
Development of Novel Immobilized Copper–Ligand Complex for Click Chemistry of Biomolecules

 ,
,
Abstract
1. Introduction
2. Results and Discussion
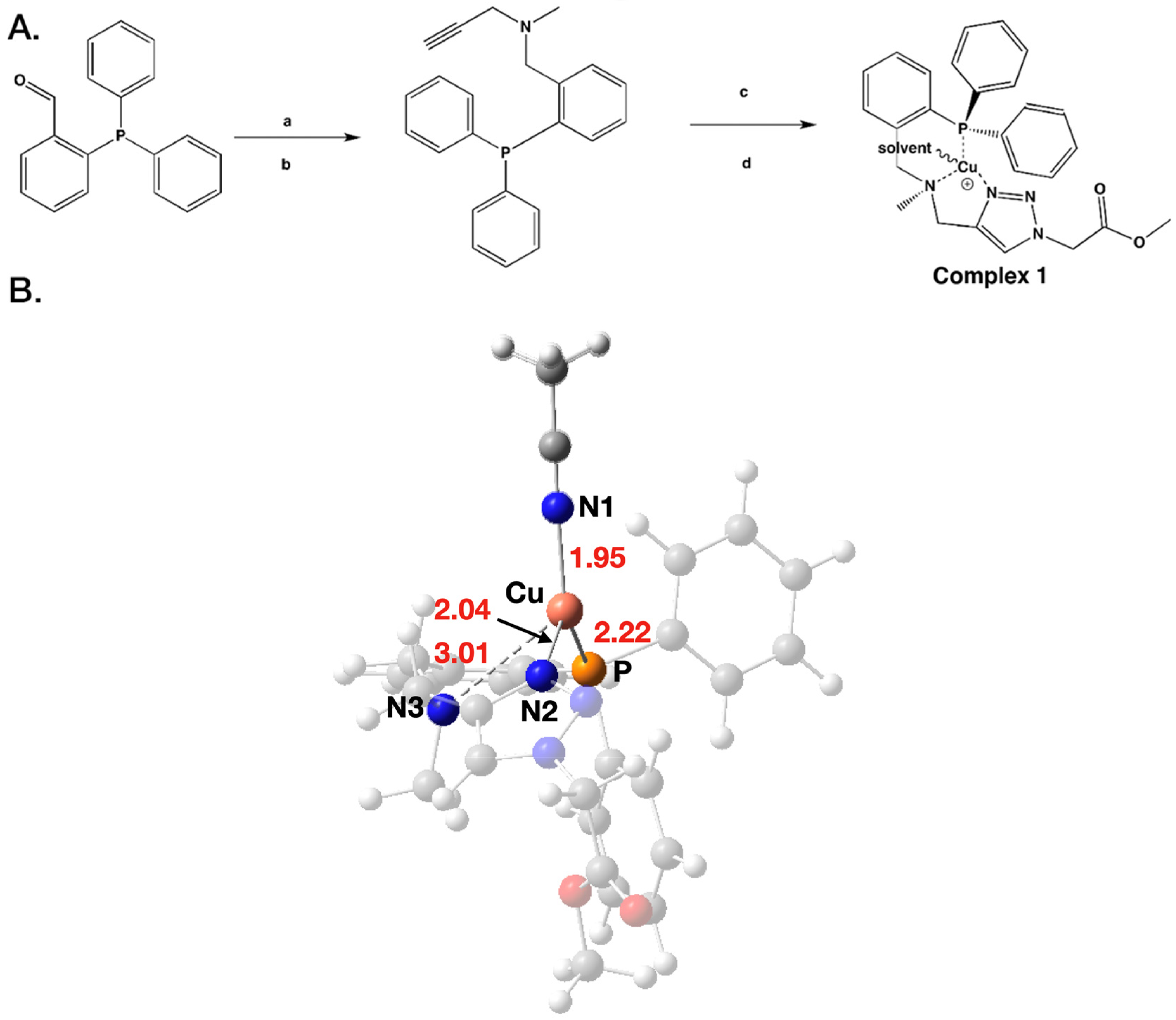
2.1. Synthesis and Characterization of PNN-Type Complex 1
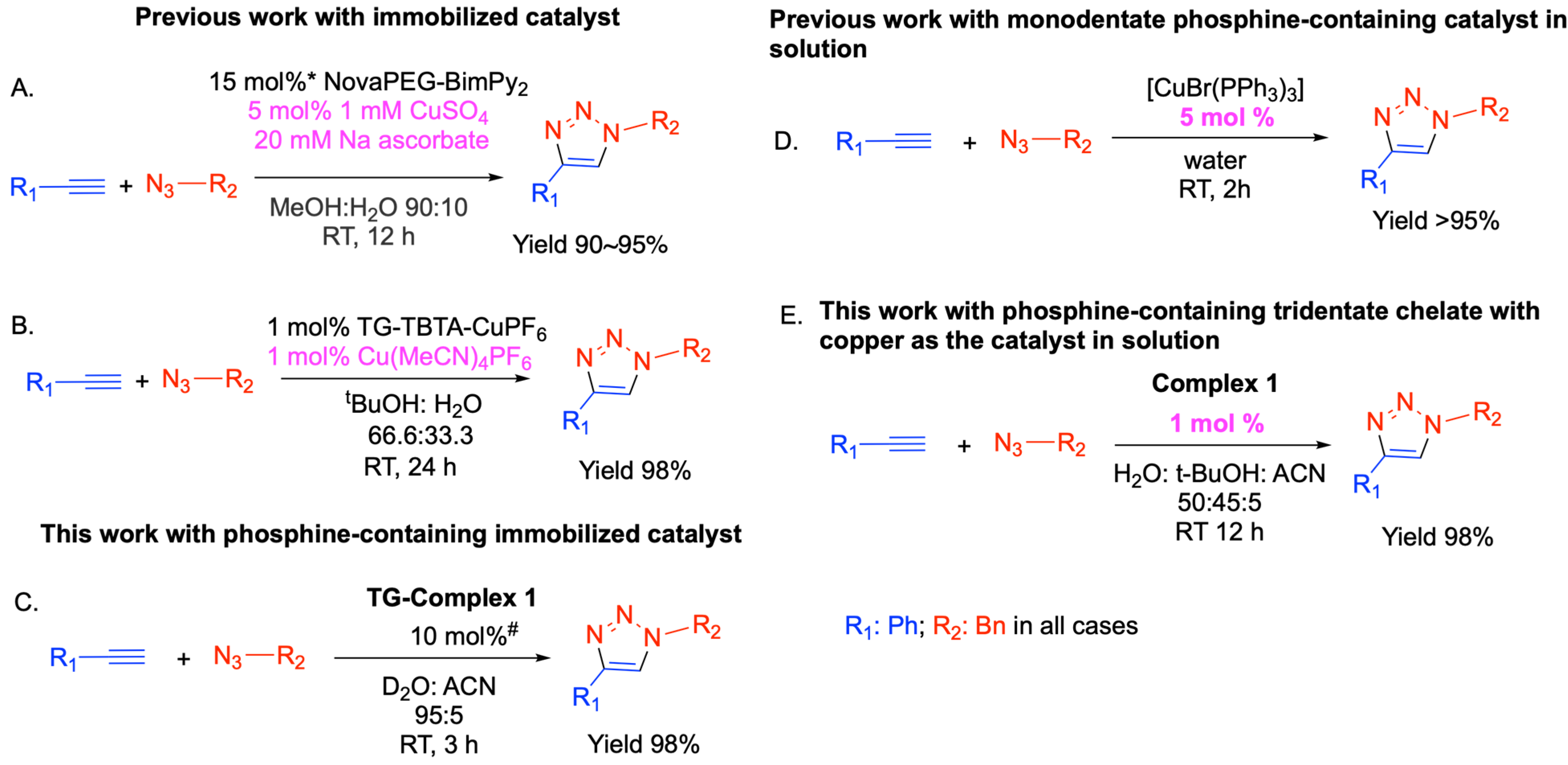
2.2. In Solution Catalysis by Complex 1
2.3. Effect of Catalyst Concentration on Reaction Yield
2.4. Effect of Deuterated Water on Reaction Yield
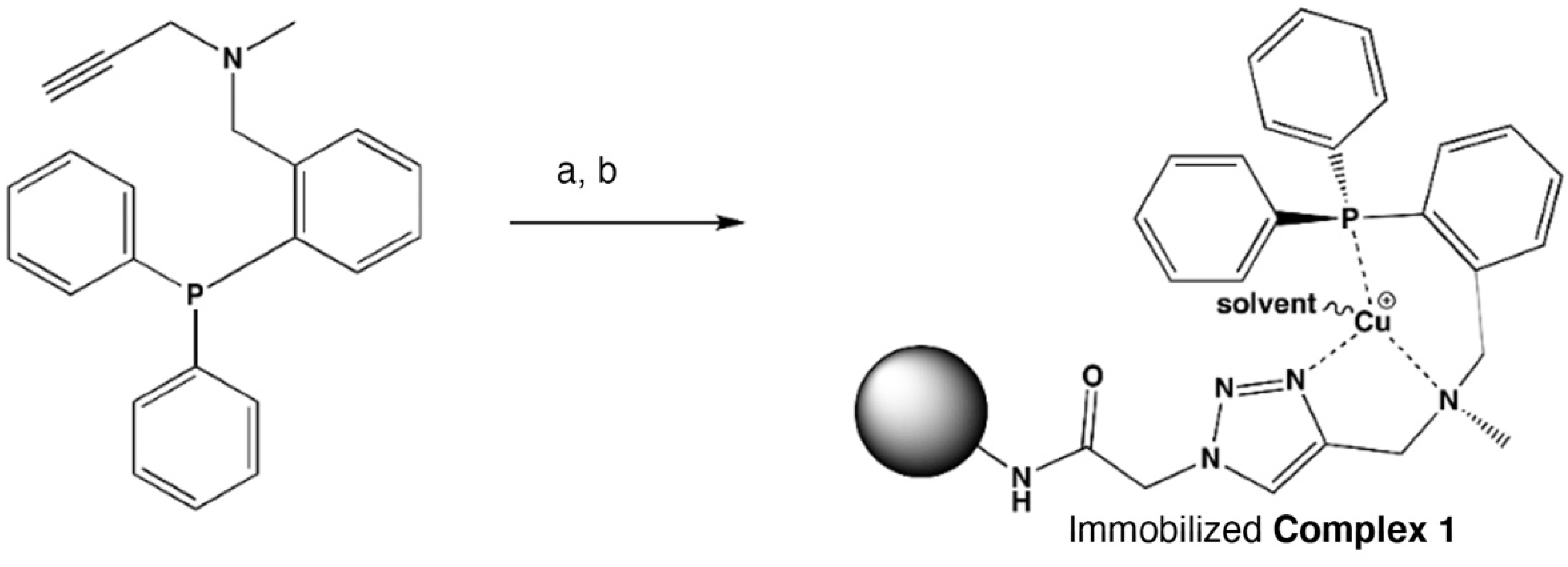
2.5. Immobilizing Complex 1 on Solid Support
2.6. Effect of Deuterated Solvent and pH Variation on CuAAC Reaction Yield for Solid Immobilized Catalyst
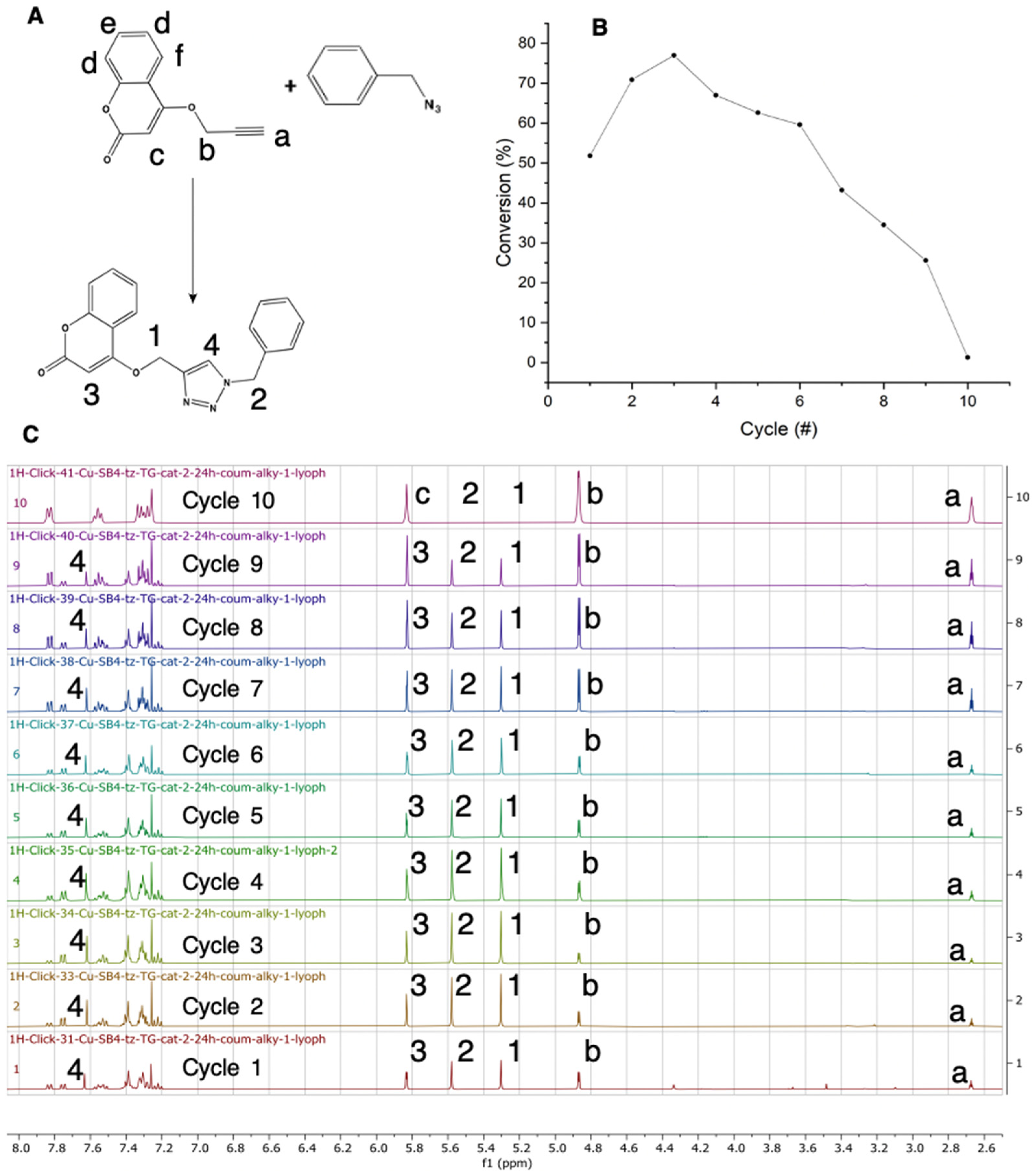
2.7. Efficiency of CuAAC Reactions with Complex 1 Catalyst Immobilized on the Solid Support
2.8. Stability of the Immobilized Complex 1 Catalyst
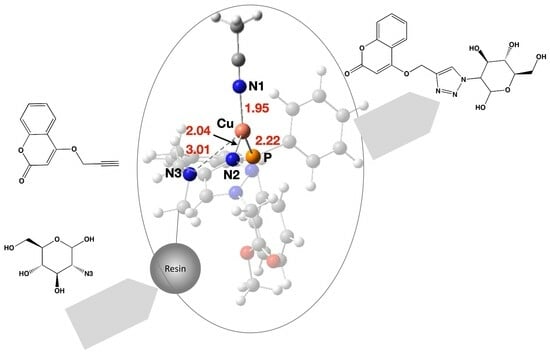
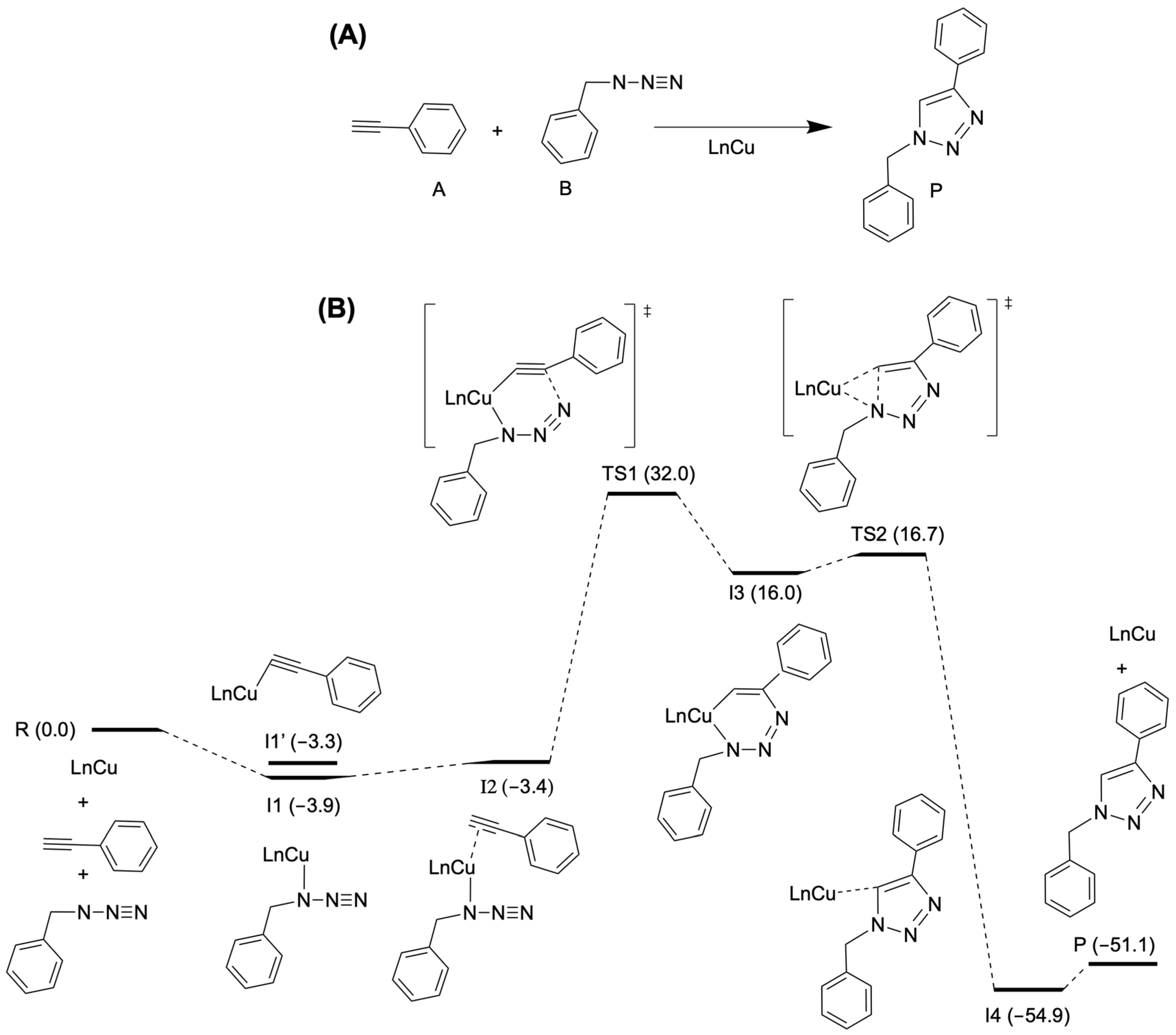
2.9. Computed Mechanism of the Catalytic Cycle
3. Experimental Section
3.1. Synthesis of Phosphine Ligand
3.2. Analysis of Ligand
3.3. Synthesis of Complex
3.4. Complexation with [Cu(CH3CN)4]+ PF6− and Immobilization of Ligand to Form Immobilized Complex 1
3.5. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castelvecchi, D.; Ledford, H. Chemists who invented revolutionary ‘click’ reactions win Nobel. Nature 2022, 610, 242–243. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow, C.K. