
Comparative Neurotoxic Effects of Doxorubicin and Sunitinib: An In Vitro Study on Human Dopaminergic Neuronal Cells

 ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
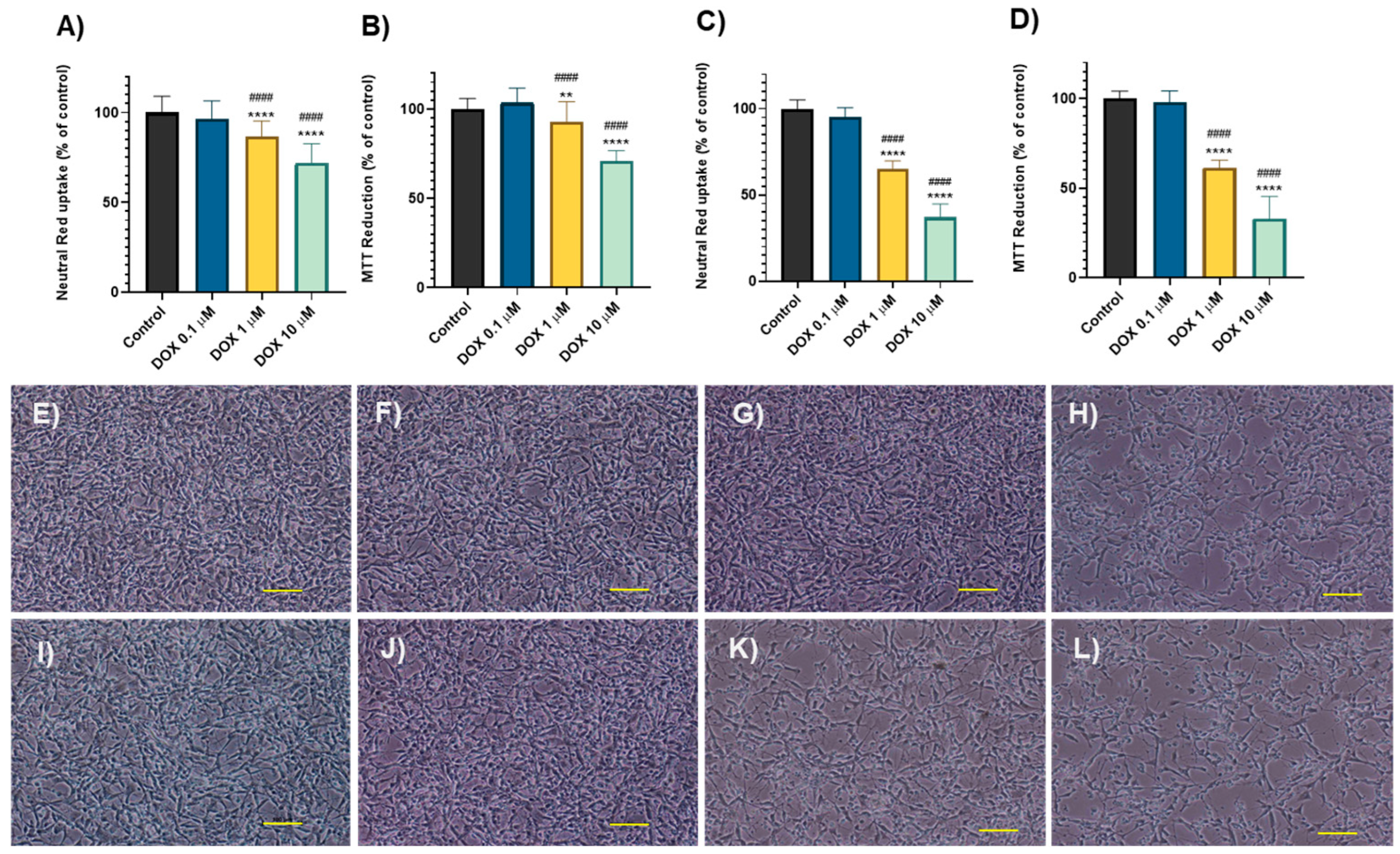
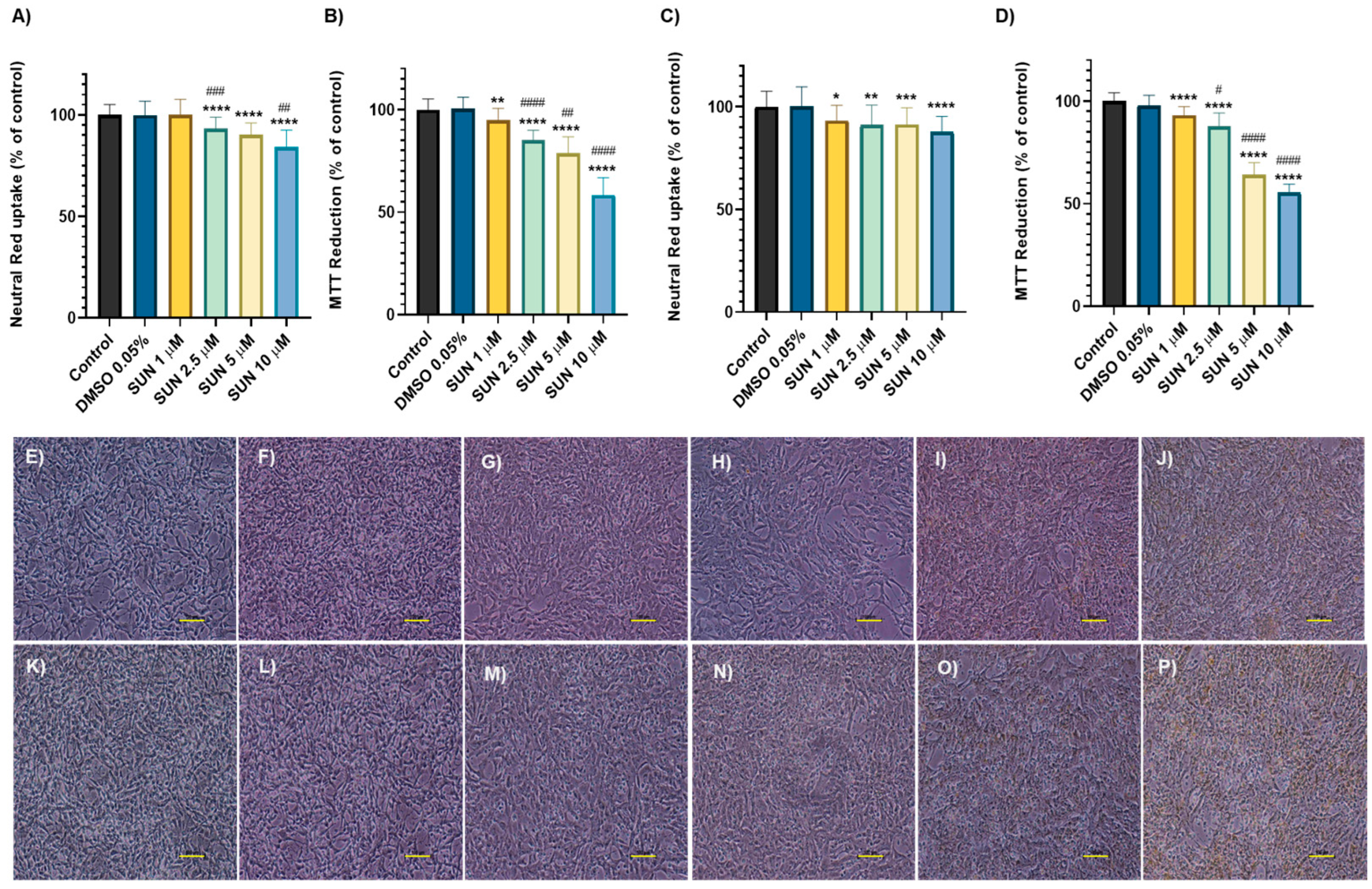
2.1. Doxorubicin and Sunitinib Caused Time- and Concentration- Dependent Cytotoxicity
2.2. Doxorubicin- and Sunitinib-Induced Mitochondrial Membrane Depolarization
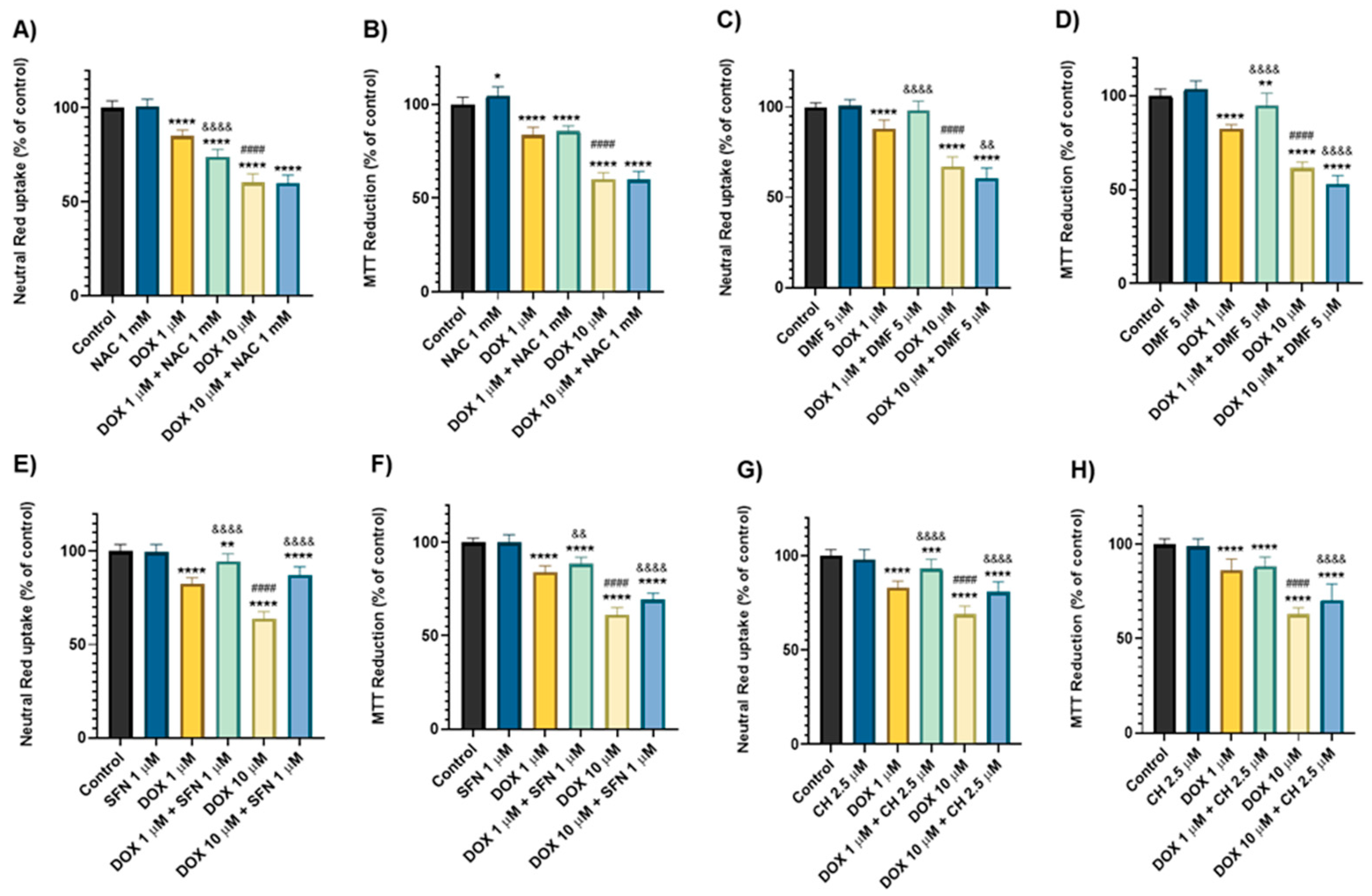
2.3. Dimethyl Fumarate, Sulphoraphane, and Cheirolin Conferred Dissimilar Outcomes Against the Toxicity Induced by Doxorubicin
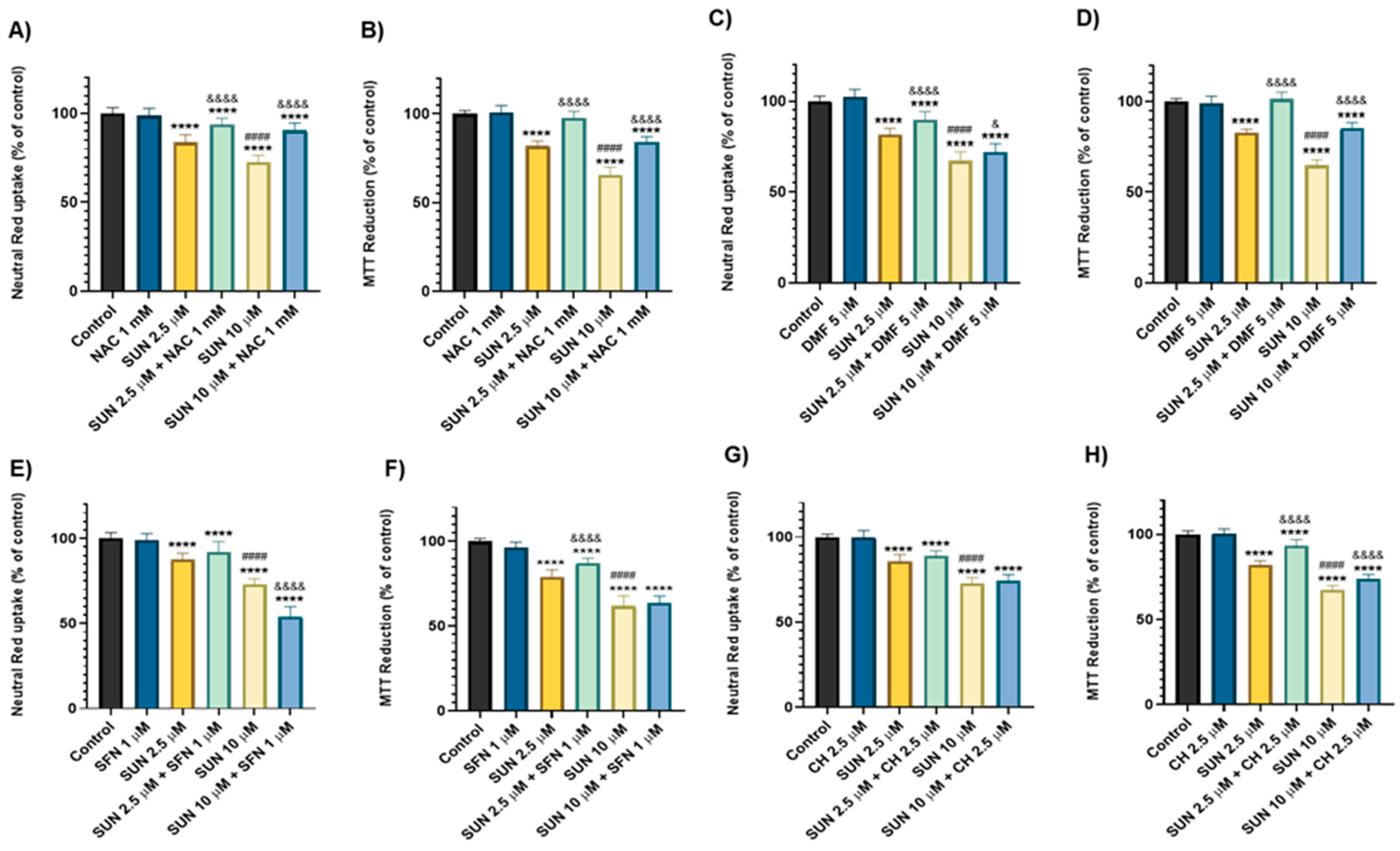
2.4. N-Acetyl Cysteine, Dimethyl Fumarate, Sulphoraphane, and Cheirolin Were Partially Protective Against Sunitinib’s Cytotoxicity
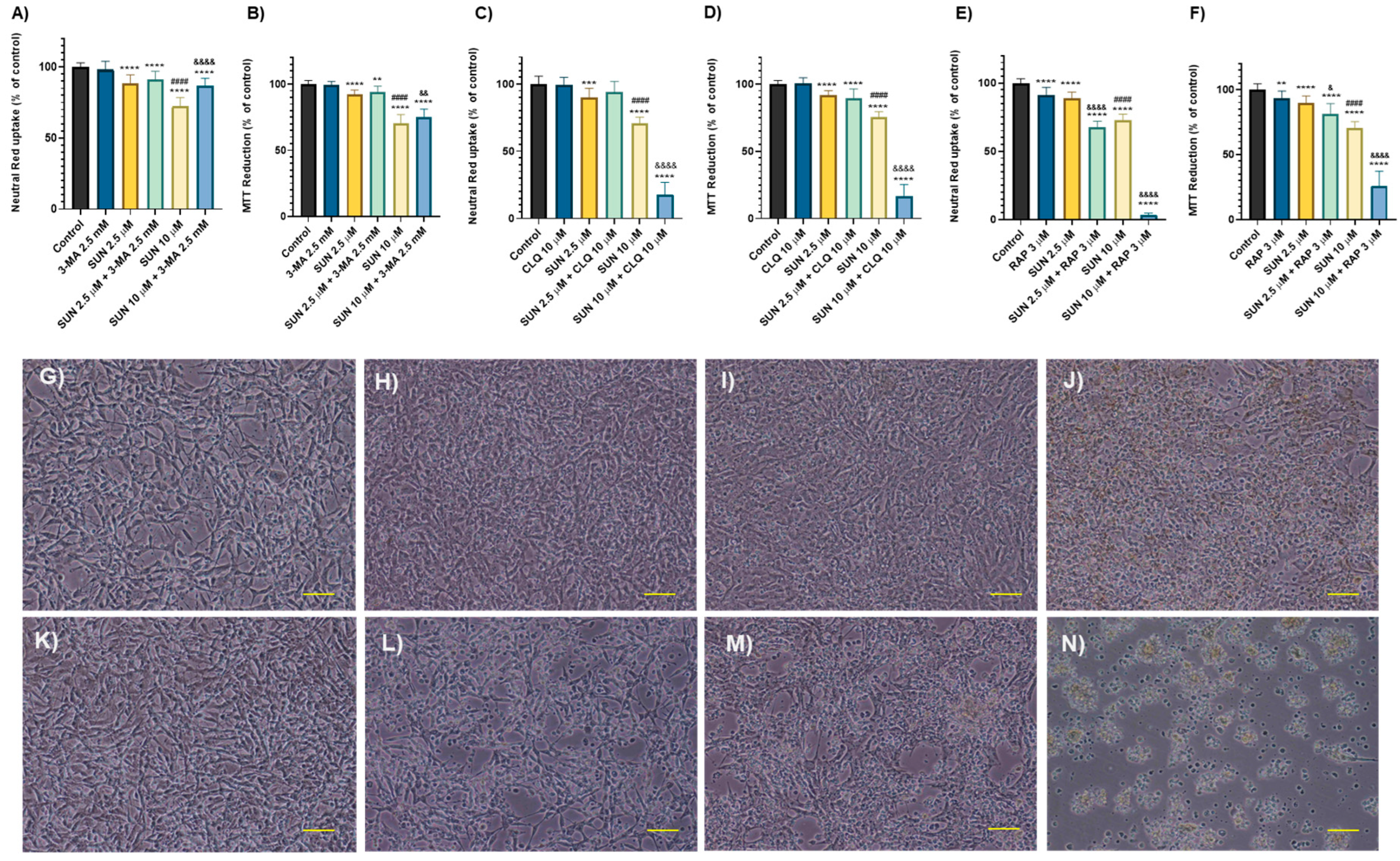
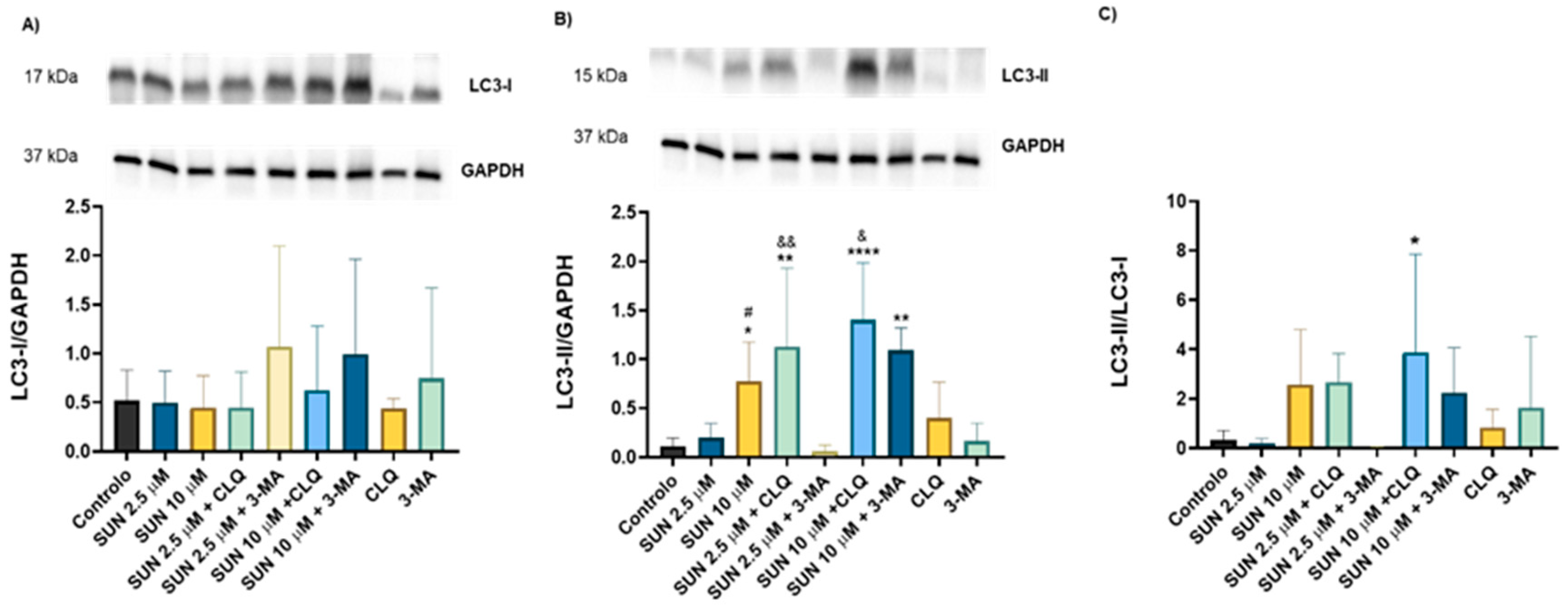
2.5. Autophagy Seems to Play a Key Role in Sunitinib’s Toxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Differentiation
4.4. Drug Exposure
4.5. Cytotoxicity and Morphological Analysis
4.5.1. Cellular Morphology
4.5.2. The Neutral Red Uptake Assay
4.5.3. The MTT Reduction Assay
4.6. Mitochondrial Membrane Potential Assay
4.7. Western Blot
4.7.1. Cell Harvest
4.7.2. Protein Quantification
4.7.3. Electrophoresis and Protein Transference
4.7.4. Incubation with the Antibodies
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| BBB | blood–brain barrier |
| CH | cheirolin |
| CLQ | chloroquine |
| CNS | central nervous system |
| DA | dopamine |
| DAT | dopamine transporter |
| DMF | dimethyl fumarate |
| DMSO | dimethyl sulfoxide |
| DOX | doxorubicin |
| HBSS | Hank’s balanced salt solution |
| LC3 | microtubule-associated protein 1a/1b-light chain 3 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| NAC | n-acetylcysteine |
| NR | neutral red |
| Nrf2 | nuclear factor (erythroid-derived 2)-related-factor 2 |
| RA | retinoic acid |
| RAP | rapamycin |
| SFN | sulforaphane |
| SUN | sunitinib |
| TPA | 12-o-tetradecanoylphorbol-13-acetate |
| VEGF | vascular endothelial growth factor |
References
- World Health Organization. Global Cancer Burden Growing, Amidst Mounting Need for Services; IARC: Lyon, France, 2024. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 25 September 2024).
- Santucci, C.; Carioli, G.; Bertuccio, P.; Malvezzi, M.; Pastorino, U.; Boffetta, P.; Negri, E.; Bosetti, C.; La Vecchia, C. Progress in cancer mortality, incidence, and survival: A global overview. Eur. J. Cancer. Prev. 2020, 29, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Simó, M.; Rifà-Ros, X.; Rodriguez-Fornells, A.; Bruna, J. Chemobrain: A systematic review of structural and functional neuroimaging studies. Neurosci. Biobehav. Rev. 2013, 37, 1311–1321. [Google Scholar] [CrossRef]
- Martins-Teixeira, M.B.; Carvalho, I. Antitumour Anthracyclines: Progress and Perspectives. ChemMedChem 2020, 15, 933–948. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Eide, S.; Feng, Z.P. Doxorubicin chemotherapy-induced “chemo-brain”: Meta-analysis. Eur. J. Pharmacol. 2020, 881, 173078. [Google Scholar] [CrossRef]
- Moretti, R.L.; Dias, E.N.; Kiel, S.G.; Augusto, M.C.M.; Rodrigues, P.S.; Sampaio, A.C.S.; Medeiros, L.S.; Martins, M.F.M.; Suffredini, I.B.; Cardoso, C.V.; et al. Behavioral and morphological effects of resveratrol and curcumin in rats submitted to doxorubicin-induced cognitive impairment. Res. Vet. Sci. 2021, 140, 242–250. [Google Scholar] [CrossRef]
