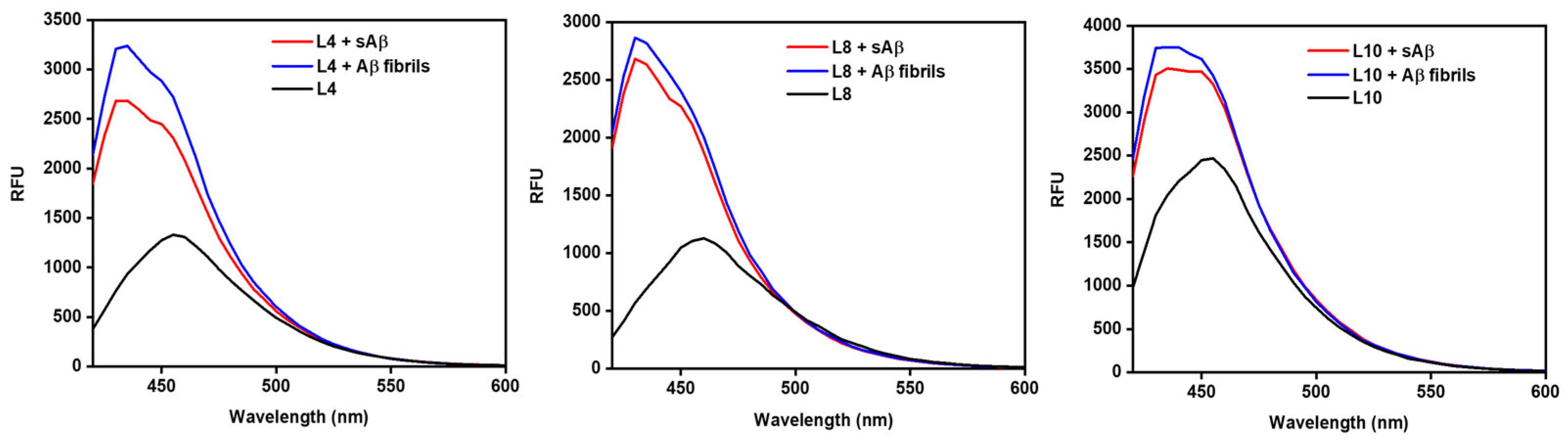
2.2. Fluorescence Turn-On Effect of Compounds Interacting with Aβ Species
2.3. Aβ Aggregation Inhibition Assays
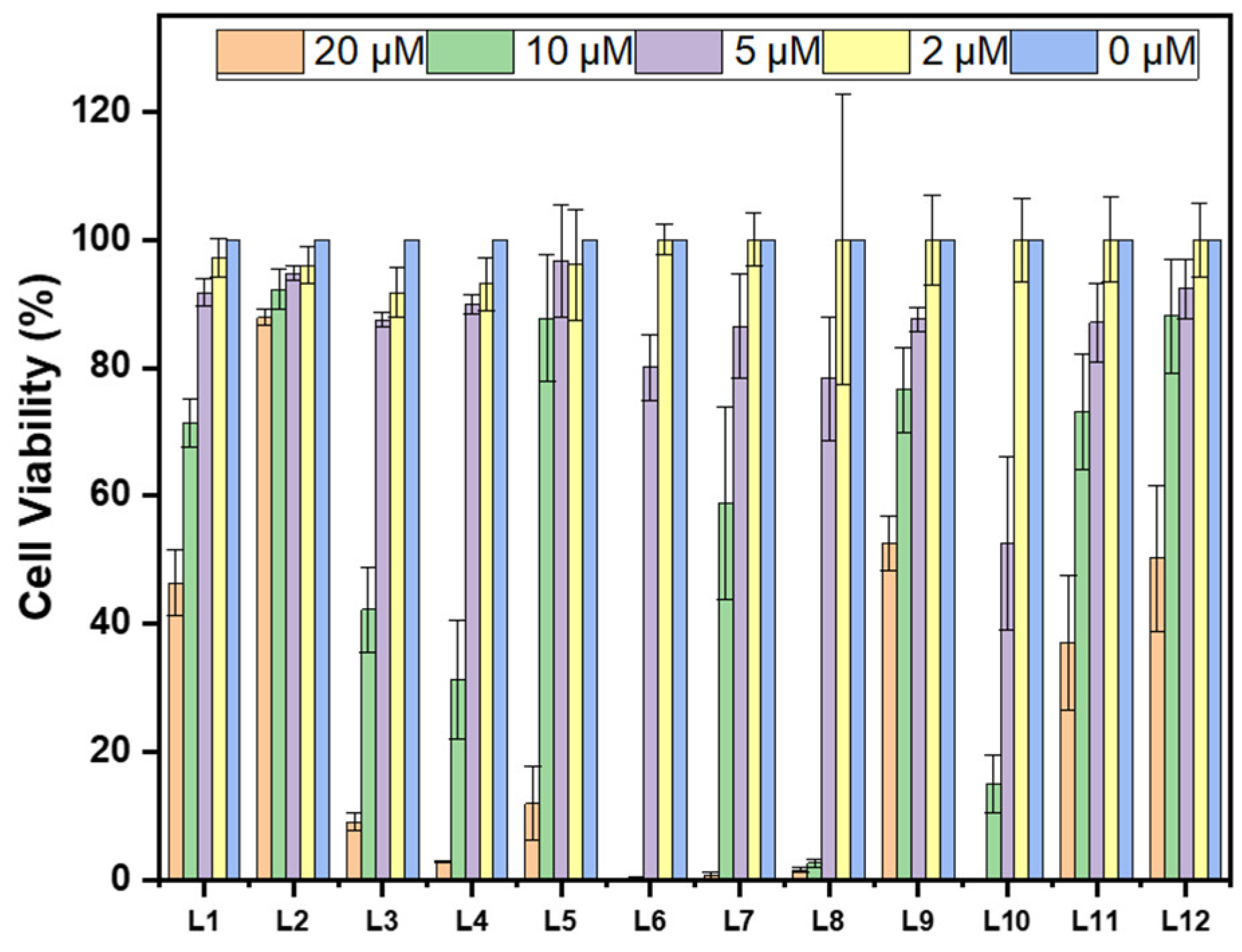
2.4. Cytotoxicity of Compounds in N2a Cells
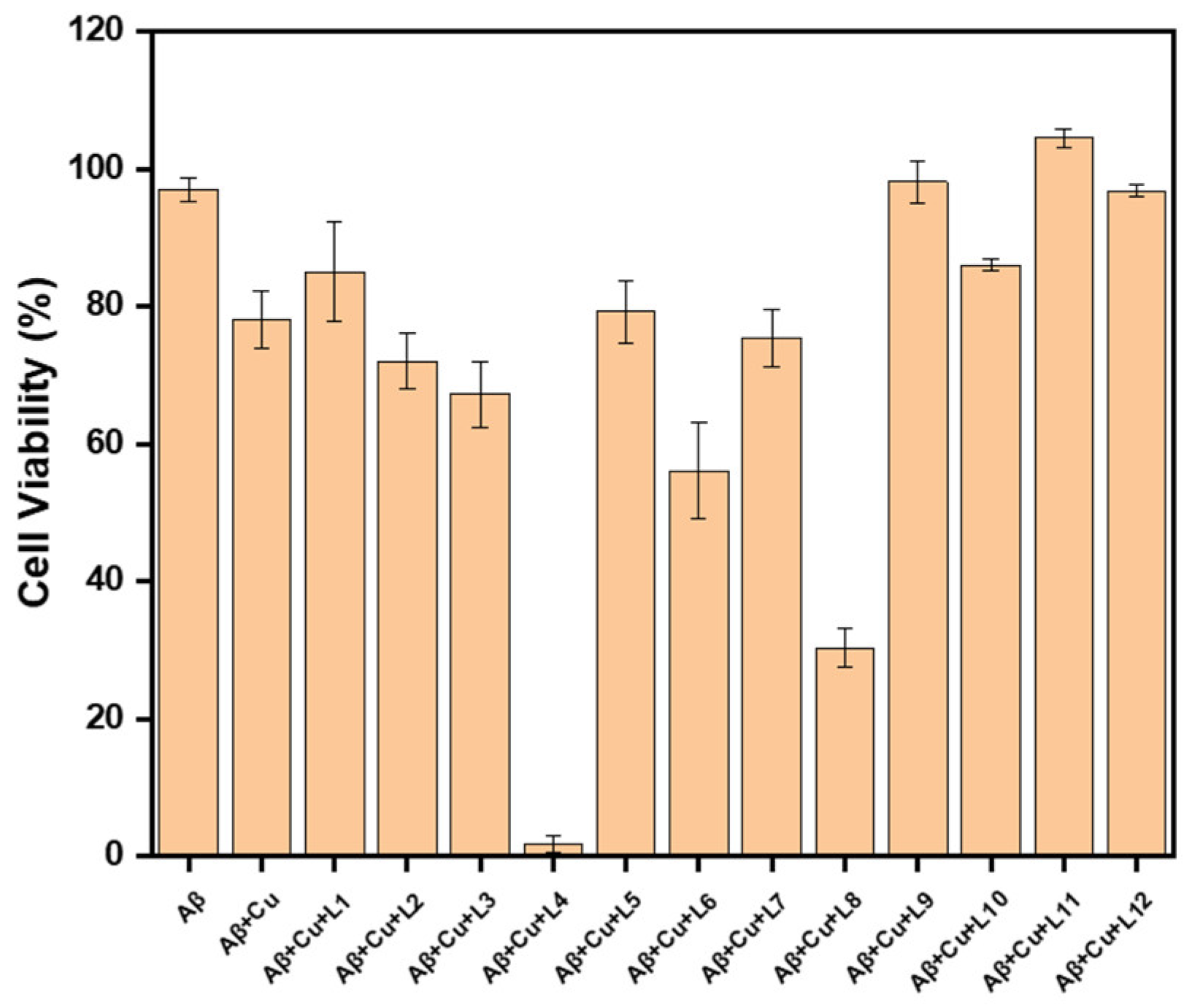
2.5. Effect of Inhibitors on Aβ42 Neurotoxicity in N2a Cells
2.6. Fluorescence Imaging of Amyloid Plaques in 5xFAD Mice Brain Sections
2.7. Docking Studies of Compounds on Aβ
3. Discussion
3.1. Application of Amyloid Inhibitors
3.2. Interpretation of Docking Results
4. Materials and Methods
4.1. General Experimental Details
4.2. Synthetic Details
4.2.1. Synthesis of Precursors: 1a, 1b, 2a, 2b, 3a, 3b, 4a, and 4b
- Compound 1a ((E)-4-(4-nitrostyryl)phenol) or 1b ((E)-2-methoxy-4-(4-nitrostyryl)phenol). A mixture of 2-(4-nitrophenyl) acetic acid (for 1a: 500 mg, 2.8 mmol)(for 1b: 2 g, 11 mmol), (for 1a) 4-hydroxybenzaldehyde (337 mg, 2.8 mmol) or (for 1b) 2-methoxy-4-vinylphenol (1.68 g, 11 mmol), and piperidine (for 1a: 271 μL)(for 1b: 1.1 mL) was added in toluene (1a: 50 mL, 1b: 100 mL). The resultant mixture was heated to reflux for 2 h. The solvent was removed, and the resulting residue was purified by silica gel column chromatography using EtOAc/Hexane (1: 3) to yield a yellow solid.
