
Expanding Predictive Capacities in Toxicology: Insights from Hackathon-Enhanced Data and Model Aggregation

 , , , ,
, , , ,
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event | Topic | Year |
|---|---|---|
| RDKit UGM [31] | An annual symposium centered around the RDKit cheminformatics library, wherein attendees engaged with presentations, facilitated dialogues, exchanged insights, and familiarized themselves with recent advancements in cheminformatics algorithms, pipelines, and databases. The concluding day was structured as a hackathon, focusing on resolving practical challenges pertinent to RDKit and KNIME. | 2012–2023 |
| D3R Grand Challenge [32] | This endeavor was designed to hone computational methodologies for estimating ligand–protein interaction energies and predicting their binding conformations. Notably, the challenge garnered support from multiple leading pharmaceutical entities, contributing data pertaining to the docking structures. | 2015–2018 |
| MATDAT18 [33] | During the hackathon, contributions spanned the development of machine learning models for material classification and crafting structure–performance correlations, complemented by advancements in computational strategies in the domain of force fields and descriptors. | 2018 |
| Nomad2018 Predicting Transparent Conductor [37] | This initiative sought to devise a strategy for the systematic engineering of highly efficient conductors based on metal sesquioxides. Participants were tasked with predicting both the band gap and atomic formation energy for conductors consisting of the combination of aluminum, gallium, and indium. | 2018 |