- McAlpin, B.R.; Mahalingam, R.; Singh, A.K.; Dharmaraj, S.; Chrisikos, T.T.; Boukelmoune, N.; Kavelaars, A.; Heijnen, C.J. HDAC6 inhibition reverses long-term doxorubicin-induced cognitive dysfunction by restoring microglia homeostasis and synaptic integrity. Theranostics 2022, 12, 603–619. [Google Scholar] [CrossRef]
- Allen, B.D.; Apodaca, L.A.; Syage, A.R.; Markarian, M.; Baddour, A.A.D.; Minasyan, H.; Alikhani, L.; Lu, C.; West, B.L.; Giedzinski, E.; et al. Attenuation of neuroinflammation reverses Adriamycin-induced cognitive impairments. Acta Neuropathol. Commun. 2019, 7, 186. [Google Scholar] [CrossRef]
- Ongnok, B.; Khuanjing, T.; Chunchai, T.; Pantiya, P.; Kerdphoo, S.; Arunsak, B.; Nawara, W.; Jaiwongkam, T.; Apaijai, N.; Chattipakorn, N.; et al. Donepezil Protects Against Doxorubicin-Induced Chemobrain in Rats via Attenuation of Inflammation and Oxidative Stress Without Interfering with Doxorubicin Efficacy. Neurotherapeutics 2021, 18, 2107–2125. [Google Scholar] [CrossRef]
- Stewart, D.J.; Grewaal, D.; Green, R.M.; Mikhael, N.; Goel, R.; A Montpetit, V.; Redmond, M.D. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer. Res. 1993, 13, 1945–1952. [Google Scholar] [PubMed]
- Sardi, I.; La Marca, G.; Cardellicchio, S.; Giunti, L.; Malvagia, S.; Genitori, L.; Massimino, M.; de Martino, M.; Giovannini, M.G. Pharmacological modulation of blood-brain barrier increases permeability of doxorubicin into the rat brain. Am. J. Cancer Res. 2013, 3, 424–432. [Google Scholar] [PubMed]
- Cardoso, C.V.; de Barros, M.P.; Bachi, A.L.L.; Bernardi, M.M.; Kirsten, T.B.; Martins, M.d.F.M.; Rocha, P.R.D.; da Silva Rodrigues, P.; Bondan, E.F. Chemobrain in rats: Behavioral, morphological, oxidative and inflammatory effects of doxorubicin administration. Behav. Brain Res. 2020, 378, 112233. [Google Scholar] [CrossRef]
- Ren, X.; Keeney, J.T.R.; Miriyala, S.; Noel, T.; Powell, D.K.; Chaiswing, L.; Bondada, S.; St Clair, D.K.; Butterfield, D.A. The triangle of death of neurons: Oxidative damage, mitochondrial dysfunction, and loss of choline-containing biomolecules in brains of mice treated with doxorubicin. Advanced insights into mechanisms of chemotherapy induced cognitive impairment (“chemobrain”) involving TNF-alpha. Free Radic. Biol. Med. 2019, 134, 1–8. [Google Scholar]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Dias-Carvalho, A.; Ferreira, M.; Reis-Mendes, A.; Ferreira, R.; Bastos, M.d.L.; Fernandes, E.; Sa, S.I.; Capela, J.P.; Carvalho, F.; Costa, V.M. Doxorubicin-induced neurotoxicity differently affects the hippocampal formation subregions in adult mice. Heliyon 2024, 10, e31608. [Google Scholar] [CrossRef]
- Dias-Carvalho, A.; Ferreira, M.; Ferreira, R.; Bastos, M.d.L.; Sá, S.I.; Capela, J.P.; Carvalho, F.; Costa, V.M. Four decades of chemotherapy-induced cognitive dysfunction: Comprehensive review of clinical, animal and in vitro studies, and insights of key initiating events. Arch. Toxicol. 2022, 96, 11–78. [Google Scholar] [CrossRef]