- Compound 2a ((E)-4-(4-aminostyryl)phenol) or 2b ((E)-4-(4-aminostyryl)-2-methoxyphenol) [55]. Stannous chloride (9.3 mol) was added to a solution of compound 1a or 1b (1.9 mmol) in ethanol (100 mL), followed by the addition of concentrated hydrochloric acid (0.75 mL). The solution was brought to reflux for 3 h and cooled to room temperature, with stirring overnight. Saturated sodium bicarbonate was added to adjust the pH to 8–9. After standard workup with ethyl acetate, crude product 2a or 2b was obtained and was used in the following step without further purifications.
- Compound 3a ((E)-4-(4-(methylamino)styryl)phenol) or 3b ((E)-2-methoxy-4-(4-(methylamino)styryl)phenol). To a mixture of 2a or 2b (0.5 mmol), paraformaldehyde (5 mmol), and sodium cyanoborohydride (1.5 mmol), acetic acid (10 mL) was added. The mixture was stirred at room temperature overnight and then poured into 100 mL of water. Saturated sodium bicarbonate was added to adjust the pH to 8–9. After standard workup with dichloromethane, the residue was purified by silica gel column chromatography using EtOAc/Hexane (1:5) to afford 3a or 3b as a white solid.
- Compound 4a ((E)-4-(4-(dimethylamino)styryl)phenol) or 4b ((E)-4-(4-(dimethylamino)styryl)-2-methoxyphenol). To a mixture of compound 2a or 2b (5.0 mmol), paraformaldehyde (50 mmol), and sodium cyanoborohydride (24 mmol), acetic acid (100 mL) was added. The mixture was stirred at room temperature overnight and then poured into 100 mL of water. Saturated sodium bicarbonate was added to adjust the pH to 8–9. After standard workup with dichloromethane (3 × 50 mL), the residue was purified by silica gel column chromatography using EtOAc/Hexane (1:10) to afford 4a or 4b as a white solid.
4.2.2. Synthesis of Compounds L1–L12
- L1, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-(dimethylamino)styryl)-6-methoxyphenol. Paraformaldehyde (5 mg, 0.1 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (18 mg, 0.1 mmol) in MeCN (20 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 4a (27 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1) to yield a light-yellow solution. The solution was neutralized by saturated NaHCO3 solution (30 mL) and extracted by dichloromethane (3 × 10 mL). The solvent was removed to yield a yellow semi-solid (13 mg, yield 30%). 1H NMR (CD3CN): δ (ppm): 7.39 (d, 2H, J = 8.8 Hz), 7.05 (d, 1H, J = 2.0 Hz), 6.95 (d, 1H, J = 16.4 Hz), 6.85 (d, 1H, J = 16.4 Hz), 6.79 (d, 1H, J = 2.0 Hz), 6.79 (d, 2H, J = 2.0 Hz), 3.86 (s, 3H), 3.83 (s, 2H), 2.95 (s, 6H), 2.91–2.80 (m, 4H), 2.67–2.60 (m, 4H), 2.55 (s, 4H), 2.36 (s, 6H). MS (ESI): Expected m/z 438.3068, found 438.3057 [M + H].
- L2, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxy-4-(4-(methylamino)styryl)phenol. Paraformaldehyde (5 mg, 0.1 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (18 mg, 0.1 mmol) in MeCN (20 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 3b (25 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1) to yield a light-yellow solution. The solution was neutralized by saturated NaHCO3 solution (30 mL) and extracted by dichloromethane (3 × 10 mL). The solvent was removed to yield a yellow semi-solid (15 mg, yield 34%). 1H NMR (CDCl3): δ (ppm): 7.33 (d, 2H, J = 8.6 Hz), 6.95 (d, 1H, J = 1.9 Hz), 6.86 (d, 1H, J = 16.3 Hz), 6.78 (d, 1H, J = 16.3 Hz), 6.74 (d, 1H, J = 1.9 Hz), 6.59 (d, 1H, J = 8.6 Hz), 3.92 (s, 3H), 3.83 (s, 2H), 3.00–2.91 (m, 4H), 2.86 (s, 3H), 2.78–2.70 (m, 4H), 2.63 (s, 4H), 2.40 (s, 6H). MS (ESI): Expected m/z 424.2911, found 424.2901 [M + H].
- L3, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-(methylamino)styryl)phenol. Paraformaldehyde (5 mg, 0.1 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (18 mg, 0.1 mmol) in MeCN (20 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 3a (22 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1) to yield a light-yellow solution. The solution was neutralized by saturated NaHCO3 solution (30 mL) and extracted by dichloromethane (3 × 10 mL). The solvent was removed to yield a yellow semi-solid (15 mg, 38%). 1H NMR (CD3CN): δ (ppm): 7.32 (d, 2H, J = 8.6 Hz), 7.28 (dd, 1H, J = 8.3, 2.3 Hz), 7.21 (d, 1H, J = 2.2 Hz), 6.91 (d, 1H, J = 16.4 Hz), 6.83 (d, 1H, J = 16.4 Hz), 6.76 (d, 1H, J = 8.3 Hz), 6.59 (d, 2H, J = 8.6 Hz), 4.51 (s, 1H), 3.86 (s, 2H), 2.93–2.83 (m, 4H), 2.78 (d, J = 3.1 Hz, 3H), 2.69–2.61 (m, 4H), 2.58 (s, 4H), 2.38 (s, 6H). MS (ESI): Expected m/z 394.2811, found 394.2802 [M + H].