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 2003, 189, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dubinsky-Davidchik, I.S.; Kluger, R. Strain-promoted azide-alkyne cycloaddition for protein-protein coupling in forming a bis-hemoglobin as a copper-free oxygen carrier. Org. Biomol. Chem. 2016, 14, 10011–10017. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Diez-Gonzalez, S. [CuBr(PPh3)3] for azide-alkyne cycloaddition reactions under strict Click conditions. J. Org. Chem. 2011, 76, 2367–2373. [Google Scholar] [CrossRef]
- Pickens, C.J.; Johnson, S.N.; Pressnall, M.M.; Leon, M.A.; Berkland, C.J. Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjug. Chem. 2018, 29, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, V.O.; Presolski, S.I.; Díaz Díaz, D.; Fokin, V.V.; Finn, M.G. Ligand-Accelerated Cu-Catalyzed Azide−Alkyne Cycloaddition: A Mechanistic Report. J. Am. Chem. Soc. 2007, 129, 12705–12712. [Google Scholar] [CrossRef]
- Presolski, S.I.; Hong, V.; Cho, S.H.; Finn, M.G. Tailored ligand acceleration of the Cu-catalyzed azide-alkyne cycloaddition reaction: Practical and mechanistic implications. J. Am. Chem. Soc. 2010, 132, 14570–14576. [Google Scholar] [CrossRef]
- Golas, P.L.; Tsarevsky, N.V.; Sumerlin, B.S.; Matyjaszewski, K. Catalyst Performance in “Click” Coupling Reactions of Polymers Prepared by ATRP: Ligand and Metal Effects. Macromolecules 2006, 39, 6451–6457. [Google Scholar] [CrossRef]