- Aluise, C.D.; Miriyala, S.; Noel, T.; Sultana, R.; Jungsuwadee, P.; Taylor, T.J.; Cai, J.; Pierce, W.M.; Vore, M.; Moscow, J.A.; et al. 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-α release: Implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free Radic. Biol. Med. 2011, 50, 1630–1638. [Google Scholar] [CrossRef]
- Gao, X.; Bayraktutan, U. TNF-alpha evokes blood-brain barrier dysfunction through activation of Rho-kinase and neurokinin 1 receptor. Immunobiology. 2023, 228, 152706. [Google Scholar] [CrossRef]
- El-Agamy, S.E.; Abdel-Aziz, A.K.; Esmat, A.; Azab, S.S. Chemotherapy and cognition: Comprehensive review on doxorubicin-induced chemobrain. Cancer Chemother. Pharmacol. 2019, 84, 1–14. [Google Scholar] [CrossRef]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy-are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Topcul, M.; Cetin, I. Endpoint of cancer treatment: Targeted therapies. Asian Pac. J. Cancer Prev. 2014, 15, 4395–4403. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Medicines Advisory Group (NCMAG) Programme NCMAG111 Sunitinib|Advice Document v1.0. Created in January 2024. Available online: https://www.healthcareimprovementscotland.scot/wp-content/uploads/2024/02/NCMAG111-Sunitinib-Advice-Document-v1.00e06.pdf (accessed on 7 May 2025).
- Jin, J.; Xie, Y.; Zhang, J.-S.; Wang, J.-Q.; Dai, S.-J.; He, W.-F.; Li, S.Y.; Ashby, C.R., Jr.; Chen, Z.S.; He, Q. Sunitinib resistance in renal cell carcinoma: From molecular mechanisms to predictive biomarkers. Drug Resist. Updat. 2023, 67, 100929. [Google Scholar] [CrossRef]
- Mena, A.C.; Pulido, E.G.; Guillén-Ponce, C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: Sunitinib. Anticancer. Drugs. 2010, 21, S3–S11. [Google Scholar] [CrossRef]
- Zou, X.; Tang, X.-Y.; Qu, Z.-Y.; Sun, Z.-W.; Ji, C.-F.; Li, Y.-J.; Guo, S.D. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef]
- Oberoi, R.K.; Mittapalli, R.K.; Elmquist, W.F. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J. Pharmacol. Exp. Ther. 2013, 347, 755–764. [Google Scholar] [CrossRef]
- Liew, M.H.; Ng, S.; Chew, C.C.; Koo, T.W.; Chee, Y.L.; Chee, E.L.-C.; Modamio, P.; Fernandez, C.; Marino, E.L.; Segarra, I. Sunitinib-paracetamol sex-divergent pharmacokinetics and tissue distribution drug-drug interaction in mice. Investig. New Drugs 2017, 35, 145–157. [Google Scholar] [CrossRef]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef]
- van der Veldt, A.A.M.; van den Eertwegh, A.J.; Hoekman, K.; Barkhof, F.; Boven, E. Reversible cognitive disorders after sunitinib for advanced renal cell cancer in patients with preexisting arteriosclerotic leukoencephalopathy. Ann. Oncol. 2007, 18, 1747–1750. [Google Scholar] [CrossRef]
- Mulder, S.F.; Bertens, D.; Desar, I.M.; Vissers, K.C.; Mulders, P.F.; Punt, C.J.; van Spronsen, D.J.; Langenhuijsen, J.F.; Kessels, R.P.; van Herpen, C.M. Impairment of cognitive functioning during Sunitinib or Sorafenib treatment in cancer patients: A cross sectional study. BMC Cancer. 2014, 14, 219. [Google Scholar] [CrossRef]