- L4, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-(dimethylamino)styryl)phenol. Paraformaldehyde (5 mg, 0.1 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (18 mg, 0.1 mmol) in MeCN (20 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 4a (24 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1) to yield a light-yellow solution. The solution was neutralized by saturated NaHCO3 solution (30 mL) and extracted by dichloromethane (3 × 10 mL). The solvent was removed to yield a yellow semi-solid (13 mg, yield 32%). 1H NMR (CD3CN): δ (ppm): 7.38 (d, 2H, J = 8.7 Hz), 7.28 (dd, 1H, J = 8.3, 2.2 Hz), 7.18 (d, J = 2.2 Hz, 1H), 6.92 (d, 1H, J = 16.4 Hz), 6.86 (d, 1H, J = 16.4 Hz), 6.74 (d, 1H, J = 8.8 Hz), 6.71 (d, 1H, J = 8.1 Hz), 3.83 (s, 2H), 2.95 (s, 6H), 2.88–2.82 (m, 4H), 2.66–2.58 (m, 4H), 2.53 (s, 4H), 2.35 (s, 6H). MS (ESI): Expected m/z 409.2962, found 408.2950 [M + H].
- L5, (E)-4-(4-(diethylamino)styryl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxyphenol. A mixture of 4-vinylphenol (200 mg, 1.7 mmol), 4-bromo-N,N-diethylaniline (388 mg, 1.7 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed and purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude product 5b. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 5b (27 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane, and dried to give a yellow semi-solid (15 mg, yield 34%). 1H NMR (CDCl3): δ (ppm): 7.34 (d, 2H, J = 8.8 Hz), 7.27 (dd, 1H, J = 7.9 Hz), 7.11 (d, 1H, J = 2.2 Hz), 6.93–6.73 (m, 3H), 6.65 (d, 2H, J = 8.9 Hz), 3.83 (s, 2H), 3.37 (q, 4H, J = 7.1 Hz), 3.04–2.82 (m, 4H), 2.58 (s, 4H), 2.41 (s, 6H), 1.17 (t, J = 7.0 Hz, 6H). MS (ESI): Expected m/z 467.3381, found 467.3372 [M + H].
- L6, (E)-4-(4-(diethylamino)styryl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)phenol. A mixture of 2-methoxy-4-vinylphenol (200 mg, 1.3 mmol), 4-bromo-N,N-diethylaniline (297 mg, 1.3 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed, and the product was purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude material 5a. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 5a (20 mg, 0.05 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane (3 × 10 mL), and dried to give a yellow semi-solid (12 mg, yield 26%). 1H NMR (CDCl3): δ (ppm): 7.34 (d, 2H, J = 8.7 Hz), 6.95 (d, 1H, J = 1.8 Hz), 6.86 (d, 1H, J = 16.2 Hz), 6.81–6.72 (m, 2H), 6.65 (d, 2H, J = 8.8 Hz), 3.92 (s, 3H), 3.83 (s, 2H), 3.37 (q, 4H, J = 7.1 Hz), 2.99–2.86 (m, 4H), 2.80–2.70 (m, 4H), 2.63 (s, 4H), 2.40 (s, 6H), 1.17 (t, J = 7.0 Hz, 6H). MS (ESI): Expected m/z 437.3275, found 437.3261 [M + H].
- L7, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxy-4-(4-(piperidin-1-yl)styryl)phenol. A mixture of 2-methoxy-4-vinylphenol (200 mg, 1.3 mmol), 1-(4-bromophenyl)piperidine (312 mg, 1.3 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed, and the product was purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude material 7a. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7a (20 mg, 0.05 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane (3 × 10 mL), and dried to give a yellow semi-solid (12 mg, yield 26%). 1H NMR (CDCl3): δ (ppm): 7.36 (d, 2H, J = 8.7 Hz), 6.95 (d, 1H, J = 1.9 Hz), 6.89 (d, 2H, J = 8.8 Hz), 6.84 (s, 2H), 6.74 (d, 1H, J = 1.9 Hz), 3.92 (s, 3H), 3.83 (s, 2H), 3.22–3.14 (m, 4H), 2.99–2.88 (m, 4H), 2.77–2.68 (m, 4H), 2.62 (s, 4H), 2.40 (s, 6H), 1.69 (m, 4H), 1.64–1.51 (m, 2H). MS (ESI): Expected m/z 479.3386, found 479.3375 [M + H].
- L8, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-(piperidin-1-yl)styryl)phenol. A mixture of 4-vinylphenol (200 mg, 1.7 mmol), 1-(4-bromophenyl)piperidine (408 mg, 1.7 mmol), triethanolamine (10 mL) and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed and purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain a crude product 7b. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7b (27 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane, and dried to give a yellow semi-solid (15 mg, yield 34%). 1H NMR (CDCl3): δ (ppm): 7.36 (d, 2H, J = 8.8 Hz), 7.29 (dd, 1H, J = 8.4, 2.2 Hz), 7.11 (d, 1H, J = 2.2 Hz), 6.95–6.78 (m, 5H), 3.83 (s, 2H), 3.26–3.14 (m, 4H), 2.98–2.84 (m, 4H), 2.72–2.62 (m, 4H), 2.56 (s, 4H), 2.39 (s, 6H), 1.71 (m, 4H), 1.65–1.51 (m, 2H). MS (ESI): Expected m/z 449.3280, found 449.3262 [M + H].