| Predicting Molecular Properties [39] | Participants were tasked with leveraging nuclear magnetic resonance data to craft an algorithm proficient in forecasting the spin–spin interaction constants between paired atoms. | 2019 |
| Bristol-Myers Squibb—Molecular Translation [40] | The focal point of this competition was the optical recognition of chemical structures, subsequently transcribed to InChI format. The synthetic datasets provided encompassed distorted images of chemical compounds. | 2021 |
| CATMOS [35] | An international coalition was commissioned to predict five distinct endpoints: EPA and GHS categorizations, dichotomous toxicological outcomes, and pinpoint estimations of LD50 for acute oral toxicity in rodents. | 2021 |
| Drugathon [34] | In this engagement, attendees were encouraged to showcase their prowess in molecular modeling and drug discovery paradigms. Submissions entailed proposals for putatively active chemical entities. Following a rigorous selection process by the orchestrating entity, BioSolveIT, the most promising submissions were synthesized to validate their biological activity. Upon successful validation, BioSolveIT extended co-authorship opportunities for a publication in a reputable, peer-reviewed journal. | 2022–2023 |
| Novozymes Enzyme Stability Prediction [38] | The challenge mandated the creation of a machine learning model adept at predicting an enzyme’s thermostability, inclusive of its single-amino acid variants. The metric of thermal stability was equated to the enzyme’s melting point. | 2022–2023 |
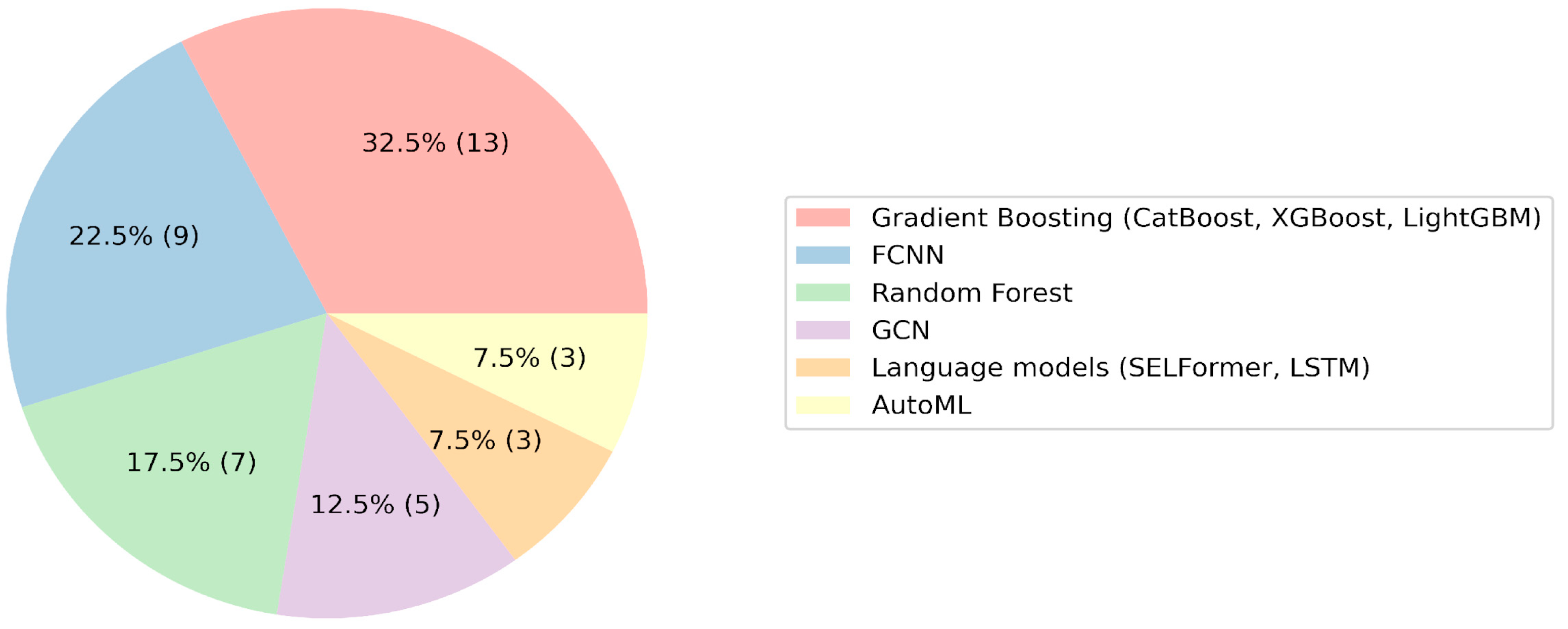
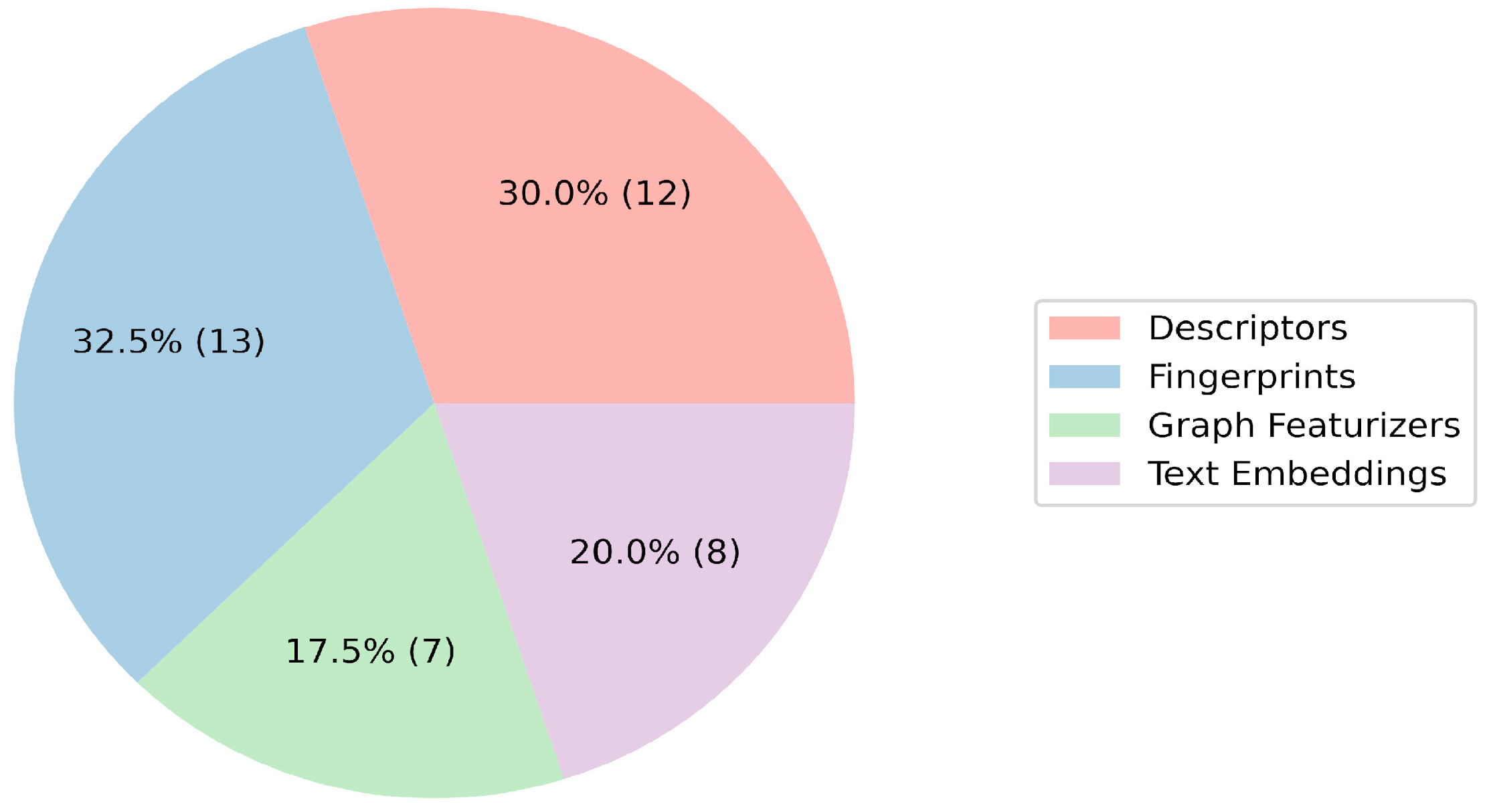
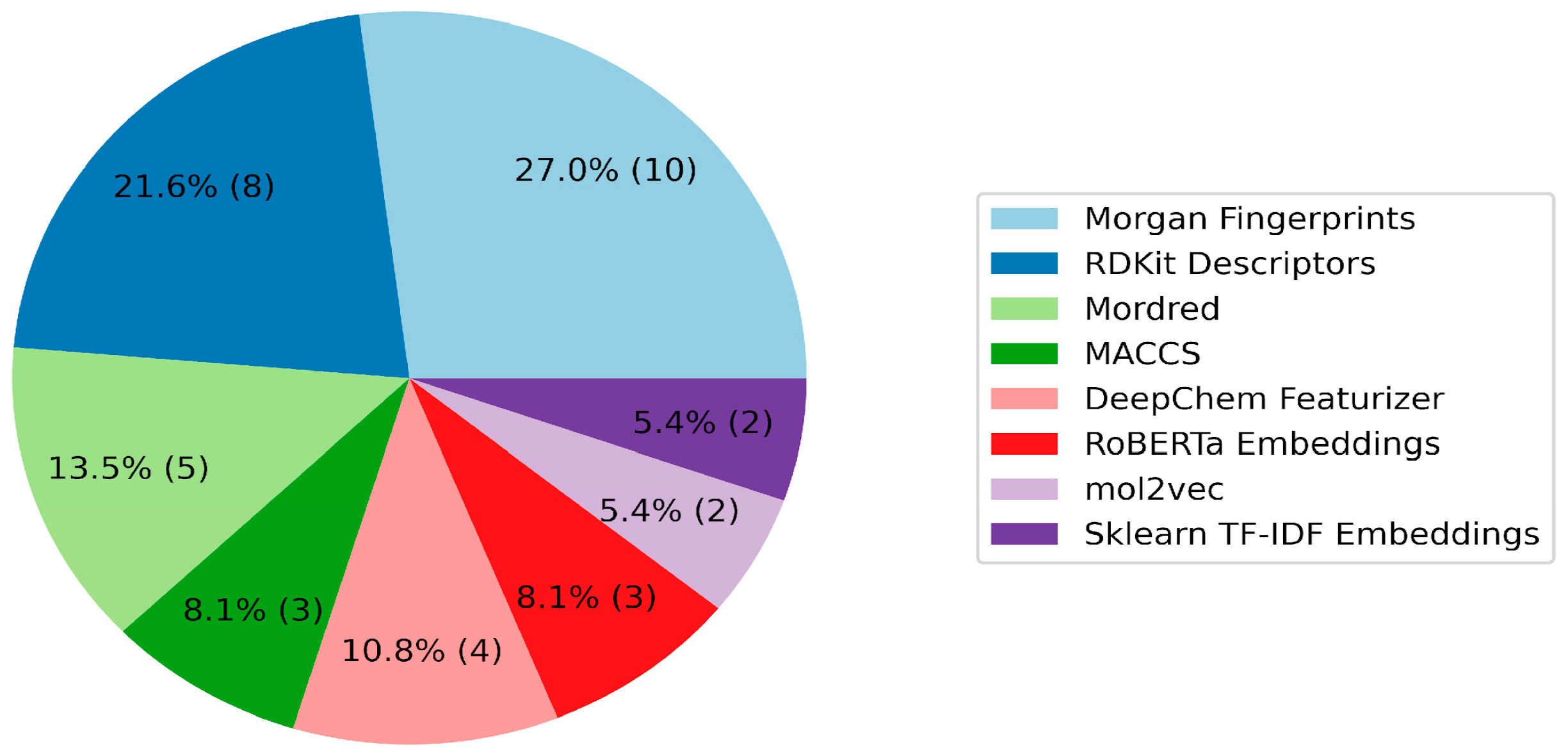
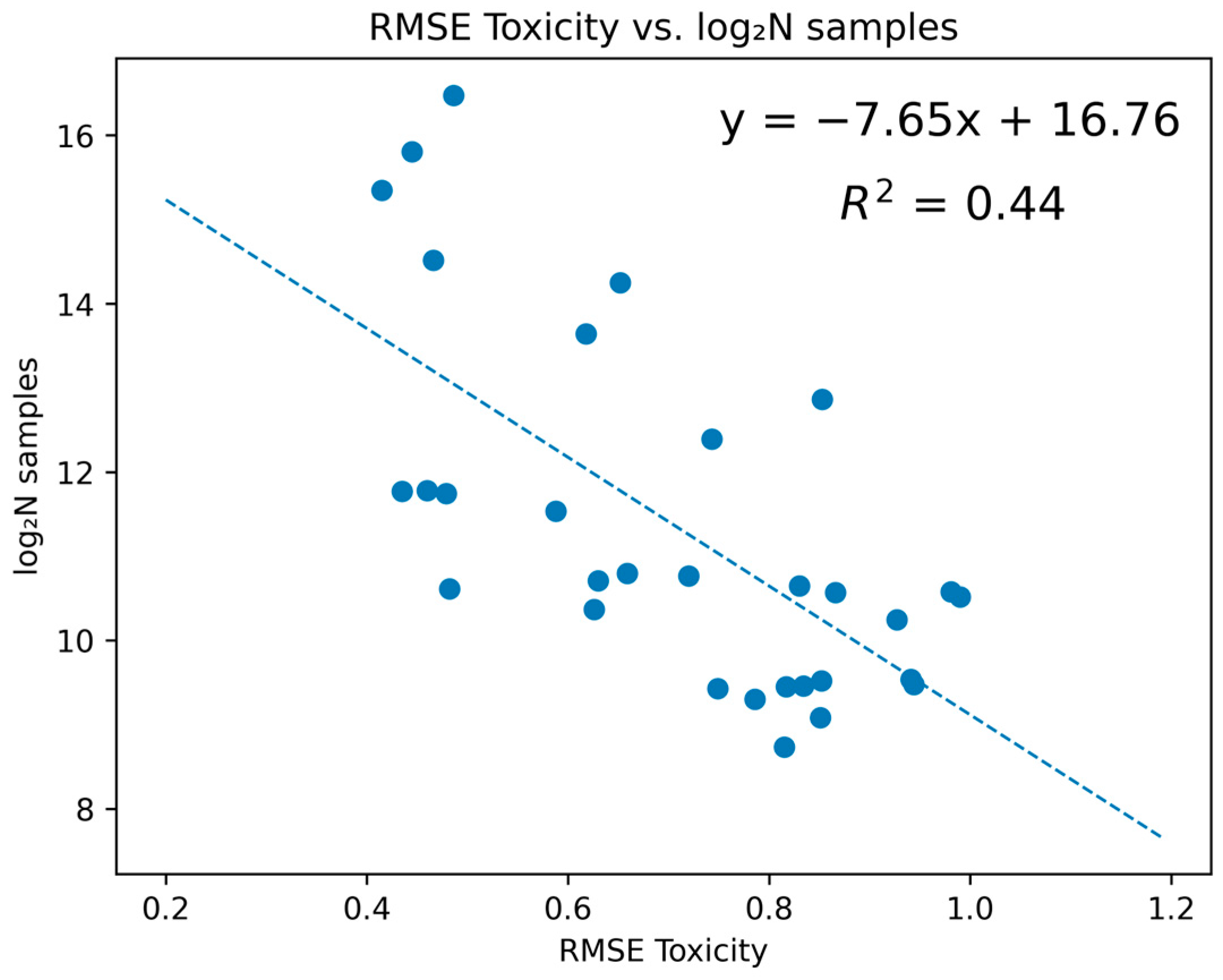
2. Results
3. Discussion
4. Materials and Methods
4.1. Organization of the Hackathon
- A presentation detailing the solution.