- Khatua, M.; Goswami, B.; Kamal; Samanta, S. Azide-Alkyne “Click” Reaction in Water Using Parts-Per-Million Amine-Functionalized Azoaromatic Cu(I) Complex as Catalyst: Effect of the Amine Side Arm. Inorg. Chem. 2021, 60, 17537–17554. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Farbiarz, K.; Mayer, R.L.; Matveenko, M.; Kählig, H.; Becker, C.F. Arginine side-chain modification that occurs during copper-catalysed azide-alkyne click reactions resembles an advanced glycation end product. Org. Biomol. Chem. 2016, 14, 6205–6211. [Google Scholar] [CrossRef] [PubMed]
- Abel, G.R., Jr.; Calabrese, Z.A.; Ayco, J.; Hein, J.E.; Ye, T. Measuring and Suppressing the Oxidative Damage to DNA During Cu(I)-Catalyzed Azide–Alkyne Cycloaddition. Bioconjug. Chem. 2016, 27, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-Y.; Jiang, N.; Zhang, J.; Wei, X.; Lin, H.-H.; Yu, X.-Q. The Oxidative Damage of Plasmid DNA by Ascorbic Acid Derivatives in vitro: The First Research on the Relationship between the Structure of Ascorbic Acid and the Oxidative Damage of Plasmid DNA. Chem. Biodivers. 2006, 3, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Presolski, S.I.; Mamidyala, S.K.; Manzenrieder, F.; Finn, M.G. Resin-supported catalysts for CuAAC click reactions in aqueous or organic solvents. ACS Comb. Sci. 2012, 14, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Eltepu, L.; Jayaraman, M.; Rajeev, K.G.; Manoharan, M. An immobilized and reusable Cu(I) catalyst for metal ion-free conjugation of ligands to fully deprotected oligonucleotides through click reaction. Chem. Commun. 