- L9, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxy-4-(4-morpholinostyryl)phenol. A mixture of 2-methoxy-4-vinylphenol (200 mg, 1.3 mmol), 4-(4-bromophenyl)morpholine (315 mg, 1.3 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed, and the product was purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude material 7c. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7c (20 mg, 0.05 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane (3 × 10 mL), and dried to give a yellow semi-solid (12 mg, yield 26%). 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 8.7 Hz, 2H), 6.90 (s, 1H), 6.82 (d, J = 8.7 Hz, 2H), 6.80 (s, 2H), 6.72 (s, 1H), 3.86 (s, 3H), 3.79 (m, 8H), 3.15–3.09 (m, 4H), 2.86 (m, 4H), 2.76 (m, 4H), 2.40 (s, 6H). MS (ESI): Expected m/z 481.3179, found 481.3162 [M + H].
- L10, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-morpholinostyryl)phenol. A mixture of 4-vinylphenol (200 mg, 1.7 mmol), 4-(4-bromophenyl)morpholine (411 mg, 1.7 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed and purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude product 7d. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7d (27 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane, and dried to give a yellow semi-solid (15 mg, yield 34%). 1H NMR (CD3CN): δ (ppm): 7.42 (d, J = 8.8 Hz, 2H), 7.31 (dd, J = 8.3, 2.3 Hz, 1H), 7.23 (d, J = 2.2 Hz, 1H), 6.93 (d, J = 7.6 Hz, 4H), 6.75 (d, J = 8.3 Hz, 1H), 3.86 (s, 2H), 3.83–3.73 (m, 4H), 3.19–3.09 (m, 4H), 2.89–2.84 (m, 4H), 2.68–2.61 (m, 4H), 2.57 (s, 4H), 2.37 (s, 6H). MS (ESI): Expected m/z 450.3073, found 451.3059 [M + H].
- L11, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-6-methoxy-4-(4-(4-methylpiperazin-1-yl)styryl)phenol. A mixture of 2-methoxy-4-vinylphenol (200 mg, 1.3 mmol), 1-(4-bromophenyl)methylpiperazine (332 mg, 1.3 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed, and the product was purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude material 7e. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7e (20 mg, 0.05 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane (3 × 10 mL), and dried to give a yellow semi-solid (12 mg, yield 26%). 1H NMR (CDCl3): δ (ppm) 7.41 (d, 2H, J = 8.7 Hz), 6.98 (d, 1H, J = 1.9 Hz), 6.93 (d, 2H, J = 10.9 Hz), 6.88 (s, 2H), 6.78 (d, 1H, J = 1.9 Hz), 3.95 (s, 3H), 3.86 (s, 2H), 3.29–3.24 (m, 4H), 3.02–2.92 (m, 4H), 2.81–2.73 (m, 4H), 2.68 (s, 4H), 2.63–2.58 (m, 4H), 2.44 (s, 6H), 2.38 (s, 3H). MS (ESI): Expected m/z 494.3495, found 494.3481 [M + H].
- L12, (E)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)methyl)-4-(4-(4-methylpiperazin-1-yl)styryl)phenol. A mixture of 4-vinylphenol (200 mg, 1.7 mmol), 1-(4-bromophenyl)methylpiperazine (433 mg, 1.7 mmol), triethanolamine (10 mL), and Pd(OAc)2 (23 mg, 0.1 mmol) was stirred under nitrogen at 100 °C for 24 h. The reaction was cooled to room temperature and quenched with water (5 mL). The resulting solution was extracted by ethyl acetate (3 × 20 mL). The solvent was removed and purified by silica gel column chromatography (ethyl acetate: hexane = 1:4) to obtain crude product 7f. Then, paraformaldehyde (6 mg, 0.2 mmol) was added to a solution of 1,4-dimethyl-1,4,7-triazacyclononane (15 mg, 0.1 mmol) in MeCN (10 mL), and the resultant mixture was heated to reflux for 30 min. A solution of compound 7f (27 mg, 0.1 mmol) in MeCN (10 mL) was added to the reaction flask, and the solution was refluxed for an additional 24 h. The solvent was removed, and the resulting residue was purified by CombiFlash (reversed-phase) using MeCN/H2O/TFA (gradient wash from 10:90:1 to 30:70:1). The solution was neutralized by saturated NaHCO3 solution (30 mL), extracted by dichloromethane, and dried to give a yellow semi-solid (15 mg, yield 34%). 1H NMR (500 MHz, CDCl3) δ 7.30 (d, 2H, J = 8.9 Hz), 7.11 (s, 1H), 6.82 (d, 2H, J = 8.5 Hz), 6.79 (s, 4H), 3.76 (s, 2H), 3.17 (m, 4H), 2.83–2.80 (m, 4H), 2.70–2.73 (m, 4H), 2.63–2.59 (m, 4H), 2.54–2.47 (m, 4H), 2.35 (s, 6H), 2.28 (s, 3H). MS (ESI): Expected m/z 464.3389, found 464.3368 [M + H].