- Hyland, K.A.; Eisel, S.L.; Hoogland, A.I.; Root, J.C.; Bowles, K.; James, B.; Nelson, A.M.; Booth-Jones, M.; Jacobsen, P.B.; Ahles, T.A.; et al. Cognition in patients treated with targeted therapy for chronic myeloid leukemia: A controlled comparison. Leuk. Lymphoma 2023, 64, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.K.; Mantawy, E.M.; Said, R.S.; Helwa, R. The tyrosine kinase inhibitor, sunitinib malate, induces cognitive impairment in vivo via dysregulating VEGFR signaling, apoptotic and autophagic machineries. Exp. Neurol. 2016, 283, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Jiao, X.; Zuzga, D.S.; Liu, Y.; Fong, D.M.; Young, D.; During, M.J. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat. Genet. 2004, 36, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-L.; Yang, Y.; Guo, L.; Xia, S.; Zhang, B.; Yan, M. Sunitinib induced hepatotoxicity in L02 cells via ROS-MAPKs signaling pathway. Front. Pharmacol. 2022, 13, 1002142. [Google Scholar] [CrossRef]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krahenbuhl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, X.; Sun, G.; Liu, Z.; Zhang, H.; Yang, Y.; Ni, Y.; Dai, J.; Zhu, S.; Chen, J.; et al. Efficacy and Safety of Individualized Schedule of Sunitinib by Drug Monitoring in Patients with Metastatic Renal Cell Carcinoma. Cancer Manag. Res. 2021, 13, 6833–6845. [Google Scholar] [CrossRef]
- van Linde, M.E.; Labots, M.; Brahm, C.G.; Hovinga, K.E.; Hamer, P.C.D.W.; Honeywell, R.J.; de Goeij-de Haas, R.; Henneman, A.A.; Knol, J.C.; Peters, G.J.; et al. Tumor Drug Concentration and Phosphoproteomic Profiles After Two Weeks of Treatment With Sunitinib in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2022, 28, 1595–1602. [Google Scholar] [CrossRef]
- Haubrich, J.; Hagena, H.; Tsanov, M.; Manahan-Vaughan, D. Editorial: Dopaminergic control of experience encoding, memory and cognition. Front. Behav. Neurosci. 2023, 17, 1230576. [Google Scholar] [CrossRef]
- Vitor, T.; Kozasa, E.H.; Bressan, R.A.; Lacerda, S.S.; Neto, G.C.C.; Batista, I.R.; Gebrim, L.H.; Cohen, L.; Amaro, E.; Felicio, A.C. Impaired brain dopamine transporter in chemobrain patients submitted to brain SPECT imaging using the technetium-99m labeled tracer TRODAT-1. Ann. Nucl. Med. 2019, 33, 269–279. [Google Scholar] [CrossRef]
- Kaplan, S.V.; Limbocker, R.A.; Gehringer, R.C.; Divis, J.L.; Osterhaus, G.L.; Newby, M.D.; Sofis, M.J.; Jarmolowicz, D.P.; Newman, B.D.; Mathews, T.A.; et al. Impaired Brain Dopamine and Serotonin Release and Uptake in Wistar Rats Following Treatment with Carboplatin. ACS Chem. Neurosci. 2016, 7, 689–699. [Google Scholar] [CrossRef]
- Garcia, K.J.; Theismann, J.T.; Schneider, T.D.; LeComte, R.S.; Jarmolowicz, D.P.; Johnson, M.A. Doxorubicin treatment has a biphasic effect over time on dopamine release and impulsive behavior in Wistar rats. Naunyn Schmiedebergs. Arch. Pharmacol. 2025, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.S.; Nogueira, T.B.; Costa, V.M.; Branco, P.S.; Ferreira, L.M.; Fernandes, E.; Bastos, M.D.L.; Meisel, A.; Carvalho, F.; Capela, J.P. Neurotoxicity of “ecstasy” and its metabolites in human dopaminergic differentiated SH-SY5Y cells. Toxicol. Lett. 2013, 216, 159–170. [Google Scholar] [CrossRef]
- Ioghen, O.C.; Ceafalan, L.C.; Popescu, B.O. SH-SY5Y Cell Line In Vitro Models for Parkinson Disease Research-Old Practice for New Trends. J. Integr. Neurosci. 2023, 22, 20. [Google Scholar] [CrossRef]