2013, 49, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Fokin, V.V. Polymer-Supported Copper(I) Catalysts for the Experimentally Simplified Azide–Alkyne Cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Bonami, L.; Van Camp, W.; Van Rijckegem, D.; Du Prez, F.E. Facile Access to an Efficient Solid-Supported Click Catalyst System Based on Poly(ethyleneimine). Macromol. Rapid Commun. 2009, 30, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.P.; Ronson, T.K.; Rizzuto, F.J.; Héliot, T.; Grice, P.; Nitschke, J.R. Incorporation of a Phosphino(pyridine) Subcomponent Enables the Formation of Cages with Homobimetallic and Heterobimetallic Vertices. J. Am. Chem. Soc. 2022, 144, 8467–8473. [Google Scholar] [CrossRef]
- Babgi, B.A. Synthetic protocols and applications of copper(I) phosphine and copper(I) phosphine/diimine complexes. J. Organomet. Chem. 2021, 956, 122124. [Google Scholar] [CrossRef]
- Wang, D.; Li, N.; Zhao, M.; Shi, W.; Ma, C.; Chen, B. Solvent-free synthesis of 1,4-disubstituted 1,2,3-triazoles using a low amount of Cu(PPh3)2NO3 complex. Green Chem. 2010, 12, 2120–2123. [Google Scholar] [CrossRef]
- Gonda, Z.; Novak, Z. Highly active copper-catalysts for azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Lukasak, B.; Morihiro, K.; Deiters, A. Aryl Azides as Phosphine-Activated Switches for Small Molecule Function. Sci. Rep. 2019, 9, 1470. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Belding, L.; Cafferty, B.J.; Mousavi, M.P.S.; Finogenova, A.M.; Cruz, R.S.; Skorb, E.V.; Whitesides, G.M. Autocatalytic Cycles in a Copper-Catalyzed Azide-Alkyne Cycloaddition Reaction. J. Am. Chem. Soc. 2018, 140, 10221–10232. [Google Scholar] [CrossRef]