- A Python-based Jupyter notebook containing:
- A module taking a test .csv file of molecular SMILES as the input and producing the corresponding predicted endpoints as the output.
- A complete model training workflow including parameter selection, hyperparameter tuning, and definition of the architecture.
- Training datasets with the molecular SMILES and the corresponding experimental values, clearly delineating data sources.
- Any supplementary files necessary for the model’s execution or evaluation purposes.
4.2. Solution Evaluation Criteria
- Dataset quality. Evaluations under this metric did not award additional points. Rather, it eliminated suboptimal submissions. Solutions were penalized for inconsistencies such as the use of synthetic data, attempts at fraud, discrepancies between the source and the data utilized, inconsistencies in the dimensions of the data, the presence of duplicate molecular entities, or a lack of source provision. This metric sought to emphasize the importance of initial data processing; a foundational step commonly practiced by researchers.
- Model evaluation. Participants were prompted to utilize evaluation metrics either from referenced publications or from the Syntelly models, available on the Syntelly website in the statistics section. For quantitative assessment, any improvement had to surpass a 5% mean value for regression (RMSE) or 3% for classification (ROC AUC) during a five-fold cross-validation. Depending on improvements in the metrics, the models’ multipliers were adjusted. For the metrics available in Syntelly, a comparison with the average values of Syntelly’s benchmarks was required. For non-Syntelly metrics, external benchmark citations were mandated. Notably, minor model improvements deemed to be statistically insignificant were not considered valuable, leading to the establishment of a minimal improvement threshold. If the model fulfilled the condition for improving the quality of the metric, then the model’s multiplier equaled (1 + M/100) for regression and equaled (1 + M/50) for classification, where M is the percentage of improvement in the metric.
- The assessment process for the models incorporated two primary factors: the extent of the dataset and the uniqueness of the samples. For the dataset’s extent, a point-based system was used, attributing 1, 0.75, and 0.5 points for the first, second, and third positions, respectively. In instances of tied positions, the points were distributed evenly among the tied participants. Zero points were assigned for datasets that were not within the top three positions.
- 4.
- Difficulty in obtaining data. The challenge associated with data acquisition was acknowledged and quantitatively assessed. Points were allocated based on the complexity of the data collection process. One point was assigned for scraping data without an API from at least one source, downloading via API from three or more sources, or manual searches of five or more sources. Downloading via API from at least one source or a manual search of at least three sources scored 0.5 points. A manual search of one source scored 0.25 points. Collecting good data is an important task as part of building a machine learning model, so we aimed to reward participants if the participants spent a large amount of time collecting good data.
- 5.
- Number of sources of experimental data. In consideration of the significant variability inherent in experimental toxicity data, models incorporating data from multiple sources were favored. A source was defined as a database or an aggregation of publications with over 1000 chemical structures. Points were allocated as per the number of sources, with a model incorporating over five sources receiving 1 point, four or five sources receiving 0.75 points, two or three sources receiving 0.5 points, and one source receiving 0 points.
- 6.
- Number of predicted toxicity models. Models were evaluated on the basis of the range of toxicity endpoints predicted. Each endpoint was scored individually, with the total points computed cumulatively, incorporating the respective multipliers.
