
The Prediction of LptA and LptC Protein–Protein Interactions and Virtual Screening for Potential Inhibitors

 , , ,
, , ,
Abstract
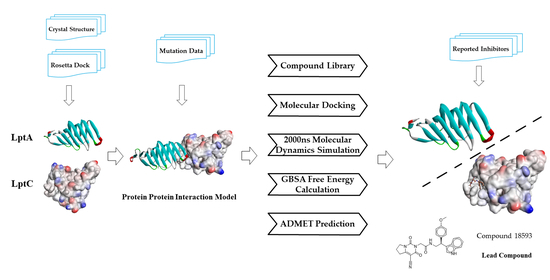
{kind=link}
{kind=link}
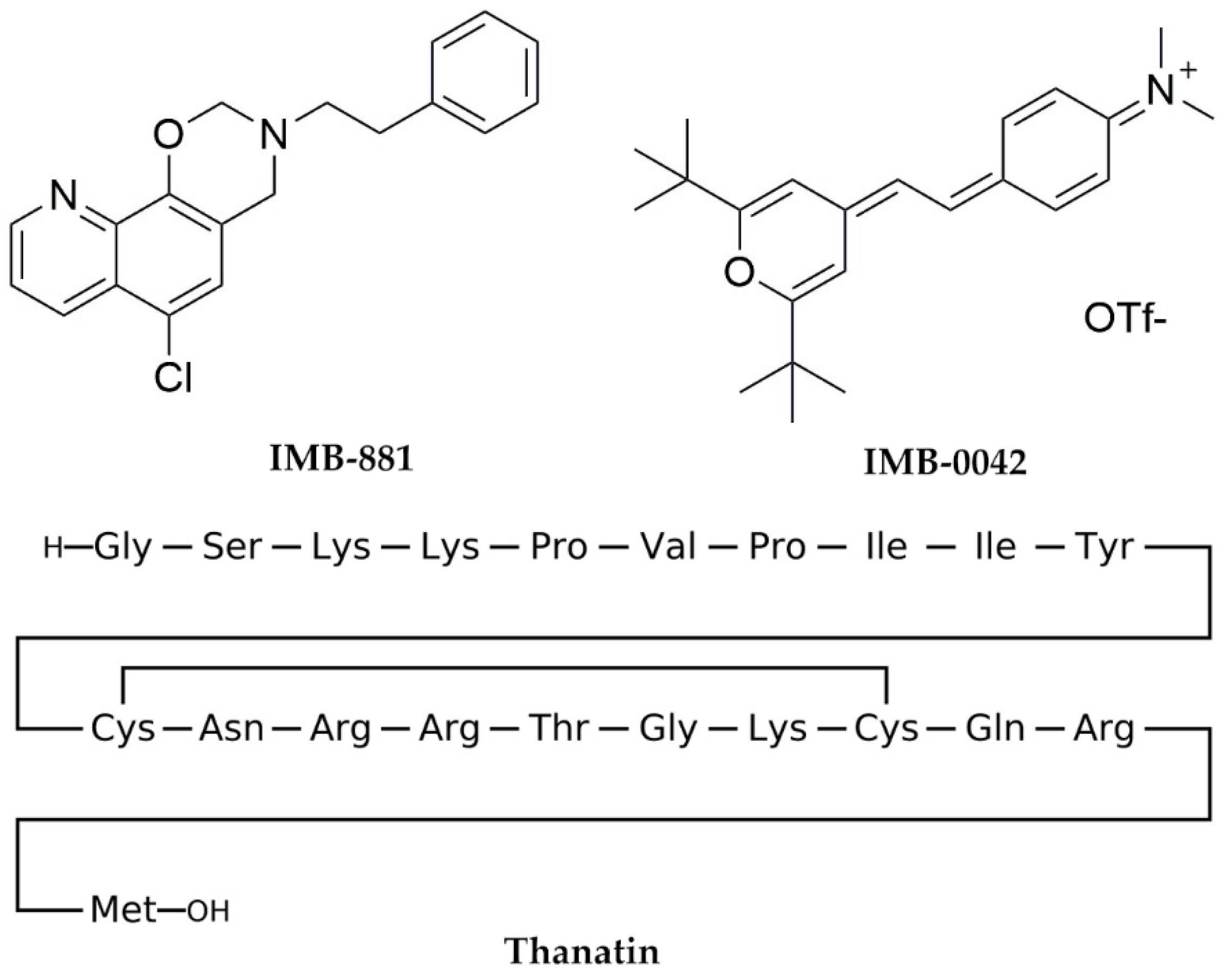{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
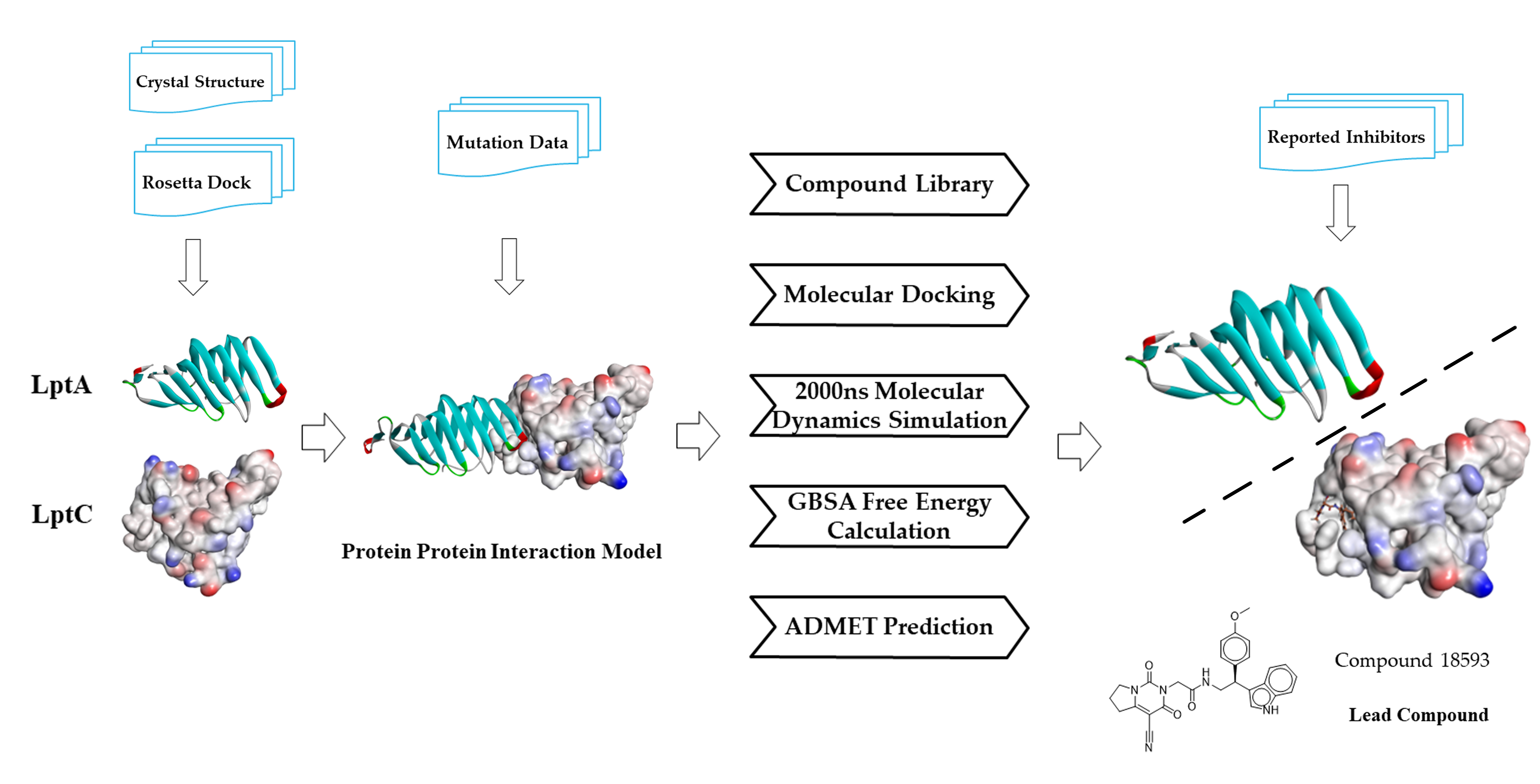
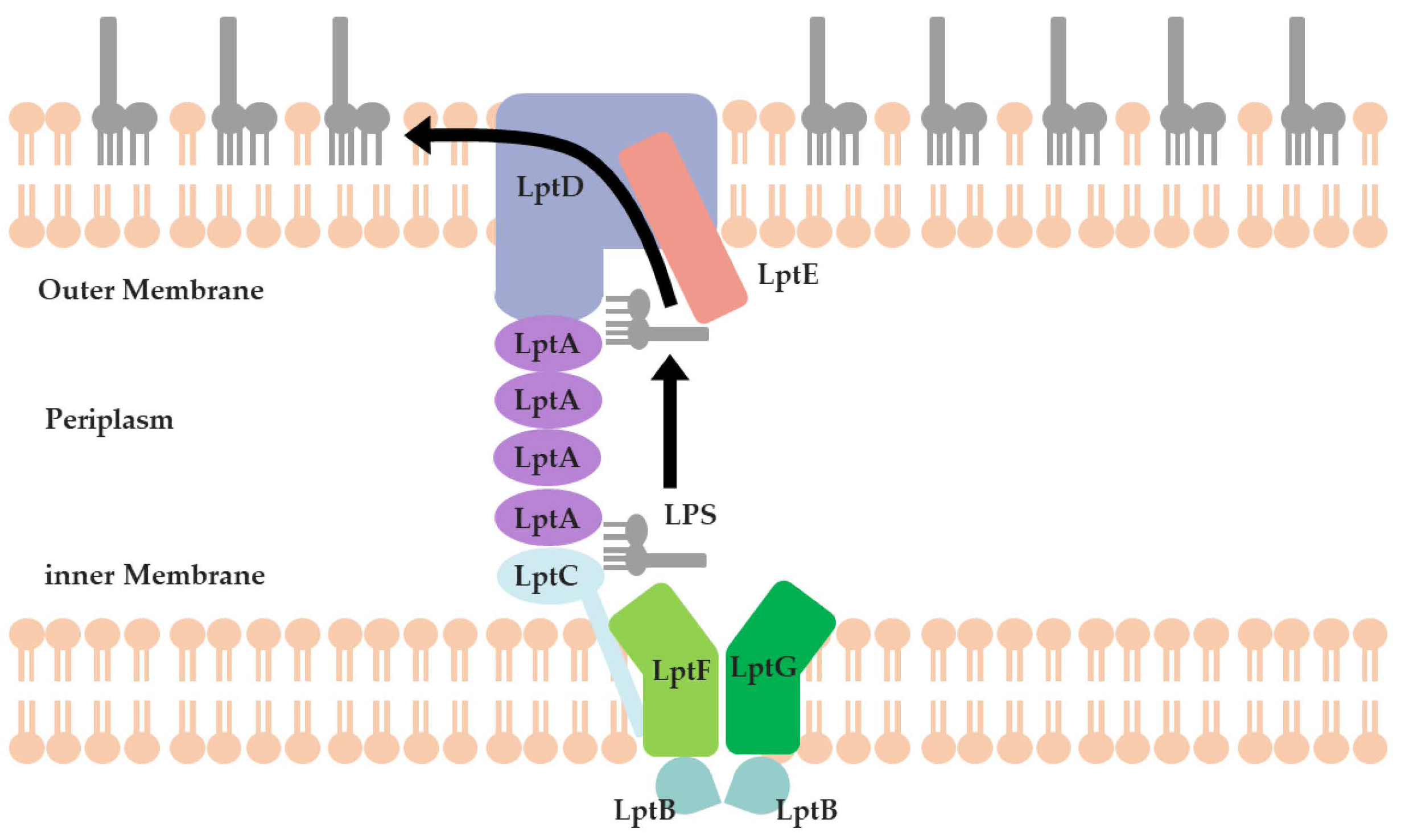
1. Introduction
2. Results and Discussion
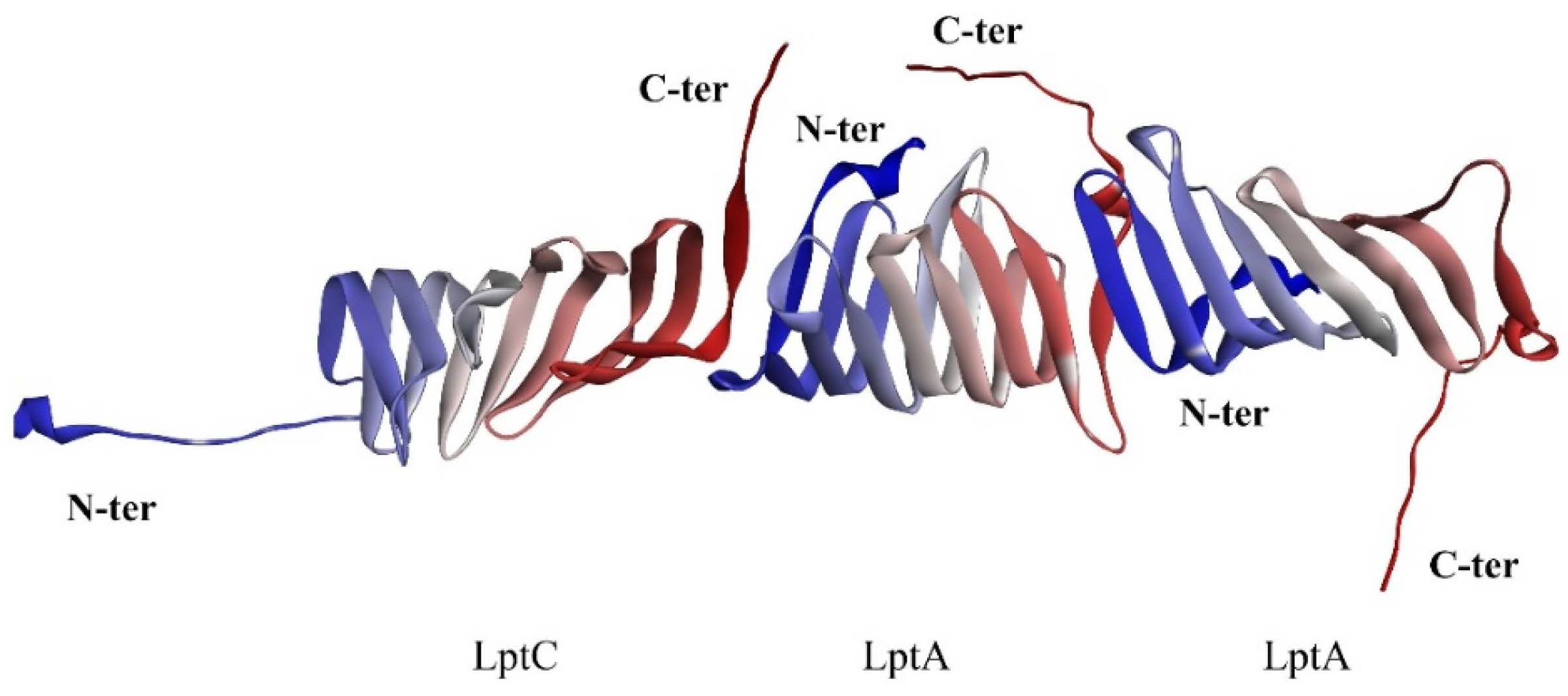
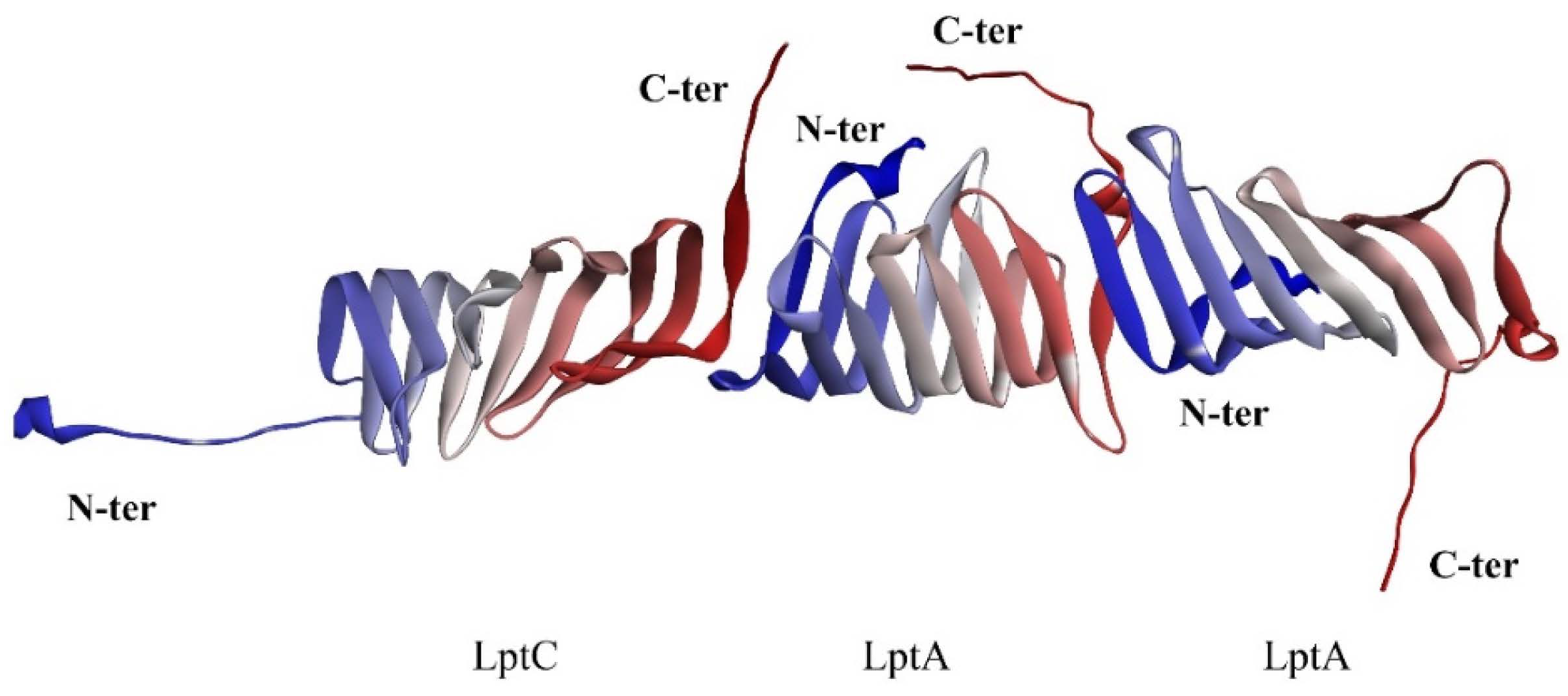
2.1. The PPI Prediction of LptA and LptC
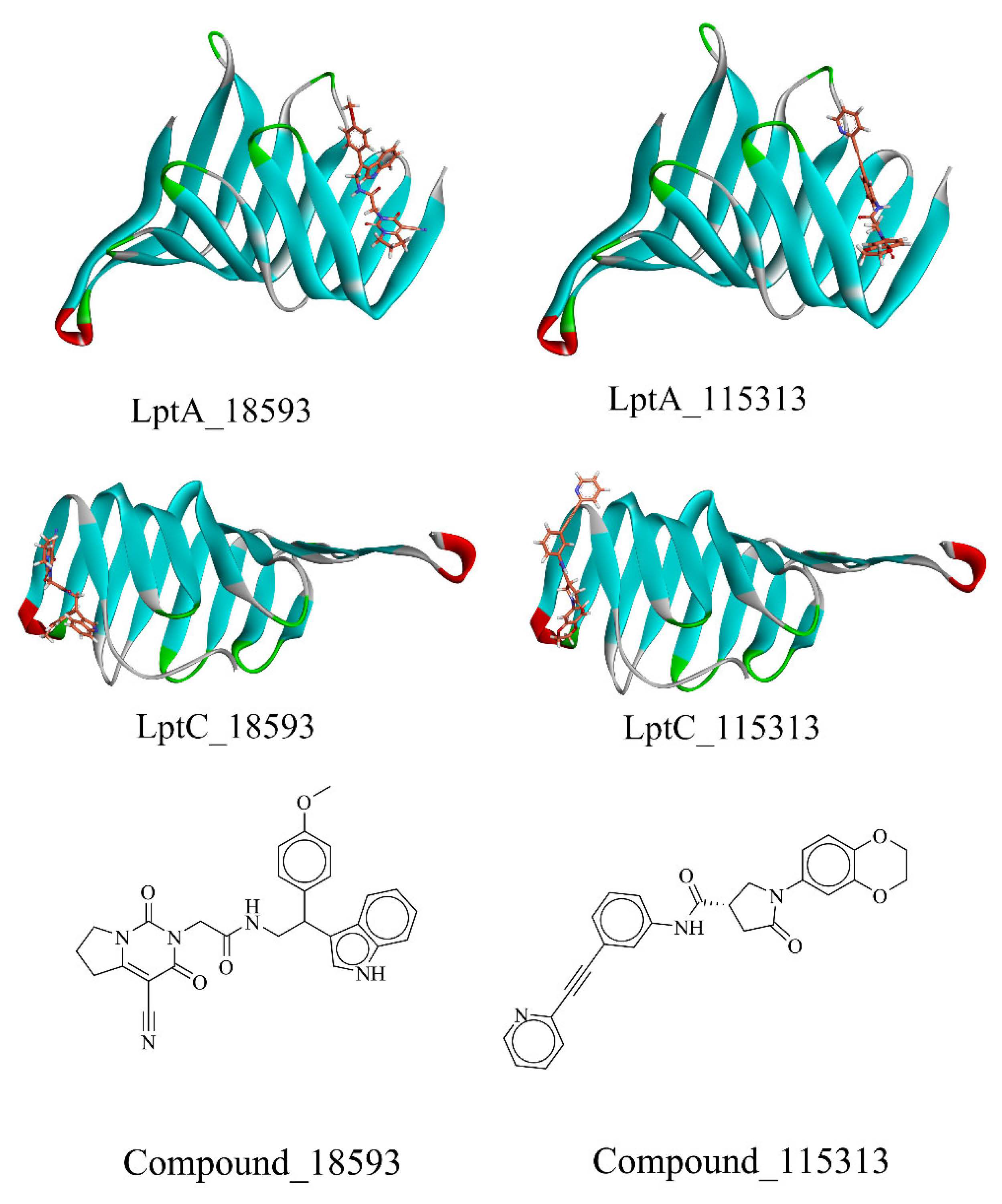
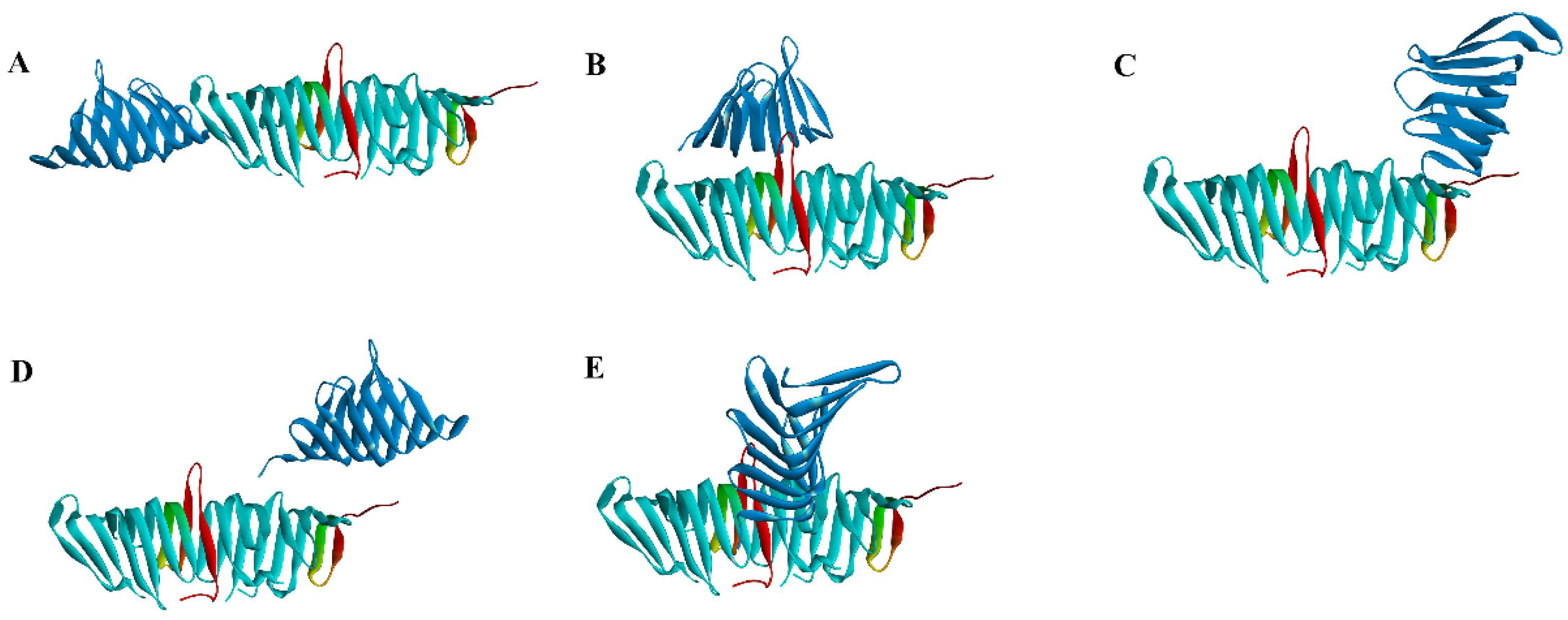
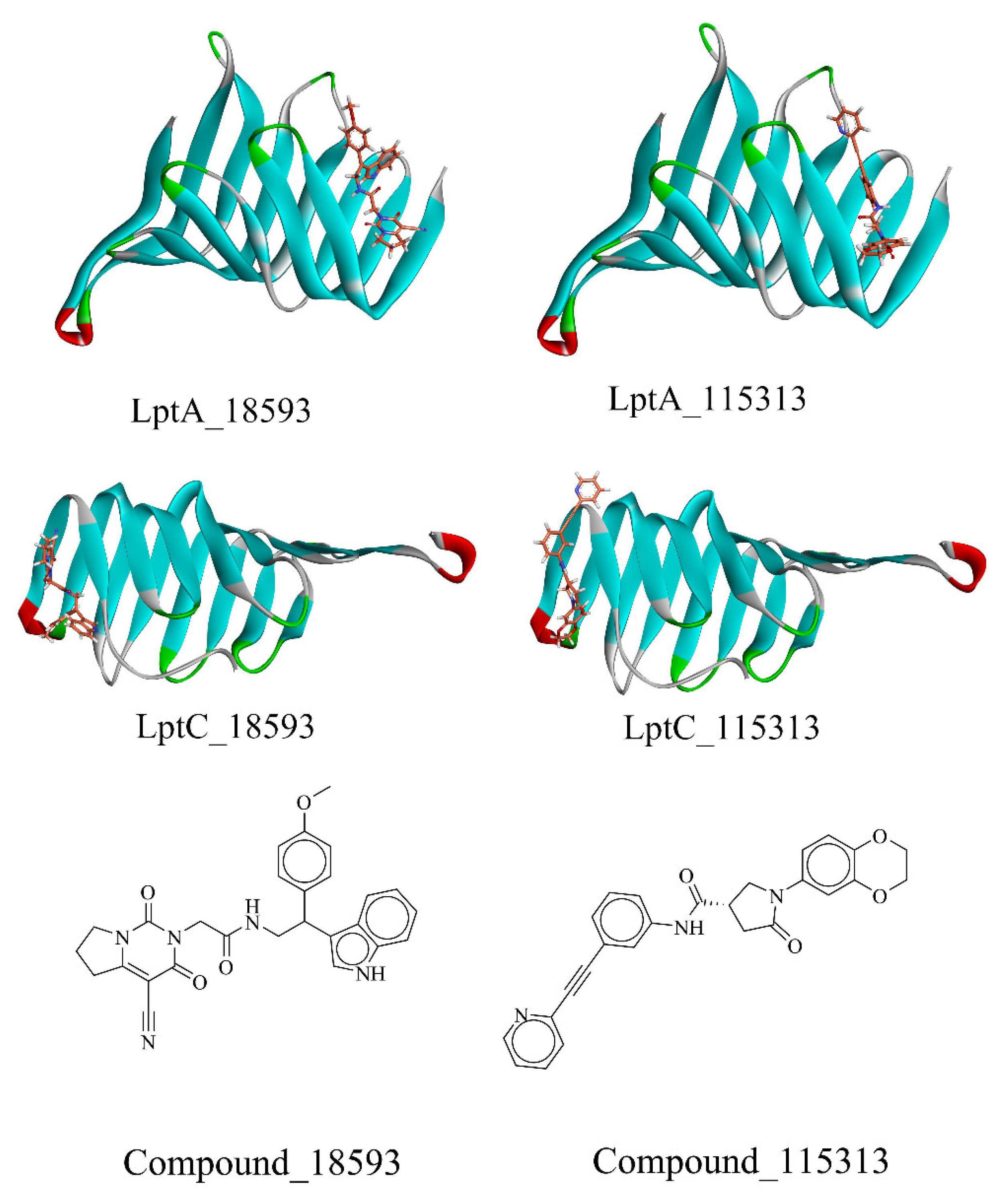
2.2. Virtual Screening of PPI Inhibitors
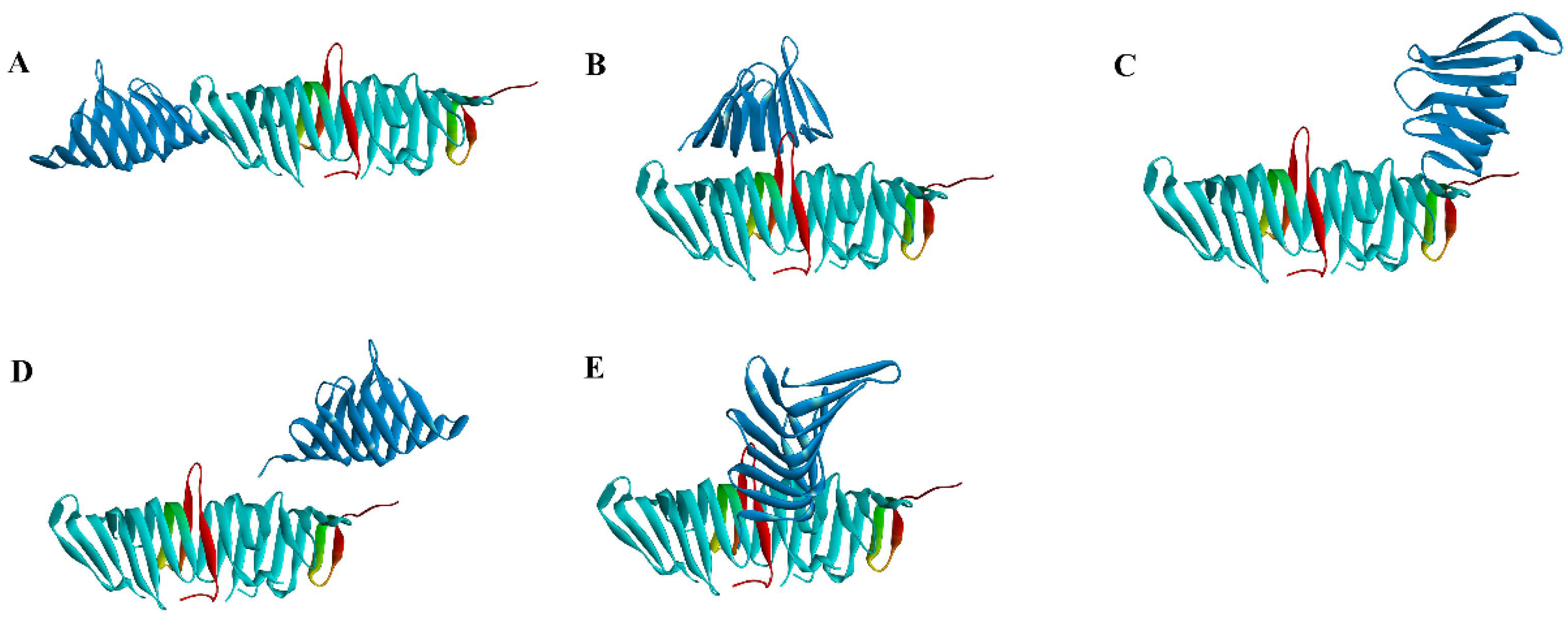
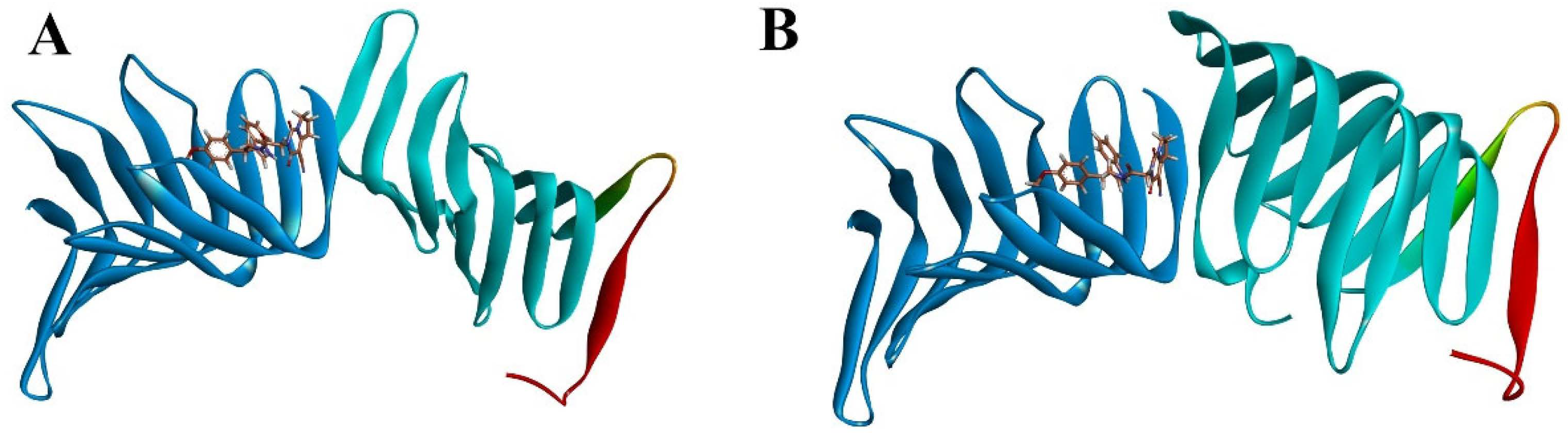
2.2.1. Molecular Docking
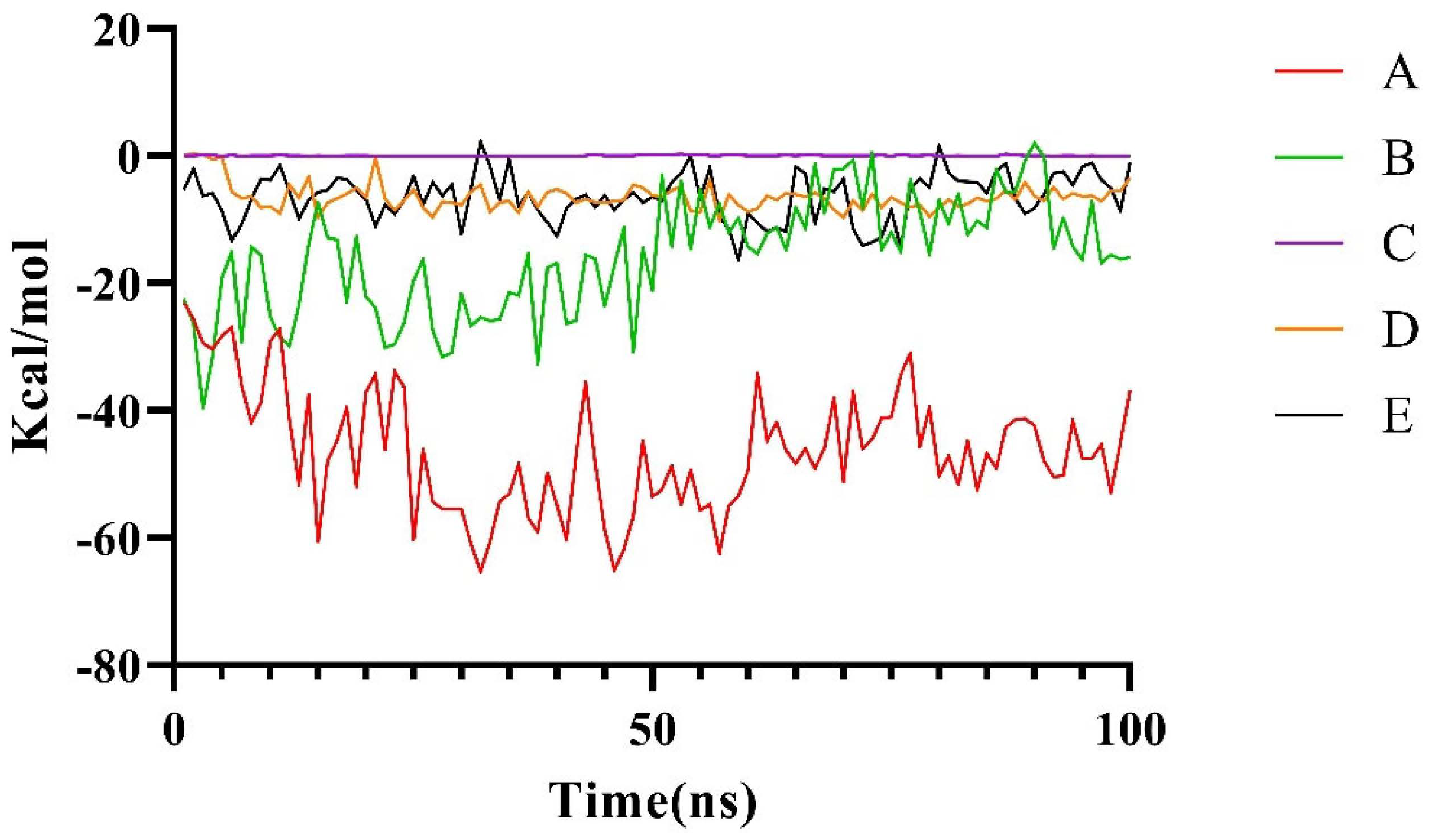
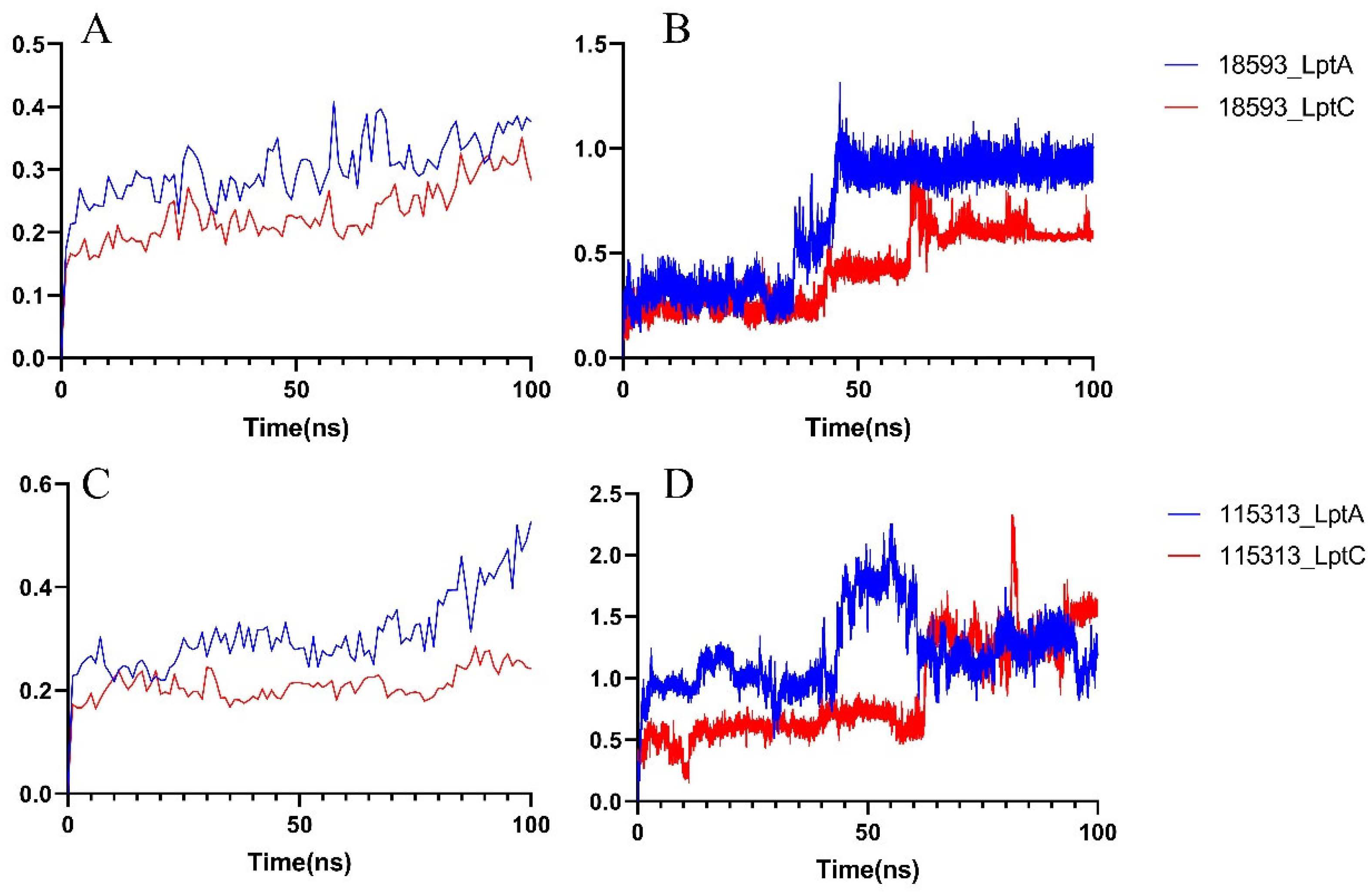
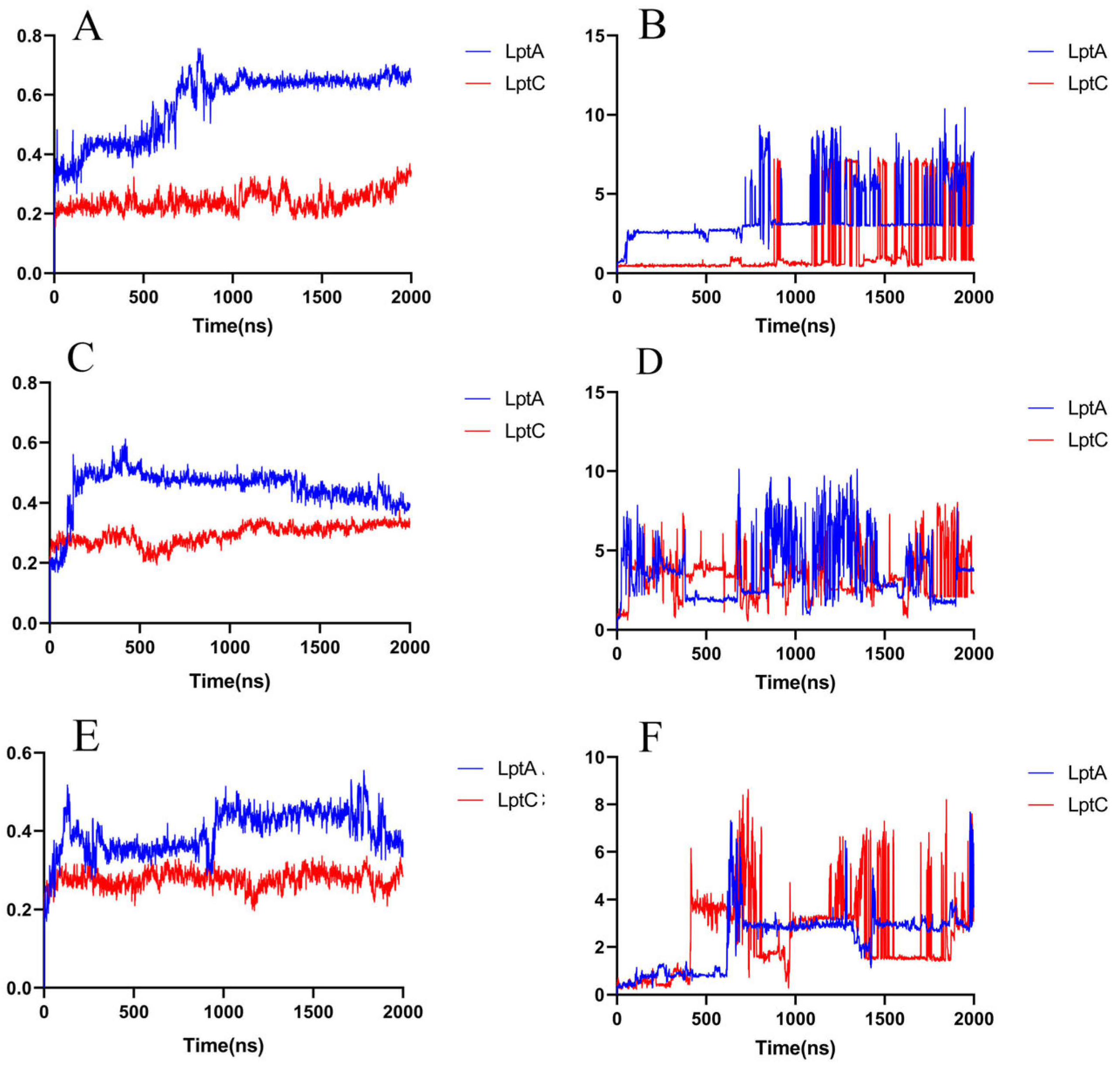