- Karagollu, O.; Gorur, M.; Gode, F.; Sennik, B.; Yilmaz, F. Phosphate ion sensors based on triazole connected ferrocene moieties. Sens. Actuators B Chem. 2014, 193, 788–798. [Google Scholar] [CrossRef]
- Kraljevic, T.G.; Harej, A.; Sedic, M.; Pavelic, S.K.; Stepanic, V.; Drenjancevic, D.; Talapko, J.; Raic-Malic, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Vagish, C.B.; Kumara, K.; Vivek, H.K.; Bharath, S.; Lokanath, N.K.; Ajay Kumar, K. Coumarin-triazole hybrids: Design, microwave-assisted synthesis, crystal and molecular structure, theoretical and computational studies and screening for their anticancer potentials against PC-3 and DU-145. J. Mol. Struct. 2021, 1230, 129899. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, C.H. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K.; Mishra, B.B.; Mishra, K.B.; Mishra, N.; Singh, A.S.; Chen, X. Cu-Catalyzed Click Reaction in Carbohydrate Chemistry. Chem. Rev. 2016, 116, 3086–3240. [Google Scholar] [CrossRef]
- Houshmand, A.; Heroux, D.; Liu, D.Y.; Zhou, W.; Linington, R.G.; Bally, M.; Warren, J.J.; Walsby, C.J. Ferrocene-appended anthraquinone and coumarin as redox-active cytotoxins. Dalton Trans. 2022, 51, 11437–11447. [Google Scholar] [CrossRef]