- 7.
- Quality of presentation of the solution. It was not enough to simply send files with the models and the metrics’ results; an essential quality for a researcher is the accurate presentation of their results. This was particularly important, considering the educational value of the project, as many of the participants were students and graduate students. The ability to clearly and concisely present the results determines how valuable the participants’ contribution will be to the scientific community. At this point, participants could receive additional points for the thoughtfulness of the solution and a competent methodology for selecting parameters and hyperparameters of the model. The Jupyter notebook should have had: (1) a selection of hyperparameters (more than 10 options were considered) or the number of layers of the neural network (more than three options were considered); (2) a selection of neural network architectures or a selection of machine learning models, additional comments describing each of the blocks; (3) the ease of perception of the laptop and convenient launch. If the laptop could not be started from start to finish, 0 points were given for the work. Each item on the Jupyter notebook was worth 1 point (the maximum number of points for a notebook was 3 points). The presentation had to include: (1) a detailed description of the data collection process, selection of and/or the search for models and descriptors, and the results obtained; (2) the provision of complete information about all models, benchmarks used and the sources, (3) the clarity, adequacy, and consistency of the information presented, free of factual errors. For the presence of factual errors, 0 points were given for the presentation. Each presentation point was worth 1 point (the maximum number of points for a presentation was 3 points).
- 8.
- Diversity of the dataset. Models benefitting from a broad chemical space were awarded a point, contingent on the demonstration of the dataset’s diversity, spanning multiple chemical classes.
- 9.
- Uniqueness of the space of toxicity indicators. An additional dimension of the evaluation lay in the variation of predicted rates amongst the participants. A point was awarded for the inclusion of unique toxicity endpoints that were absent in other submissions and directly pertinent to molecular toxicity.
- 10.
- Interpretability of the model and descriptors used. The capacity for the models to elucidate the toxic effects of molecules, offering insights into the underlying mechanisms, was rewarded with an additional point, accentuating the importance of the model’s interpretability in the context of scientific discovery.
4.3. Model Preparation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hornberg, J.J.; Laursen, M.; Brenden, N.; Persson, M.; Thougaard, A.V.; Toft, D.B.; Mow, T. Exploratory toxicology as an integrated part of drug discovery. Part I: Why and how. Drug Discov. Today 2014, 19, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-X.; Li, X.-J.; Zhang, H.-Y. Where is the hope for drug discovery? Let history tell the future. Drug Discov. Today 2009, 14, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Taglang, G.; Jackson, D.B. Use of “big data” in drug discovery and clinical trials. Gynecol. Oncol. 2016, 141, 17–23. [Google Scholar] [CrossRef]
- Kell, D.B. Finding novel pharmaceuticals in the systems biology era using multiple effective drug targets, phenotypic screening and knowledge of transporters: Where drug discovery went wrong and how to fix it. FEBS J. 2013, 280, 5957–5980. [Google Scholar] [CrossRef]
- Thomas, C.E.; Will, Y. The impact of assay technology as applied to safety assessment in reducing compound attrition in drug discovery. Expert. Opin. Drug Discov. 2012, 7, 109–122. [Google Scholar] [CrossRef]
- Aghila Rani, K.G.; Hamad, M.A.; DZaher, M.; Sieburth, S.M.; Madani, N.; Al-Tel, T.H. Drug development post COVID-19 pandemic: Toward a better system to meet current and future global health challenges. Expert. Opin. Drug Discov. 2021, 16, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Amir-Aslani, A. Toxicogenomic predictive modeling: Emerging opportunities for more efficient drug discovery and development. Technol. Forecast. Social. Change 2008, 75, 905–932. [Google Scholar] [CrossRef]