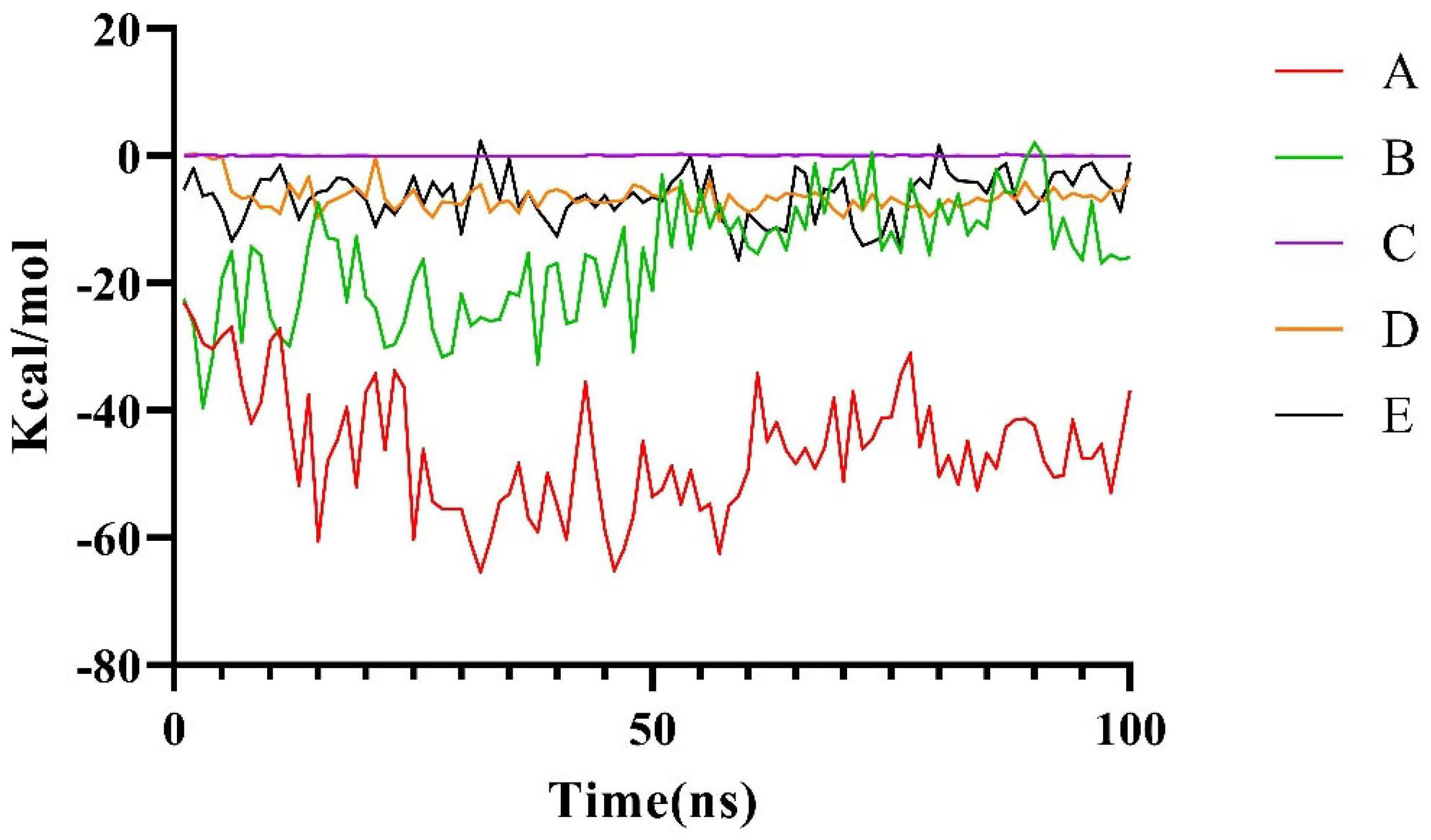
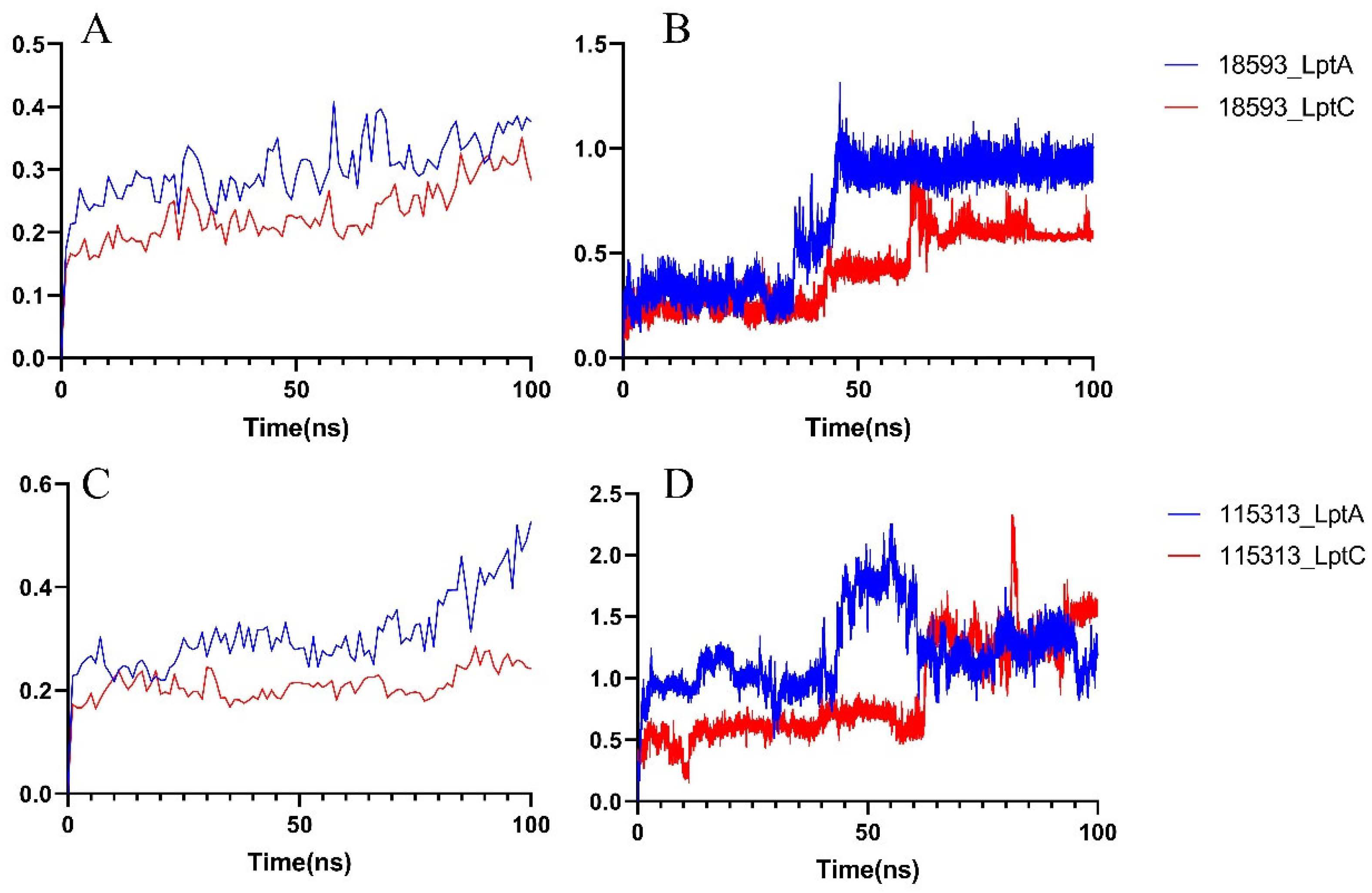
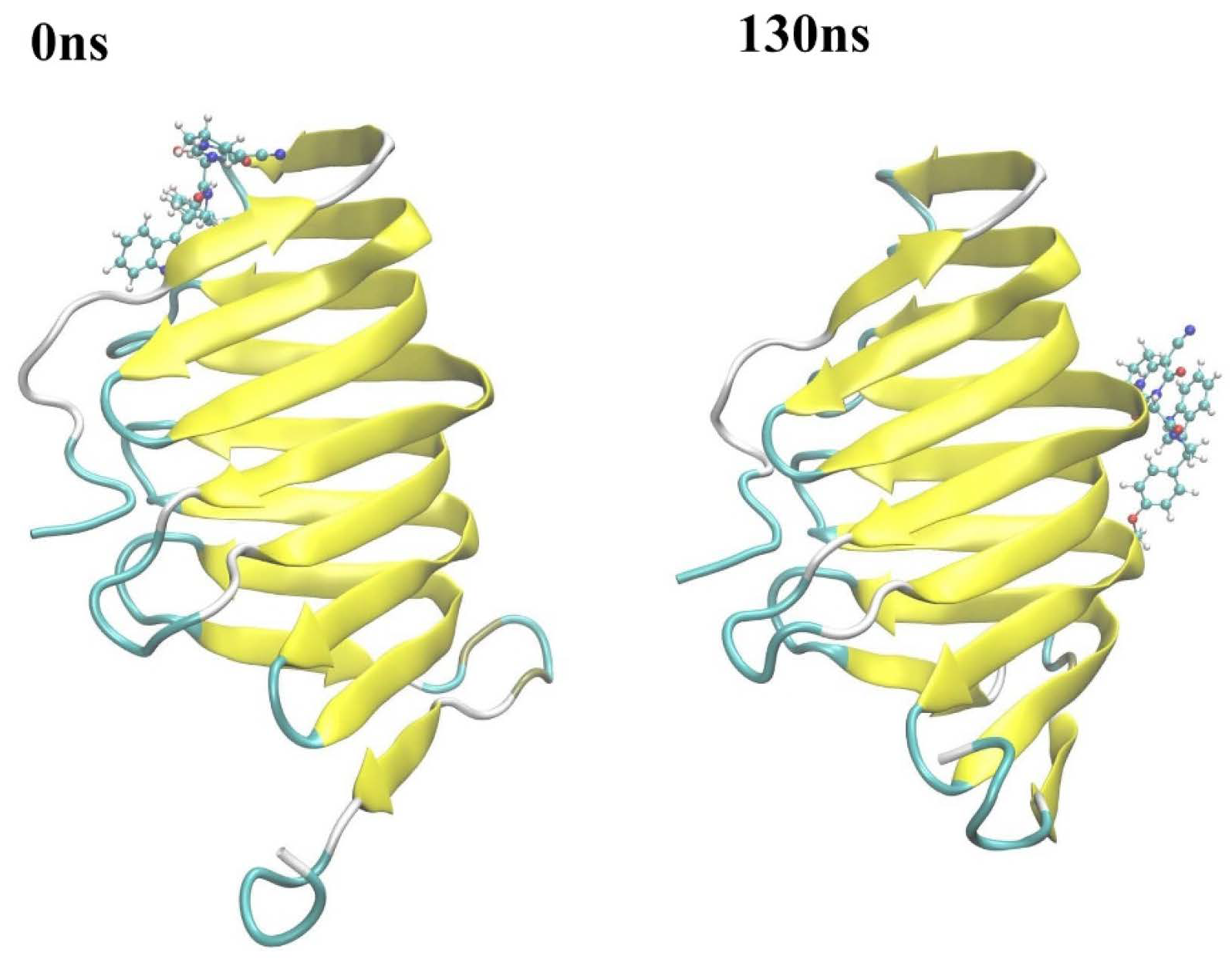
2.2.2. Molecular Dynamics Simulation
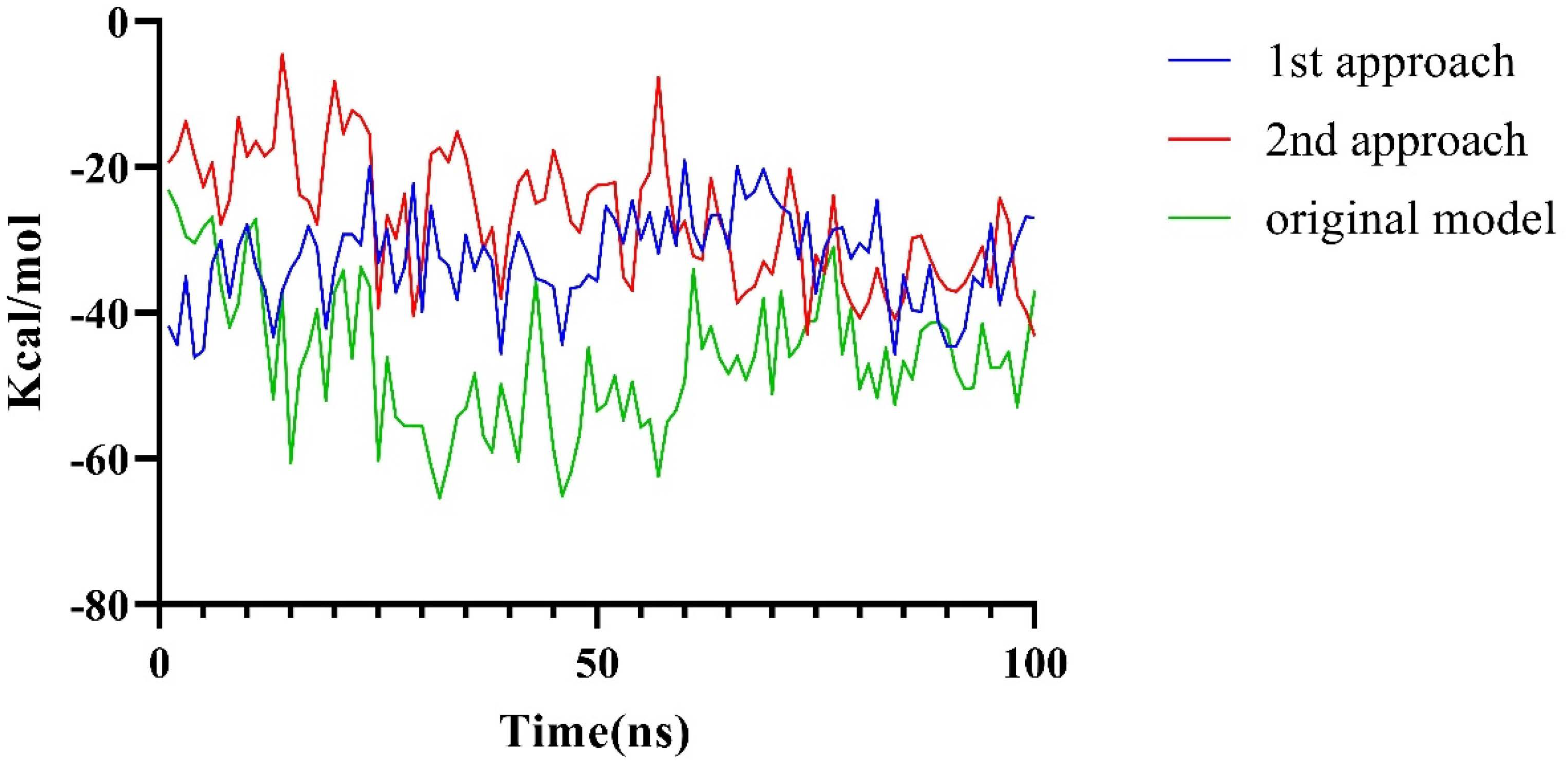
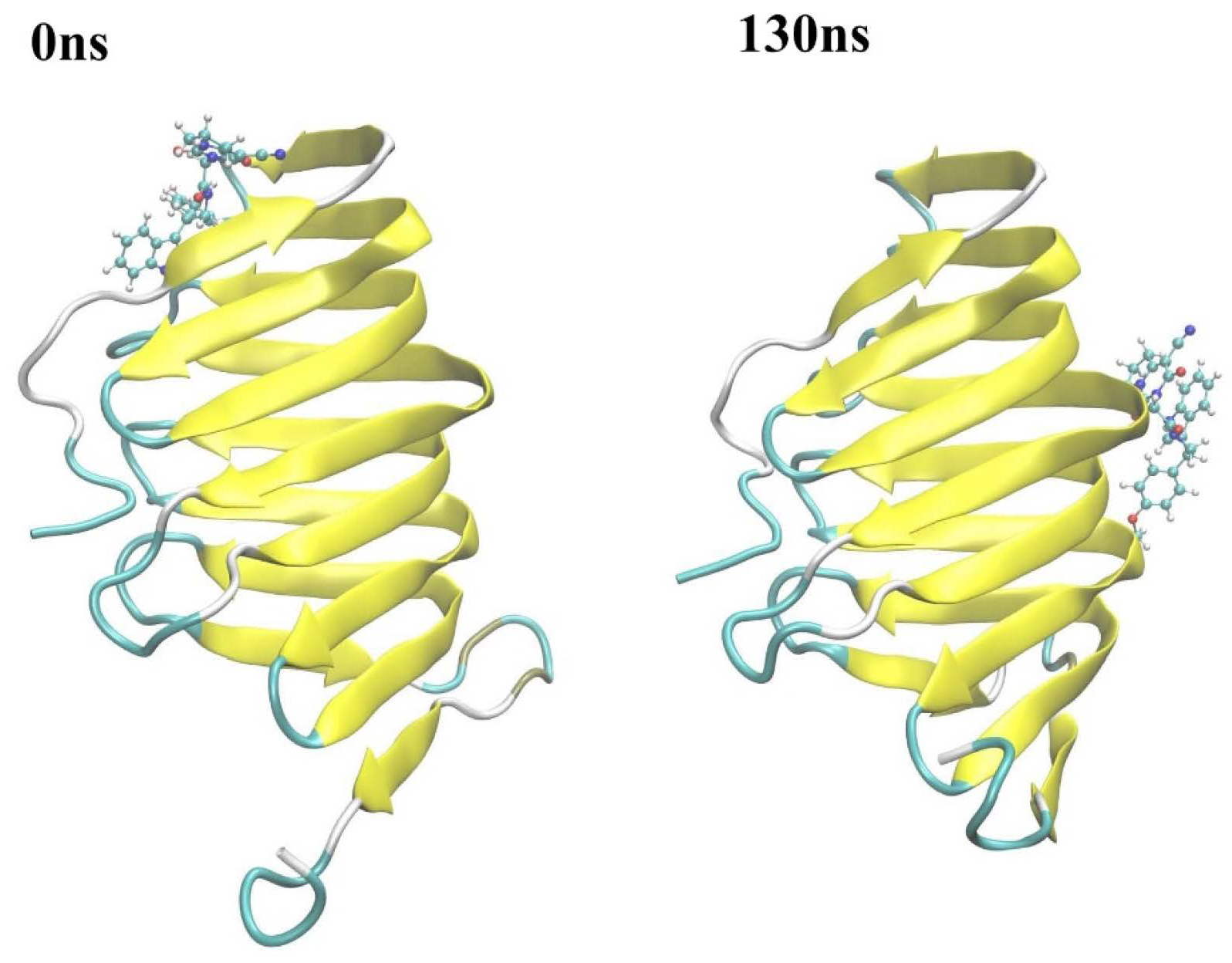
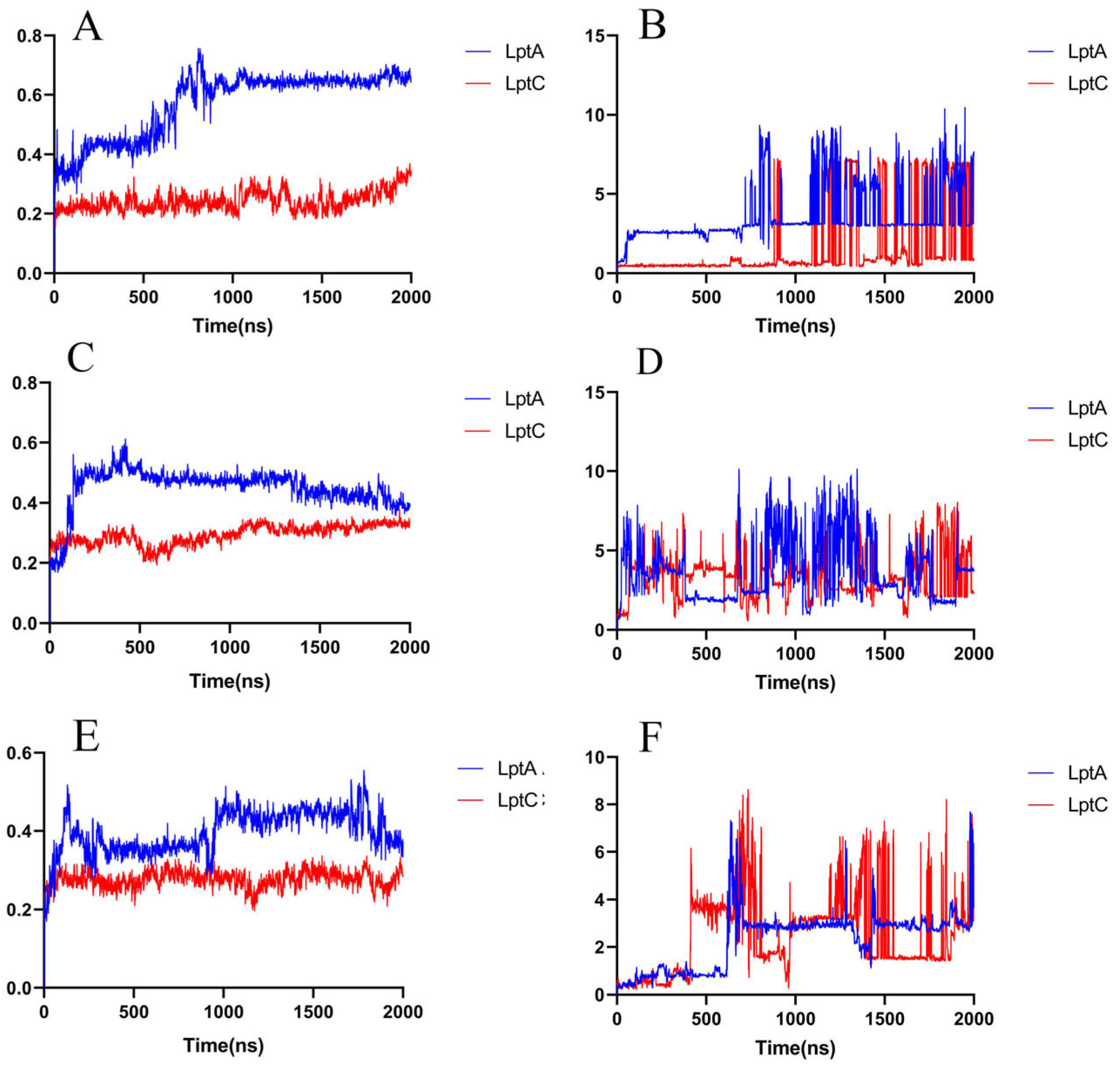
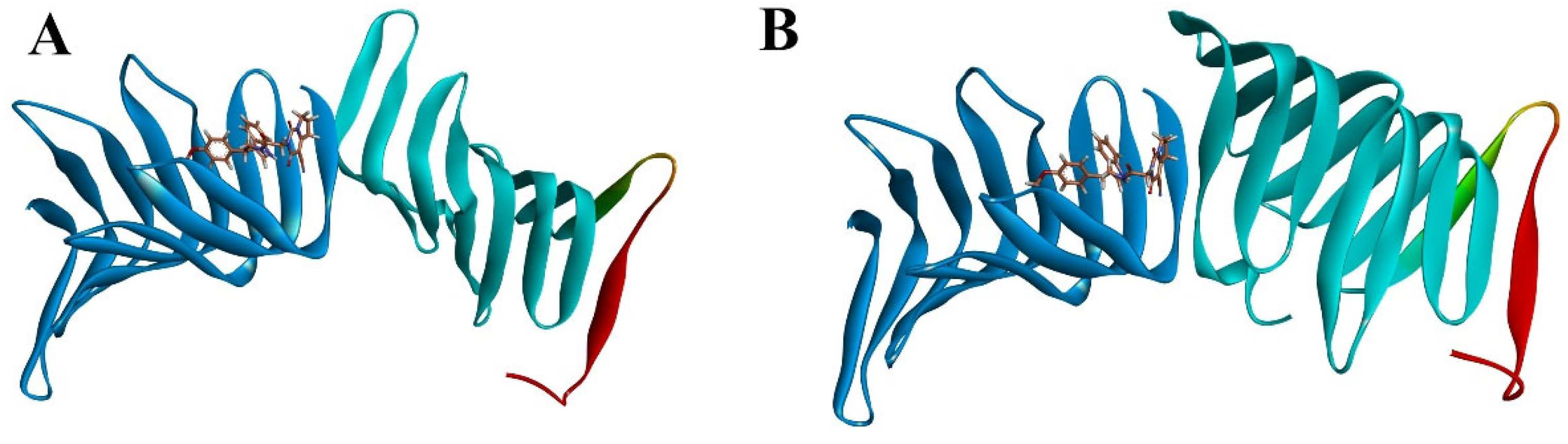
2.2.3. The PPI Blocking Ability of Compound 18593
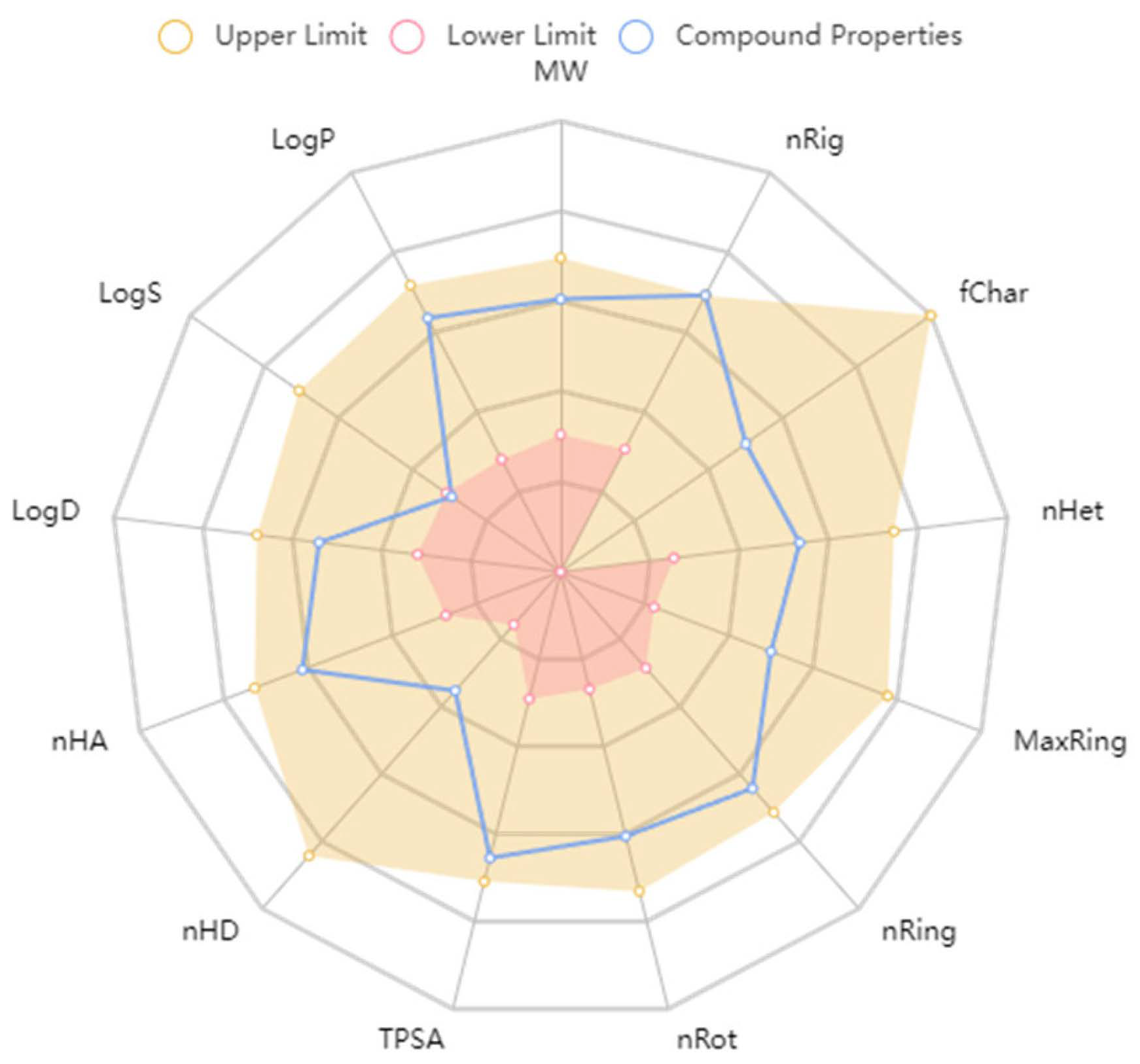
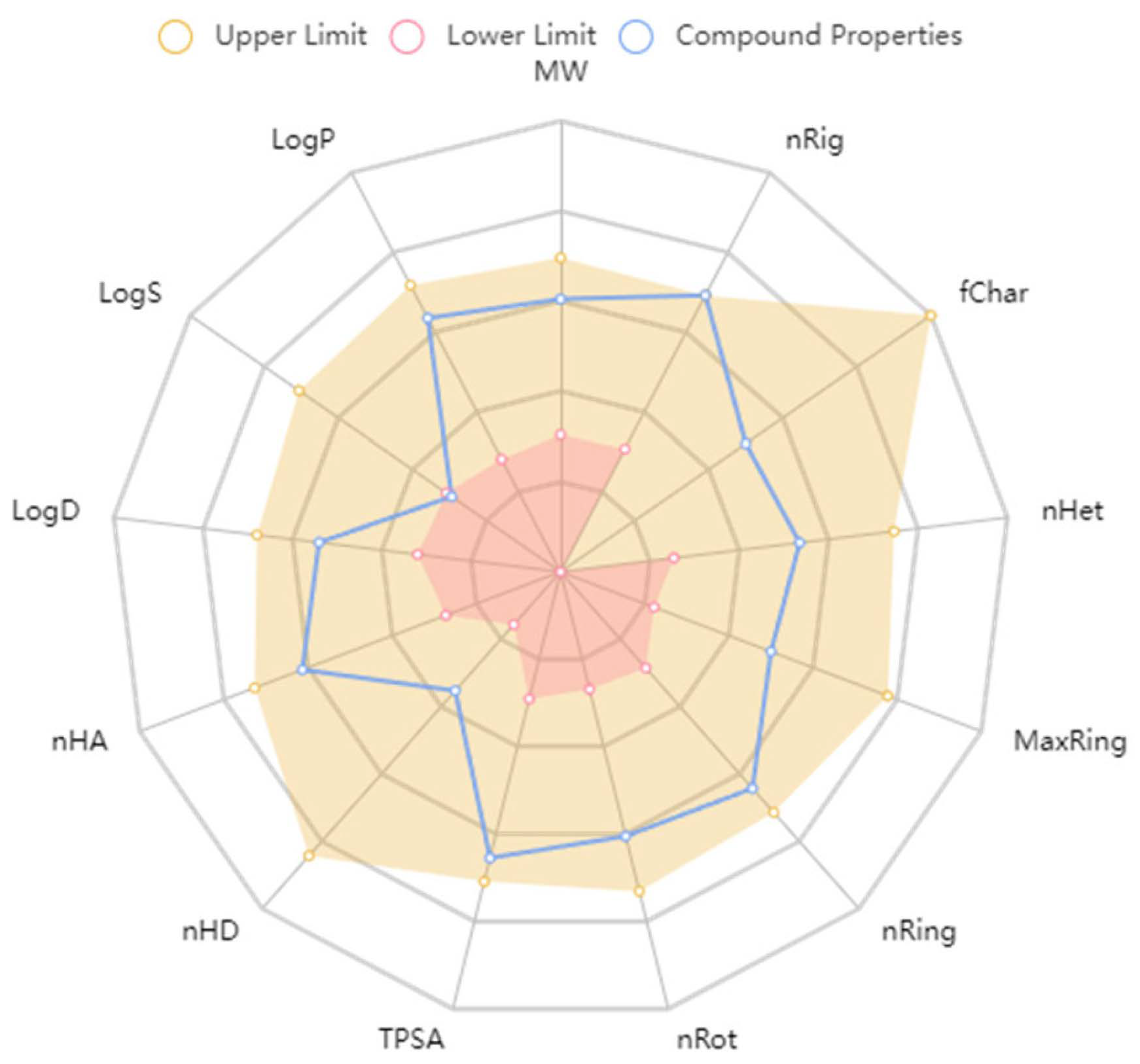
2.2.4. ADMET Predictions
3. Materials and Methods
3.1. Protein Preparation
3.2. Ligand Preparation
3.3. Molecular Docking
3.4. Molecular Dynamics Simulations
3.5. ADMET Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jim, O.N. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- Smoke, S.M.; Brophy, A.; Reveron, S.; Iovleva, A.; Kline, E.G.; Marano, M.; Miller, L.P.; Shields, R.K. Evolution and transmission of cefiderocol-resistant