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Noll, C.A.; Betz, L.D. Determination of Copper Ion by Modified Sodium Diethyldithiocarbamate Procedure. Anal. Chem. 1952, 24, 1894–1895. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; HPC SYSTEMS Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction. J. Chem. Phys. 1992, 97, 9173–9177. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, P.; Preuss, H.; Stoll, H.; Von Szentpály, L. A proper account of core-polarization with pseudopotentials: Single valence-electron alkali compounds. Chem. Phys. Lett. 1982, 89, 418–422. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory; Schaefer, H.F., Ed.; Springer: Boston, MA, USA, 1977; pp. 1–27. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Mamidyala, S.K.; Dutta, S.; Chrunyk, B.A.; Préville, C.; Wang, H.; Withka, J.M.; McColl, A.; Subashi, T.A.; Hawrylik, S.J.; Griffor, M.C.; et al. Glycomimetic Ligands for the Human Asialoglycoprotein Receptor. J. Am. Chem. Soc. 2012, 134, 1978–1981. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Yu, F.; Zhu, Z.; Shobaki, D.; Chen, H.; Wang, M.; Wang, J.; Qin, G.; Erasquin, U.J.; et al. Copper-Catalyzed Click Reaction on/in Live Cells. Chem. Sci. 2017, 8, 2107–2114. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) |
| A | ACN | 100 | 114 | 71.5 |
| B | ACN:H2O | 80:20 | 120 | 83.8 |
| C | ACN:tBuOH | 50:50 | 120 | 91.5 |
| D | ACN:tBuOH | 5:95 | 48 | 95.8 |
| E | H2O:tBuOH:ACN | 20:75:5 | 12 | 93.5 |
| F | H2O:tBuOH:ACN | 50:45:5 | 12 | 98.1 |
| G | H2O:tBuOH:ACN | 90:5:5 | 6 | 90.9 |
| H * | H2O:tBuOH:ACN | 50:45:5 | 24 | 31.8 |
 | ||||
| Condition | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) |
| A | H2O:tBuOH:ACN | 45:50:5 | 12 | 73.5 |