- Barbosa, D.J.; Capela, J.P.; Silva, R.; Vilas-Boas, V.; Ferreira, L.M.; Branco, P.S.; Fernandes, E.; Bastos, M.D.L.; Carvalho, F. The mixture of “ecstasy” and its metabolites is toxic to human SH-SY5Y differentiated cells at in vivo relevant concentrations. Arch. Toxicol. 2014, 88, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Pytel, M.; Mozrzymas, J.; Leskiewicz, M.; Regulska, M.; Antkiewicz-Michaluk, L.; Lason, W. The attenuating effect of memantine on staurosporine-, salsolinol- and doxorubicin-induced apoptosis in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 2008, 52, 864–877. [Google Scholar] [CrossRef]
- Almeida, D.; Pinho, R.; Correia, V.; Soares, J.; Bastos, M.D.L.; Carvalho, F.; Capela, J.P.; Costa, V.M. Mitoxantrone is More Toxic than Doxorubicin in SH-SY5Y Human Cells: A ‘Chemobrain’ In Vitro Study. Pharmaceuticals 2018, 11, 41. [Google Scholar] [CrossRef]
- Lopes, M.A.; Meisel, A.; Dirnagl, U.; Carvalho, F.D.; Bastos Mde, L. Doxorubicin induces biphasic neurotoxicity to rat cortical neurons. Neurotoxicology 2008, 29, 286–293. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Guo, L.; Gong, H.; Tang, T.-L.; Zhang, B.-K.; Zhang, L.-Y.; Yan, M. Crizotinib and Sunitinib Induce Hepatotoxicity and Mitochondrial Apoptosis in L02 Cells via ROS and Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 620934. [Google Scholar] [CrossRef]
- Bates, S.E.; Mickley, L.A.; Chen, Y.-N.; Richert, N.; Rudick, J.; Biedler, J.L.; Fojo, A.T. Expression of a drug resistance gene in human neuroblastoma cell lines: Modulation by retinoic acid-induced differentiation. Mol. Cell. Biol. 1989, 9, 4337–4344. [Google Scholar] [CrossRef]
- Dalzell, A.M.; Mistry, P.; Wright, J.; Williams, F.M.; Brown, C.D. Characterization of multidrug transporter-mediated efflux of avermectins in human and mouse neuroblastoma cell lines. Toxicol. Lett. 2015, 235, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Menze, E.T.; Tadros, M.G.; Tolba, M.F. Caffeic acid phenethyl ester counteracts doxorubicin-induced chemobrain in Sprague-Dawley rats: Emphasis on the modulation of oxidative stress and neuroinflammation. Neuropharmacology 2020, 181, 108334. [Google Scholar] [CrossRef]
- Ibrahim, S.S.; Elseoud, O.G.A.; Mohamedy, M.H.; Amer, M.M.; Mohamed, Y.Y.; Elmansy, S.A.; Kadry, M.M.; Attia, A.A.; Fanous, R.A.; Kamel, M.S.; et al. Nose-to-brain delivery of chrysin transfersomal and composite vesicles in doxorubicin-induced cognitive impairment in rats: Insights on formulation, oxidative stress and TLR4/NF-kB/NLRP3 pathways. Neuropharmacology 2021, 197, 108738. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-kappaB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2021, 9, 809952. [Google Scholar]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.V.; Hushmandi, K.; Ashrafizadeh, M.; et al. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef]
- Brum, G.; Carbone, T.; Still, E.; Correia, V.; Szulak, K.; Calianese, D.; Best, C.; Cammarata, G.; Higgins, K.; Ji, F.; et al. N-acetylcysteine potentiates doxorubicin-induced ATM and p53 activation in ovarian cancer cells. Int. J. Oncol. 2013, 42, 211–218. [Google Scholar] [CrossRef]
- Lin, X.; Li, Q.; Wang, Y.-J.; Ju, Y.-W.; Chi, Z.-Q.; Wang, M.-W.; Liu, J.G. Morphine inhibits doxorubicin-induced reactive oxygen species generation and nuclear factor kappaB transcriptional activation in neuroblastoma SH-SY5Y cells. Biochem. J. 2007, 406, 215–221. [Google Scholar] [CrossRef]