Acinetobacter baumannii during an outbreak in the burn intensive care unit. Clin. Infect. Dis. 2023, 76, e1261–e1265. [Google Scholar] [CrossRef] [PubMed]
- Simner, P.J.; Beisken, S.; Bergman, Y.; Posch, A.E.; Cosgrove, S.E.; Tamma, P.D. Cefiderocol Activity Against Clinical Pseudomonas aeruginosa Isolates Exhibiting Ceftolozane-Tazobactam Resistance. Open Forum Infect. Dis. 2021, 8, ofab311. [Google Scholar] [CrossRef] [PubMed]
- Sperandeo, P.; Deho, G.; Polissi, A. The lipopolysaccharide transport system of Gram-negative bacteria. Biochim. Biophys. Acta 2009, 1791, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Freinkman, E.; Kahne, D. Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 2012, 338, 1214–1217. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Martorana, A.M.; Fernández-Piñar, R.; Mancone, C.; Polissi, A.; Imperi, F. Pseudomonas aeruginosa LptE is crucial for LptD assembly, cell envelope integrity, antibiotic resistance and virulence. Virulence 2018, 9, 1718–1733. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N.; Gronenberg, L.S.; Kahne, D.; Silhavy, T.J. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 5537–5542. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.P.; Tefsen, B.; Geurtsen, J.; Tommassen, J. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. USA 2004, 101, 9417–9422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Wang, W.; Zhang, J.; Lin, Y.; Hong, B.; You, X.; Song, D.; Wang, Y.; Jiang, J.; et al. Identification of an anti-Gram-negative bacteria agent disrupting the interaction between lipopolysaccharide transporters LptA and LptC. Int. J. Antimicrob. Agents 2019, 53, 442–448. [Google Scholar] [CrossRef]
- Dai, X.; Yuan, M.; Lu, Y.; Zhu, X.; Liu, C.; Zheng, Y.; Si, S.; Yuan, L.; Zhang, J.; Li, Y. Identification of a small molecule that inhibits the interaction of LPS transporters LptA and LptC. Antibiotics 2022, 11, 1385. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Fehlbaum, P.; Bulet, P.; Chernysh, S.; Briand, J.P.; Roussel, J.P.; Letellier, L.; Hetru, C.; Hoffmann, J.A. Structure-activity analysis of thanatin, a 21-residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc. Natl. Acad. Sci. USA 1996, 93, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Moura, E.C.; Baeta, T.; Romanelli, A.; Laguri, C.; Martorana, A.M.; Erba, E.; Simorre, J.-P.; Sperandeo, P.; Polissi, A. Thanatin impairs lipopolysaccharide transport complex assembly by targeting LptC–LptA interaction and decreasing LptA stability. Front. Microbiol. 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Vetterli, S.U.; Zerbe, K.; Muller, M.; Urfer, M.; Mondal, M.; Wang, S.Y.; Moehle, K.; Zerbe, O.; Vitale, A.; Pessi, G.; et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 2018, 4, eaau2634. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Orlando, B.J.; Liao, M. Structural basis of lipopolysaccharide extraction by the LptB(2)FGC complex. Nature 2019, 567, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Sperandeo, P.; Villa, R.; Martorana, A.M.; Samalikova, M.; Grandori, R.; Deho, G.; Polissi, A. New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J. Bacteriol. 2011, 193, 1042–1053. [Google Scholar] [CrossRef]
- Chaudhury, S.; Gray, J.J. Conformer selection and induced fit in flexible backbone protein–protein docking using computational and NMR ensembles. J. Mol. Biol. 2008, 381, 1068–1087. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Rotili, D.; Mai, A.; Bolla, J.R.; Robinson, C.V. Mass spectrometry enables the discovery of inhibitors of an LPS transport assembly via disruption of protein–protein interactions. Chem. Commun. 2021, 57, 10747–10750. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Open Babel, Version 3.0.0. Available online: http://openbabel.org (accessed on 17 October 2022).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. Sobtop, Version 1.0(dev3.2). Available online: http://sobereva.com/soft/Sobtop (accessed on 17 February 2024).
- Lu, J.; Qiu, Y.; Baron, R.; Molinero, V. Coarse-Graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to Monatomic Anisotropic Water Models Using Relative Entropy Minimization. J. Chem. Theory Comput. 2014, 10, 4104–4120. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., 3rd. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; Dong, W.; Li, Y.; Cao, W.; Xiao, Z.; Zhou, Y.; Teng, Y.; You, X.; Yang, X.; Huang, H.; et al. The Prediction of LptA and LptC Protein–Protein Interactions and Virtual Screening for Potential Inhibitors. Molecules 2024, 29, 1827. https://doi.org/10.3390/molecules29081827
Ren Y, Dong W, Li Y, Cao W, Xiao Z, Zhou Y, Teng Y, You X, Yang X, Huang H, et al. The Prediction of LptA and LptC Protein–Protein Interactions and Virtual Screening for Potential Inhibitors. Molecules. 2024; 29(8):1827. https://doi.org/10.3390/molecules29081827
Chicago/Turabian StyleRen, Yixin, Wenting Dong, Yan Li, Weiting Cao, Zengshuo Xiao, Ying Zhou, Yun Teng, Xuefu You, Xinyi Yang, Huoqiang Huang, and et al. 2024. "The Prediction of LptA and LptC Protein–Protein Interactions and Virtual Screening for Potential Inhibitors" Molecules 29, no. 8: 1827. https://doi.org/10.3390/molecules29081827
APA StyleRen, Y., Dong, W., Li, Y., Cao, W., Xiao, Z., Zhou, Y., Teng, Y., You, X., Yang, X., Huang, H., & Wang, H. (2024). The Prediction of LptA and LptC Protein–Protein Interactions and Virtual Screening for Potential Inhibitors. Molecules, 29(8), 1827. https://doi.org/10.3390/molecules29081827