 | ||||
| Condition | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) |
| B | H2O:tBuOH:ACN | 25:25:50 | 30 | 61.0 |
 | ||||
|---|---|---|---|---|
| Condition | Solvent (mol% Catalyst) | Composition % (v:v:v) | Time (h) | Conversion (%) |
| A | H2O:tBuOH:ACN (2) | 90:5:5 | 6 | 90.9 |
| B | H2O:tBuOH:ACN (1) | 90:5:5 | 6 | 43.0 |
| C | H2O:tBuOH:ACN (0.5) | 90:5:5 | 6 | 13.6 |
| D | D2O:tBuOH:ACN (1) | 90:5:5 | 6 | 89.2 |
| E | D2O:tBuOH:ACN (0.5) | 90:5:5 | 6 | 87.1 |
 | ||||
|---|---|---|---|---|
| Condition | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) |
| On silica | H2O:tBuOH:ACN | 50:45:5 | 120 | 62.0 |
| On Tentagel | H2O:tBuOH:ACN | 50:45:5 | 120 | 94.7 |
 | |||||
|---|---|---|---|---|---|
| Condition | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) | pH |
| A | H2O:tBuOH | 95:5 | 12 | 59.3 a/52.1 b | 6.2 |
| B | H2O:tBuOH | 95:5 | 3 | 21.0 b | 6.2 |
| C | D2O:tBuOH | 95:5 | 12 | 98.0 | 6.9 |
| D | D2O:tBuOH | 95:5 | 3 | 98.0 a/98.0 b | 6.9 |
| E | H2O:tBuOH | 95:5 | 12 | 98.0 a/90.0 b | 7.0 |
| F | H2O:tBuOH | 95:5 | 3 | 63.6 a/58.7 b,c | 7.0 |
| Condition | Product | Solvent | Composition % (v:v:v) | Time (h) | Conversion (%) |
|---|---|---|---|---|---|
| A | Triazole 1 | D2O:t-BuOH:ACN | 50:45:5 | 12 | 98.1 |
| B | Triazole 1 | D2O:ACN | 95:5 | 3 | 98.0 |
| C | Triazole 2 | D2O:tBuOH:ACN | 50:45:5 | 12 | 90.0 |
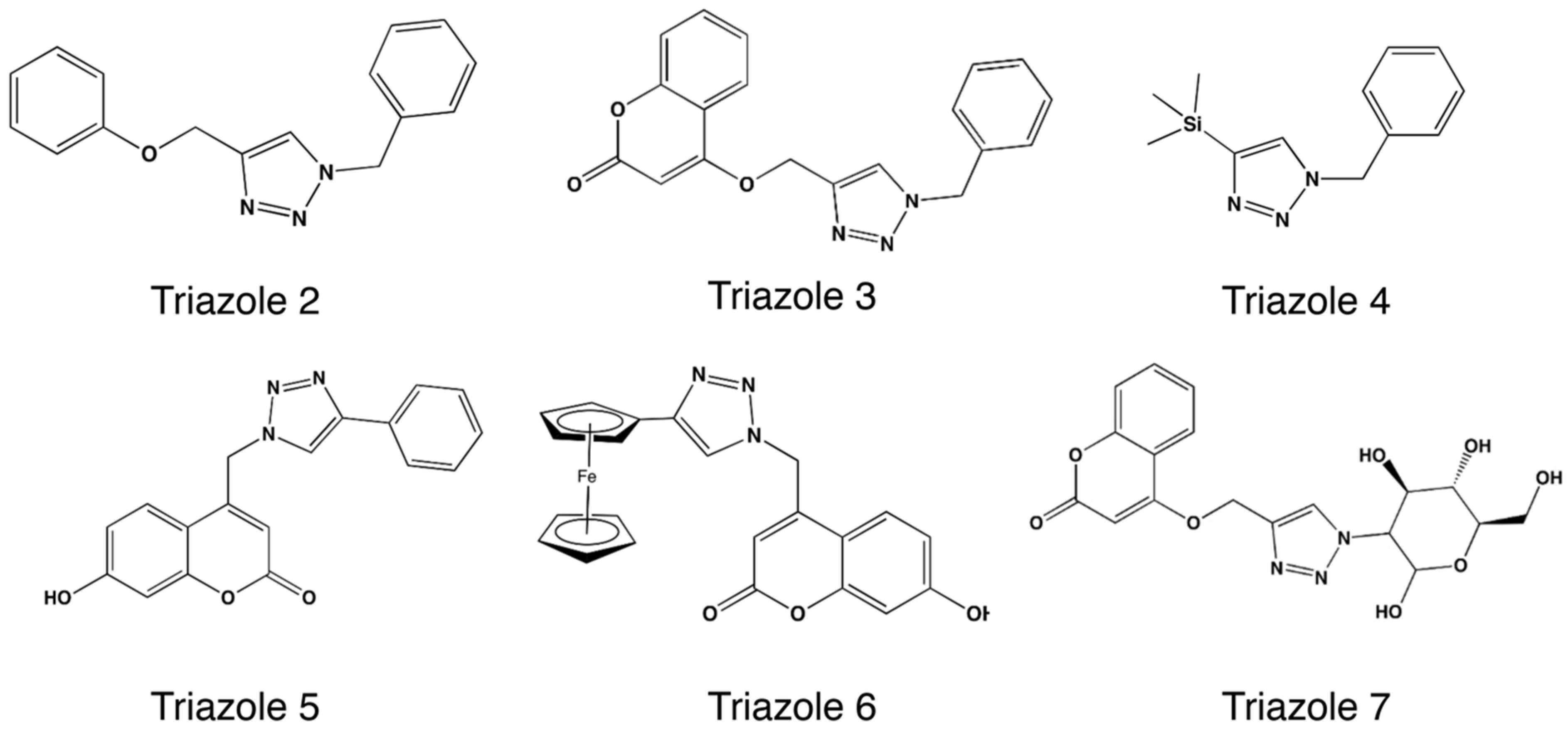
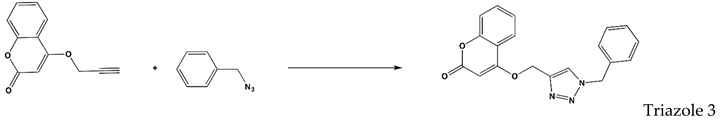
| D | Triazole 3 | H2O:tBuOH:ACN | 25:25:50 | 24 | 67.4 |