- Kitamura, Y.; Ushio, S.; Sumiyoshi, Y.; Wada, Y.; Miyazaki, I.; Asanuma, M.; Sendo, T. N-Acetylcysteine Attenuates the Anxiety-Like Behavior and Spatial Cognition Impairment Induced by Doxorubicin and Cyclophosphamide Combination Treatment in Rats. Pharmacology 2020, 106, 286–293. [Google Scholar] [CrossRef]
- Mohammed, W.I.; Radwan, R.A.; Elsayed, H.M. Prophylactic and Ameliorative Effect of N-Acetylcysteine on Doxorubicin-Induced Neurotoxicity in Wister Rats. Egypt. J. Basic Clin. Pharmacol. 2019, 9, 101396. [Google Scholar] [CrossRef]
- Hwang, H.J.; Hong, S.H.; Moon, H.S.; Yoon, Y.E.; Park, S.Y. Ginsenoside Rh2 sensitizes the anti-cancer effects of sunitinib by inducing cell cycle arrest in renal cell carcinoma. Sci. Rep. 2022, 12, 19752. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, T.; Miyagi, A.; Shimizu, S.; Shigematsu, N.; Kobayashi, D.; Shimazoe, T. Dimethyl fumarate ameliorates chemotherapy agent-induced neurotoxicity in vitro. J. Pharmacol. Sci. 2018, 137, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sobue, A.; Watanabe, S.; Komine, O.; Saido, T.C.; Saito, T.; Yamanaka, K. Dimethyl fumarate improves cognitive impairment and neuroinflammation in mice with Alzheimer’s disease. J. Neuroinflamm. 2024, 21, 55. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, R.; McElhanon, K.; Allen, C.D.; Megyesi, J.K.; Benes, H.; Singh, S.P. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic. Biol. Med. 2015, 86, 90–101. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Q.; Sun, Y.; Wang, X.; Lv, L.; Zhang, L.; Liu, J.S.; Zhao, S.; Wang, X.L. Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovasc. Ther. 2017, 35, e12277. [Google Scholar] [CrossRef]
- Li, B.; Kim, D.S.; Yadav, R.K.; Kim, H.R.; Chae, H.J. Sulforaphane prevents doxorubicin-induced oxidative stress and cell death in rat H9c2 cells. Int. J. Mol. Med. 2015, 36, 53–64. [Google Scholar] [CrossRef]
- Piroli, G.G.; Manuel, A.M.; Patel, T.; Walla, M.D.; Shi, L.; Lanci, S.A.; Wang, J.; Galloway, A.; Ortinski, P.I.; Smith, D.S.; et al. Identification of Novel Protein Targets of Dimethyl Fumarate Modification in Neurons and Astrocytes Reveals Actions Independent of Nrf2 Stabilization. Mol. Cell. Proteom. 2019, 18, 504–519. [Google Scholar] [CrossRef]
- Ali, M.; Bonay, M.; Vanhee, V.; Vinit, S.; Deramaudt, T.B. Comparative effectiveness of 4 natural and chemical activators of Nrf2 on inflammation, oxidative stress, macrophage polarization, and bactericidal activity in an in vitro macrophage infection model. PLoS ONE 2020, 15, e0234484. [Google Scholar] [CrossRef]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Wear, D.; Bhagirath, E.; Balachandar, A.; Vegh, C.; Pandey, S. Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma. Int. J. Mol. Sci. 2023, 24, 12052. [Google Scholar] [CrossRef]