4.3. Fluorescence Spectra Measurements
4.4. Amyloid β Peptide Experiments
4.4.1. Fluorescence Turn-On and Cell Studies
4.4.2. Inhibition Assay
4.5. Alamar Blue Assay
4.5.1. Cytotoxicity Studies
4.5.2. N2a Cell Rescue Studies
4.6. Histological Staining of 5xFAD Mice Brain Sections
4.7. Molecular Docking
4.8. Log D Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta |
| CR | Congo Red |
| EAG | Electron accepting group |
| EDG | Electron donating group |
| NMR | Nuclear magnetic resonance |
| sAβ | Soluble amyloid beta |
| tacn | 1,4,7-triazacyclononane |
| ThT | Thioflavin T |
| TICT | Twisted Intramolecular Charge Transfer |
References
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure–neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed]
- Tew, D.J.; Bottomley, S.P.; Smith, D.P.; Ciccotosto, G.D.; Babon, J.; Hinds, M.G.; Masters, C.L.; Cappai, R.; Barnham, K.J. Stabilization of Neurotoxic Soluble β-Sheet-Rich Conformations of the Alzheimer’s Disease Amyloid-β Peptide. Biophys. J. 2008, 94, 2752–2766. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wu, W.-H.; Li, Q.-Y.; Zhao, Y.-F.; Li, Y.-M. Copper inducing Aβ42 rather than Aβ40 nanoscale oligomer formation is the key process for Aβ neurotoxicity. Nanoscale 2011, 3, 4746–4751. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zeng, F.; Li, X.; Ran, C.; Xu, Y.; Li, Y. Highly Specific Detection of Aβ Oligomers in Early Alzheimer’s Disease by a Near-Infrared Fluorescent Probe with a “V-shaped” Spatial Conformation. Chem. Commun. 2020, 56, 583–586. [Google Scholar] [CrossRef]
- Matsumura, K.; Ono, M.; Kitada, A.; Watanabe, H.; Yoshimura, M.; Iikuni, S.; Kimura, H.; Okamoto, Y.; Ihara, M.; Saji, H. Structure–Activity Relationship Study of Heterocyclic Phenylethenyl and Pyridinylethenyl Derivatives as Tau-Imaging Agents That Selectively Detect Neurofibrillary Tangles in Alzheimer’s Disease Brains. J. Med. Chem. 2015, 58, 7241–7257. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.-G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies—Still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Vadukul, D.M.; Maina, M.; Franklin, H.; Nardecchia, A.; Serpell, L.C.; Marshall, K.E. Internalisation and toxicity of amyloid-beta 1–42 are influenced by its conformation and assembly state rather than size. FEBS Lett. 2020, 594, 3490–3503. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Ciaglia, T.; Miranda, M.R.; Di Micco, S.; Vietri, M.; Smaldone, G.; Musella, S.; Di Sarno, V.; Auriemma, G.; Sardo, C.; Moltedo, O.; et al. Neuroprotective Potential of Indole-Based Compounds: A Biochemical Study on Antioxidant Properties and Amyloid Disaggregation in Neuroblastoma Cells. Antioxidants 2024, 13, 1585. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, X.; Zhang, R.; Zhao, C. Flavonoids with Potential Anti-Amyloidogenic Effects as Therapeutic Drugs for Treating Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 505–533. [Google Scholar] [CrossRef]
- Radbakhsh, S.; Barreto, G.E.; Bland, A.R.; Sahebkar, A. Curcumin: A small molecule with big functionality against amyloid aggregation in neurodegenerative diseases and type 2 diabetes. BioFactors 2021, 47, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Hilt, S.; Liu, R.; Maezawa, I.; Rojalin, T.; Aung, H.H.; Budamagunta, M.; Slez, R.; Gong, Q.; Carney, R.P.; Voss, J.C. Novel Stilbene-Nitroxyl Hybrid Compounds Display Discrete Modulation of Amyloid Beta Toxicity and Structure. Front. Chem. 2022, 10, 896386. [Google Scholar] [CrossRef] [PubMed]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Guo, W.; Patel, S.; Cho, H.-J.; Sun, L.; Mirica, L.M. Amphiphilic stilbene derivatives attenuate the neurotoxicity of soluble Aβ42 oligomers by controlling their interactions with cell membranes. Chem. Sci. 2022, 13, 12818–12830. [Google Scholar] [CrossRef]
- Sun, L.; Cho, H.-J.; Sen, S.; Arango, A.S.; Huynh, T.T.; Huang, Y.; Bandara, N.; Rogers, B.E.; Tajkhorshid, E.; Mirica, L.M. Amphiphilic Distyrylbenzene Derivatives as Potential Therapeutic and Imaging Agents for the Soluble Amyloid-β Oligomers in Alzheimer’s Disease. J. Am. Chem. Soc. 2021, 143, 10462–10476. [Google Scholar] [CrossRef]
- Fu, Z.; Yang, J.; Wei, Y.; Li, J. Effects of piceatannol and pterostilbene against β-amyloid-induced apoptosis on the PI3K/Akt/Bad signaling pathway in PC12 cells. Food Funct. 2016, 7, 1014–1023. [Google Scholar] [CrossRef]
- Yuan, W.; Shang, Z.; Qiang, X.; Tan, Z.; Deng, Y. Synthesis of pterostilbene and resveratrol carbamate derivatives as potential dual cholinesterase inhibitors and neuroprotective agents. Res. Chem. Intermed. 2014, 40, 787–800. [Google Scholar] [CrossRef]