| E | Triazole 3 | D2O:tBuOH:ACN | 25:25:50 | 3 | 77.5 |
| F | Triazole 4 | D2O:ACN | 95:5 | 12 | 54.5 |
| G a | Triazole 5 | D2O:tBuOH:ACN | 33.33:33:33.33 | 24 | 98.0 |
| H | Triazole 6 | D2O:tBuOH:ACN | 33.33:33:33.33 | 24 | 43.0 |
| I b,c | Triazole 7 | D2O:tBuOH:ACN | 25:25:50 | 24 | 24.5 |
| J b,c | Triazole 7 | D2O:tBuOH:ACN | 25:25:50 | 48 * | 78.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandler, R.; Benaragama, Y.; Bera, M.; Wang, C.; Samiha, R.A.; Sameera, W.M.C.; Das, S.; Nag, A. Development of Novel Immobilized Copper–Ligand Complex for Click Chemistry of Biomolecules. Molecules 2024, 29, 2148. https://doi.org/10.3390/molecules29092148
Kandler R, Benaragama Y, Bera M, Wang C, Samiha RA, Sameera WMC, Das S, Nag A. Development of Novel Immobilized Copper–Ligand Complex for Click Chemistry of Biomolecules. Molecules. 2024; 29(9):2148. https://doi.org/10.3390/molecules29092148
Chicago/Turabian StyleKandler, Rene, Yomal Benaragama, Manoranjan Bera, Caroline Wang, Rasheda Aktar Samiha, W. M. C. Sameera, Samir Das, and Arundhati Nag. 2024. "Development of Novel Immobilized Copper–Ligand Complex for Click Chemistry of Biomolecules" Molecules 29, no. 9: 2148. https://doi.org/10.3390/molecules29092148
APA StyleKandler, R., Benaragama, Y., Bera, M., Wang, C., Samiha, R. A., Sameera, W. M. C., Das, S., & Nag, A. (2024). Development of Novel Immobilized Copper–Ligand Complex for Click Chemistry of Biomolecules. Molecules, 29(9), 2148. https://doi.org/10.3390/molecules29092148