- Chicote, J.; Yuste, V.J.; Boix, J.; Ribas, J. Cell Death Triggered by the Autophagy Inhibitory Drug 3-Methyladenine in Growing Conditions Proceeds With DNA Damage. Front. Pharmacol. 2020, 11, 580343. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhou, Z.; Li, L.; Gu, J.; Wang, C.; Xu, F.; Dong, Q.; Zhou, X. Intrathecal Injection of 3-Methyladenine Reduces Neuronal Damage and Promotes Functional Recovery via Autophagy Attenuation after Spinal Cord Ischemia/Reperfusion Injury in Rats. Biol. Pharm. Bull. 2016, 39, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Xu, Y.Z.; Lu, W.J.; Li, Y.H.; Tan, S.S.; Lin, H.J.; Wu, T.M.; Li, Y.; Wang, S.Y.; Zhao, Y.L. Chloroquine potentiates the anticancer effect of sunitinib on renal cell carcinoma by inhibiting autophagy and inducing apoptosis. Oncol. Lett. 2018, 15, 2839–2846. [Google Scholar]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The role of interaction between autophagy and apoptosis in tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Jasutkar, H.G.; Wasserlein, E.M.; Ishola, A.; Litt, N.; Staniszewski, A.; Arancio, O.; Yamamoto, A. Adult-onset deactivation of autophagy leads to loss of synapse homeostasis and cognitive impairment, with implications for Alzheimer disease. Autophagy 2024, 20, 2540–2555. [Google Scholar] [CrossRef]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar] [CrossRef]
- Tanemura, M.; Ohmura, Y.; Deguchi, T.; Machida, T.; Tsukamoto, R.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Ito, T.; et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am. J. Transplant. 2012, 12, 102–114. [Google Scholar] [CrossRef]
- Xie, D.; Zhao, T.; Zhang, X.; Kui, L.; Wang, Q.; Wu, Y.; Zheng, T.; Ma, P.; Zhang, Y.; Molteni, H.; et al. Autophagy Contributes to the Rapamycin-Induced Improvement of Otitis Media. Front. Cell Neurosci. 2021, 15, 753369. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef]
- Cvetko, F.; Caldwell, S.T.; Higgins, M.; Suzuki, T.; Yamamoto, M.; Prag, H.A.; Hartley, R.C.; Dinkova-Kostova, A.T.; Murphy, M.P. Nrf2 is activated by disruption of mitochondrial thiol homeostasis but not by enhanced mitochondrial superoxide production. J. Biol. Chem. 2021, 296, 100169. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. NAC, NAC, Knockin’ on Heaven’s door: Interpreting the mechanism of action of N-acetylcysteine in tumor and immune cells. Redox Biol. 2022, 57, 102497. [Google Scholar] [CrossRef] [PubMed]
- Jannatifar, R.; Parivar, K.; Roodbari, N.H.; Nasr-Esfahani, M.H. The Effect of N-Acetyl-Cysteine on NRF2 Antioxidant Gene Expression in Asthenoteratozoospermia Men: A Clinical Trial Study. Int. J. Fertil. Steril. 2020, 14, 171–175. [Google Scholar]
- Aala, J.; Harchegani, A.B.; Monsef, H.A.; Mohsenifar, Z.; Ebrahimi, P.; Parvizi, M.R. N-Acetyl cysteine mitigates histopathological changes and inflammatory genes expressions in the liver of cadmium exposed rats. Environ. Anal. Health Toxicol. 2021, 36, e2021024-0. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myślińska, D.; Grembecka, B.; Ruciński, J.; Plucinska, K.; Jerzemowska, G.; Wrona, D. Dimethyl fumarate attenuates intracerebroventricular streptozotocin-induced spatial memory impairment and hippocampal neurodegeneration in rats. Behav. Brain Res. 2016, 308, 24–37. [Google Scholar] [CrossRef]
- Yadav, S.K.; Soin, D.; Ito, K.; Dhib-Jalbut, S. Insight into the mechanism of action of dimethyl fumarate in multiple sclerosis. J. Mol. Med. 2019, 97, 463–472. [Google Scholar] [CrossRef]
- Vanduchova, A.; Anzenbacher, P.; Anzenbacherova, E. Isothiocyanate from Broccoli, Sulforaphane, and Its Properties. J. Med. Food. 2019, 22, 121–126. [Google Scholar] [CrossRef]