- Garcia, G.X.; Larsen, S.W.; Pye, C.; Galbreath, M.; Isovitsch, R.; Fradinger, E.A. The functional group on (E)-4,4′–disubstituted stilbenes influences toxicity and antioxidative activity in differentiated PC-12 cells. Bioorg. Med. Chem. Lett. 2013, 23, 6355–6359. [Google Scholar] [CrossRef]
- Breitung, E.M.; Shu, C.-F.; McMahon, R.J. Thiazole and Thiophene Analogues of Donor−Acceptor Stilbenes: Molecular Hyperpolarizabilities and Structure−Property Relationships. J. Am. Chem. Soc. 2000, 122, 1154–1160. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Wu, Q.; Wang, S.; Yang, S.; Li, X.; Chen, N.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int. Immunopharmacol. 2021, 99, 108002. [Google Scholar] [CrossRef] [PubMed]
- Freyssin, A.; Page, G.; Fauconneau, B.; Rioux Bilan, A. Natural stilbenes effects in animal models of Alzheimer’s disease. Neural Reg. Res. 2020, 15, 843–849. [Google Scholar] [CrossRef]
- Gao, D.; Hao, J.-p.; Li, B.-y.; Zheng, C.-c.; Miao, B.-b.; Zhang, L.; Li, Y.-l.; Li, L.; Li, X.-j.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates neuroinflammation for Alzheimer’s disease via cGAS-STING. Eur. J. Pharmacol. 2023, 953, 175809. [Google Scholar] [CrossRef] [PubMed]
- Firdoos, S.; Dai, R.; Tahir, R.A.; Khan, Z.Y.; Li, H.; Zhang, J.; Ni, J.; Quan, Z.; Qing, H. In silico identification of novel stilbenes analogs for potential multi-targeted drugs against Alzheimer’s disease. J. Mol. Mod. 2023, 29, 209. [Google Scholar] [CrossRef]
- Yu, Z.; Moshood, Y.; Wozniak, M.K.; Patel, S.; Terpstra, K.; Llano, D.A.; Dobrucki, L.W.; Mirica, L.M. Amphiphilic Molecules Exhibiting Zwitterionic Excited-State Intramolecular Proton Transfer and Near-Infrared Emission for the Detection of Amyloid β Aggregates in Alzheimer’s Disease. Chem. Eur. J. 2023, 29, e202302408. [Google Scholar] [CrossRef]
- Cho, H.-J.; Huynh, T.T.; Rogers, B.E.; Mirica, L.M. Design of a multivalent bifunctional chelator for diagnostic 64Cu PET imaging in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 30928–30933. [Google Scholar] [CrossRef]
- Cui, M.; Ono, M.; Watanabe, H.; Kimura, H.; Liu, B.; Saji, H. Smart Near-Infrared Fluorescence Probes with Donor–Acceptor Structure for in Vivo Detection of β-Amyloid Deposits. J. Am. Chem. Soc. 2014, 136, 3388–3394. [Google Scholar] [CrossRef]
- Kung, H.F.; Choi, S.R.; Qu, W.; Zhang, W.; Skovronsky, D. 18F Stilbenes and Styrylpyridines for PET Imaging of Aβ Plaques in Alzheimer’s Disease: A Miniperspective. J. Med. Chem. 2010, 53, 933–941. [Google Scholar] [CrossRef]
- Martí-Centelles, R.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Inhibitory effect of cytotoxic stilbenes related to resveratrol on the expression of the VEGF, hTERT and c-Myc genes. Eur. J. Med. Chem. 2015, 103, 488–496. [Google Scholar] [CrossRef]
- Kung, H.F.; Kung, M.-P.; Zhuang, Z.-P. Stilbene Derivatives and Their Use for Binding and Imaging Amyloid Plaques. International Patent no. 05854410.7, 5 October 2005. [Google Scholar]
- Xiao, G.; Li, Y.; Qiang, X.; Xu, R.; Zheng, Y.; Cao, Z.; Luo, L.; Yang, X.; Sang, Z.; Su, F.; et al. Design, synthesis and biological evaluation of 4′-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1030–1041. [Google Scholar] [CrossRef]
- Lennol, M.P.; Canelles, S.; Guerra-Cantera, S.; Argente, J.; García-Segura, L.M.; de Ceballos, M.L.; Chowen, J.A.; Frago, L.M. Amyloid-β1-40 differentially stimulates proliferation, activation of oxidative stress and inflammatory responses in male and female hippocampal astrocyte cultures. Mech. Ageing Dev. 2021, 195, 111462. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Kim, J.; Mirica, L.M. The effect of Cu2+ and Zn2+ on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 2013, 5, 1529–1536. [Google Scholar] [CrossRef]
- Sun, L.; Sharma, A.K.; Han, B.-H.; Mirica, L.M. Amentoflavone: A Bifunctional Metal Chelator that Controls the Formation of Neurotoxic Soluble Aβ42 Oligomers. ACS Chem. Neurosci. 2020, 11, 2741–2752. [Google Scholar] [CrossRef]
- Cho, H.-J.; Sharma, A.K.; Zhang, Y.; Gross, M.L.; Mirica, L.M. A Multifunctional Chemical Agent as an Attenuator of Amyloid Burden and Neuroinflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2020, 11, 1471–1481. [Google Scholar] [CrossRef]
- Raju, S.K.; Sundhararajan, N.; Sekar, P.; Nagalingam, Y. Therapeutic aspects of biologically potent vanillin derivatives: A critical review. J. Drug Deliv. Therap. 2023, 13, 177–189. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Liccardo, M.; Sirangelo, I. Overview of the Role of Vanillin in Neurodegenerative Diseases and Neuropathophysiological Conditions. Int. J. Mol. Sci. 2023, 24, 1817. [Google Scholar] [CrossRef]
- Kumar, S.N.; Nair, H.R.; Kumar, B.P. Comparative analysis of anti-oxidant potential of vanillin and ferulic acid invitro. Food Humanit. 2023, 1, 1206–1212. [Google Scholar] [CrossRef]
- Terpstra, K.; Wang, Y.; Huynh, T.T.; Bandara, N.; Cho, H.-J.; Rogers, B.E.; Mirica, L.M. Divalent 2-(4-Hydroxyphenyl)benzothiazole Bifunctional Chelators for 64Cu Positron Emission Tomography Imaging in Alzheimer’s Disease. Inorg. Chem. 2022, 61, 20326–20336. [Google Scholar] [CrossRef]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Mahan, T.E.; Wang, C.; Bao, X.; Choudhury, A.; Ulrich, J.D.; Holtzman, D.M. Selective reduction of astrocyte apoE3 and apoE4 strongly reduces Aβ accumulation and plaque-related pathology in a mouse model of amyloidosis. Mol. Neurodegener. 2022, 17, 13. [Google Scholar] [CrossRef]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Qian, Z.; Chen, Y.; Qian, H.; Wei, G.; Zhang, Q. Norepinephrine Inhibits Alzheimer’s Amyloid-β Peptide Aggregation and Destabilizes Amyloid-β Protofibrils: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2019, 10, 1585–1594. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. [Google Scholar] [CrossRef]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat. Commun. 2020, 11, 3014. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Dong, X.; Qiao, Q.; Qian, Z.; Wei, G. Recent computational studies of membrane interaction and disruption of human islet amyloid polypeptide: Monomers, oligomers and protofibrils. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1826–1839. [Google Scholar] [CrossRef]
- Gao, D.; Wan, J.; Zou, Y.; Gong, Y.; Dong, X.; Xu, Z.; Tang, J.; Wei, G.; Zhang, Q. The destructive mechanism of Aβ1–42 protofibrils by norepinephrine revealed via molecular dynamics simulations. Phys. Chem. Chem. Phys. 2022, 24, 19827–19836. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-L.; Singh, P.K.; Calvano, M.; Norris, E.H.; Strickland, S. A possible mechanism for the enhanced toxicity of beta-amyloid protofibrils in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2309389120. [Google Scholar] [CrossRef] [PubMed]
- Vangala, V.R.; Bhogala, B.R.; Dey, A.; Desiraju, G.R.; Broder, C.K.; Smith, P.S.; Mondal, R.; Howard, J.A.K.; Wilson, C.C. Correspondence between Molecular Functionality and Crystal Structures. Supramolecular Chemistry of a Family of Homologated Aminophenols. J. Am. Chem. Soc. 2003, 125, 14495–14509. [Google Scholar] [CrossRef] [PubMed]
- Jameson, L.P.; Smith, N.W.; Dzyuba, S.V. Dye-Binding Assays for Evaluation of the Effects of Small Molecule Inhibitors on Amyloid (Aβ) Self-Assembly. ACS Chem. Neurosci. 2012, 3, 807–819. [Google Scholar] [CrossRef]
- Terpstra, K.; Huang, Y.; Na, H.; Sun, L.; Gutierrez, C.; Yu, Z.; Mirica, L.M. 2-Phenylbenzothiazolyl iridium complexes as inhibitors and probes of amyloid β aggregation. Dalton Trans. 2024, 53, 14258–14264. [Google Scholar] [CrossRef]
- Ran, C.; Xu, X.; Raymond, S.B.; Ferrara, B.J.; Neal, K.; Bacskai, B.J.; Medarova, Z.; Moore, A. Design, Synthesis, and Testing of Difluoroboron-Derivatized Curcumins as Near-Infrared Probes for in Vivo Detection of Amyloid-β Deposits. J. Am. Chem. Soc. 2009, 131, 15257–15261. [Google Scholar] [CrossRef]
- Xue, C.; Lee, Y.K.; Tran, J.; Chang, D.; Guo, Z. A mix-and-click method to measure amyloid-β concentration with sub-micromolar sensitivity. R. Soc. Open Sci. 2017, 4, 170325. [Google Scholar] [CrossRef]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils—Current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef]
- Ertl, P. A Web Tool for Calculating Substituent Descriptors Compatible with Hammett Sigma Constants. Chem. Methods 2022, 2, e202200041. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez, C.; Sun, L.; Huang, Y.; Gui, K.; Terpstra, K.; Mirica, L.M. N-Alkylamino Stilbene Compounds as Amyloid β Inhibitors for Alzheimer’s Disease Research. Molecules 2025, 30, 2471. https://doi.org/10.3390/molecules30112471
Gutiérrez C, Sun L, Huang Y, Gui K, Terpstra K, Mirica LM. N-Alkylamino Stilbene Compounds as Amyloid β Inhibitors for Alzheimer’s Disease Research. Molecules. 2025; 30(11):2471. https://doi.org/10.3390/molecules30112471
Chicago/Turabian StyleGutiérrez, Citlali, Liang Sun, Yiran Huang, Kai Gui, Karna Terpstra, and Liviu M. Mirica. 2025. "N-Alkylamino Stilbene Compounds as Amyloid β Inhibitors for Alzheimer’s Disease Research" Molecules 30, no. 11: 2471. https://doi.org/10.3390/molecules30112471
APA StyleGutiérrez, C., Sun, L., Huang, Y., Gui, K., Terpstra, K., & Mirica, L. M. (2025). N-Alkylamino Stilbene Compounds as Amyloid β Inhibitors for Alzheimer’s Disease Research. Molecules, 30(11), 2471. https://doi.org/10.3390/molecules30112471