- Brasil, F.B.; de Almeida, F.J.S.; Luckachaki, M.D.; Dall’oglio, E.L.; de Oliveira, M.R. The isothiocyanate sulforaphane prevents mitochondrial impairment and neuroinflammation in the human dopaminergic SH-SY5Y and in the mouse microglial BV2 cells: Role for heme oxygenase-1. Metab. Brain Dis. 2023, 38, 419–435. [Google Scholar] [CrossRef]
- Janczewski, L. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules 2022, 27, 1750. [Google Scholar] [CrossRef]
- Ernst, I.M.; Palani, K.; Esatbeyoglu, T.; Schwarz, K.; Rimbach, G. Synthesis and Nrf2-inducing activity of the isothiocyanates iberverin, iberin and cheirolin. Pharmacol. Res. 2013, 70, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.M.; Stinckens, M.; Ates, G.; Vanhaecke, T. Neutral Red Uptake Assay to Assess Cytotoxicity In Vitro. Methods Mol. Biol. 2023, 2644, 237–245. [Google Scholar] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb-prot095505. [Google Scholar] [CrossRef]
- Dionísio, F.; Araújo, A.M.; Duarte-Araújo, M.; Bastos, M.d.L.; Guedes de Pinho, P.; Carvalho, F.; Costa, V.M. Cardiotoxicity of cyclophosphamide’s metabolites: An in vitro metabolomics approach in AC16 human cardiomyocytes. Arch. Toxicol. 2022, 96, 653–671. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Prasertsuksri, P.; Kraokaew, P.; Pranweerapaiboon, K.; Sobhon, P.; Chaithirayanon, K. Neuroprotection of Andrographolide against Neurotoxin MPP(+)-Induced Apoptosis in SH-SY5Y Cells via Activating Mitophagy, Autophagy, and Antioxidant Activities. Int. J. Mol. Sci. 2023, 24, 8528. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Vitorino-Oliveira, C.; Ferreira, M.; Carvalho, F.; Remião, F.; Sousa, E.; Bastos, M.d.L.; Costa, V.M. Comparative In Vitro Study of the Cytotoxic Effects of Doxorubicin’s Main Metabolites on Cardiac AC16 Cells Versus the Parent Drug. Cardiovasc. Toxicol. 2024, 24, 266–279. [Google Scholar] [CrossRef]
- Tie, F.; Fu, Y.; Hu, N.; Wang, H. Silibinin Protects against H2O2-Induced Oxidative Damage in SH-SY5Y Cells by Improving Mitochondrial Function. Antioxidants 2022, 11, 1101. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
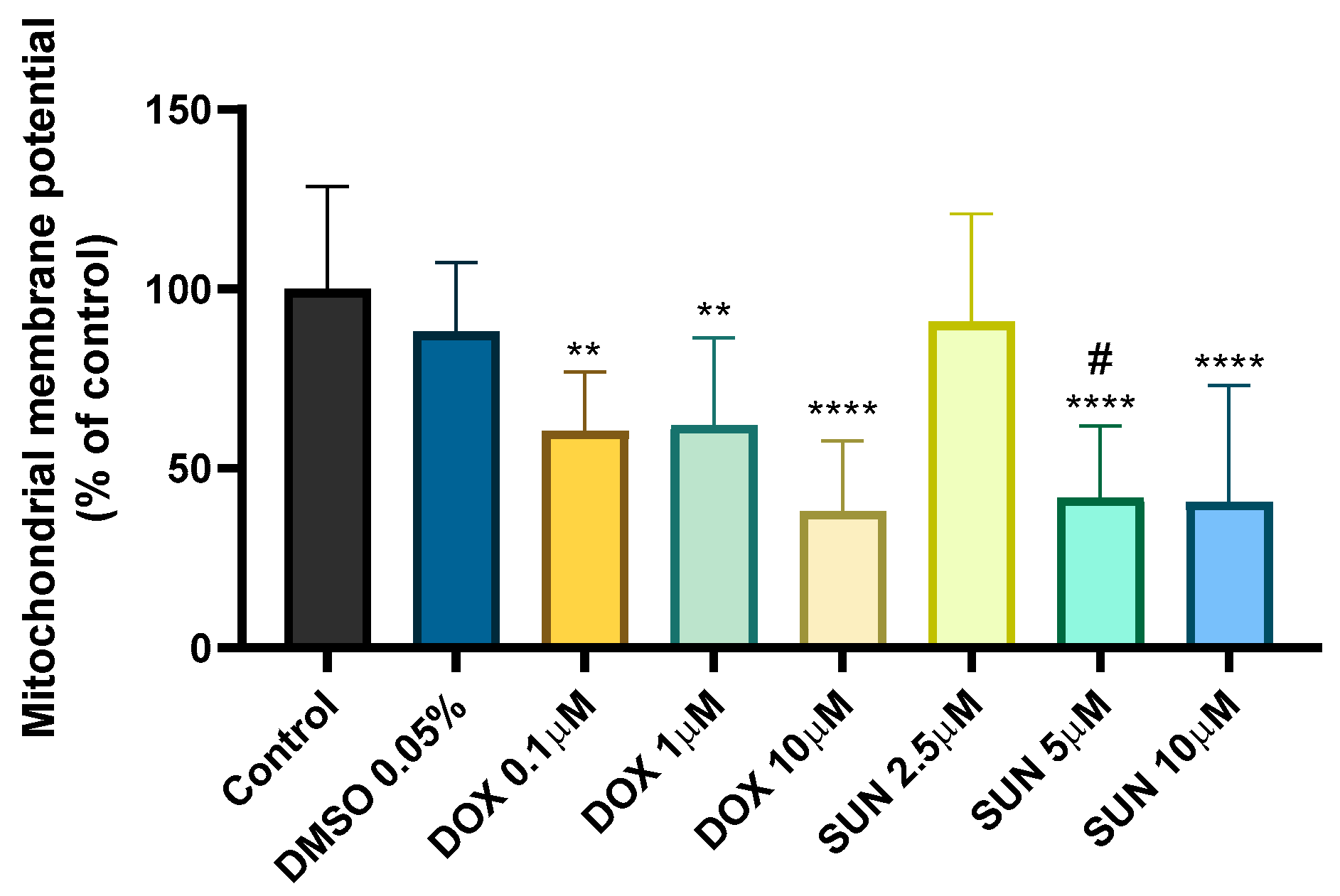
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa-Azevedo, M.; Igreja-Cardoso, M.B.; Carvalho, F.; Costa, V.M. Comparative Neurotoxic Effects of Doxorubicin and Sunitinib: An In Vitro Study on Human Dopaminergic Neuronal Cells. Molecules 2025, 30, 2342. https://doi.org/10.3390/molecules30112342
Barbosa-Azevedo M, Igreja-Cardoso MB, Carvalho F, Costa VM. Comparative Neurotoxic Effects of Doxorubicin and Sunitinib: An In Vitro Study on Human Dopaminergic Neuronal Cells. Molecules. 2025; 30(11):2342. https://doi.org/10.3390/molecules30112342
Chicago/Turabian StyleBarbosa-Azevedo, Maria, Maria B. Igreja-Cardoso, Félix Carvalho, and Vera Marisa Costa. 2025. "Comparative Neurotoxic Effects of Doxorubicin and Sunitinib: An In Vitro Study on Human Dopaminergic Neuronal Cells" Molecules 30, no. 11: 2342. https://doi.org/10.3390/molecules30112342
APA StyleBarbosa-Azevedo, M., Igreja-Cardoso, M. B., Carvalho, F., & Costa, V. M. (2025). Comparative Neurotoxic Effects of Doxorubicin and Sunitinib: An In Vitro Study on Human Dopaminergic Neuronal Cells. Molecules, 30(11), 2342. https://doi.org/10.3390/molecules30112342