- Li, A.P. Overview: Evaluation of metabolism-based drug toxicity in drug development. Chem. Biol. Interact. 2009, 179, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Svensson, F.; Zoufir, A.; Bender, A. eMolTox: Prediction of molecular toxicity with confidence. Bioinformatics 2018, 34, 2508–2509. [Google Scholar] [CrossRef]
- Roncaglioni, A.; Toropov, A.A.; Toropova, A.P.; Benfenati, E. In silico methods to predict drug toxicity. Curr. Opin. Pharmacol. 2013, 13, 802–806. [Google Scholar] [CrossRef]
- Wathieu, H.; Ojo, A.; Dakshanamurthy, S. Prediction of Chemical Multi-target Profiles and Adverse Outcomes with Systems Toxicology. Curr. Med. Chem. 2017, 24, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lu, J.; Zhang, J.; Feng, K.-R.; Zheng, M.-Y.; Cai, Y.-D. Predicting chemical toxicity effects based on chemical-chemical interactions. PLoS ONE 2013, 8, e56517. [Google Scholar]
- Jain, S.; Siramshetty, V.B.; Alves, V.M.; Muratov, E.N.; Kleinstreuer, N.; Tropsha, A.; Nicklaus, M.C.; Simeonov, A.; Zakharov, A.V. Large-Scale Modeling of Multispecies Acute Toxicity End Points Using Consensus of Multitask Deep Learning Methods. J. Chem. Inf. Model. 2021, 61, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.W.H.; Goodman, J.M.; Allen, T.E.H. Machine Learning in Predictive Toxicology: Recent Applications and Future Directions for Classification Models. Chem. Res. Toxicol. 2021, 34, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tamura, I.; Abe, R.; Takanobu, H.; Nakamura, A.; Suzuki, T.; Hirose, A.; Nishimura, T.; Tatarazako, N. Chronic toxicity of an environmentally relevant mixture of pharmaceuticals to three aquatic organisms (alga, daphnid, and fish). Environ. Toxicol. Chem. 2016, 35, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Phillips, J.; Sedykh, A.; Tandon, A.; Sprankle, C.; Morefield, S.Q.; Shapiro, A.; Allen, D.; Shah, R.; Maull, E.A.; et al. An Integrated Chemical Environment to Support 21st-Century Toxicology. Environ. Health Perspect. 2017, 125, 054501. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the Human Hazard Characterization of Chemicals: A Tox21 Update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.S.; Houck, K.A.; Kavlock, R.J.; Knudsen, T.B.; Martin, M.T.; Mortensen, H.M.; Reif, D.M.; Rotroff, D.M.; Shah, I.; Richard, A.M.; et al. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ. Health Perspect. 2010, 118, 485–492. [Google Scholar] [CrossRef]
- Ginsberg, G.L.; Fedinick, K.P.; Solomon, G.M.; Elliott, K.C.; Vandenberg, J.J.; Barone, S.; Bucher, J.R. New Toxicology Tools and the Emerging Paradigm Shift in Environmental Health Decision-Making. Environ. Health Perspect. 2019, 127, 125002. [Google Scholar] [CrossRef]
- Kirchhübel, N.; Fantke, P. Getting the chemicals right: Toward characterizing toxicity and ecotoxicity impacts of inorganic substances. J. Clean. Prod. 2019, 227, 554–565. [Google Scholar] [CrossRef]
- Olker, J.H.; Elonen, C.M.; Pilli, A.; Anderson, A.; Kinziger, B.; Erickson, S.; Skopinski, M.; Pomplun, A.; LaLone, C.A.; Russom, C.L.; et al. The ECOTOXicology Knowledgebase: A Curated Database of Ecologically Relevant Toxicity Tests to Support Environmental Research and Risk Assessment. Enviro Toxic. Chem. 2022, 41, 1520–1539. [Google Scholar] [CrossRef]
- Wignall, J.A.; Muratov, E.; Sedykh, A.; Guyton, K.Z.; Tropsha, A.; Rusyn, I.; Chiu, W.A. Conditional Toxicity Value (CTV) Predictor: An In Silico Approach for Generating Quantitative Risk Estimates for Chemicals. Environ. Health Perspect. 2018, 126, 57008. [Google Scholar] [CrossRef]
- LeBlanc, G.A.; Olmstead, A.W. Evaluating the Toxicity of Chemical Mixtures. Environ. Health Perspect. 2004, 112, A729–A730. [Google Scholar] [CrossRef][Green Version]
- Kramer, C.; Dahl, G.; Tyrchan, C.; Ulander, J. A comprehensive company database analysis of biological assay variability. Drug Discov. Today 2016, 2, 1213–1221. [Google Scholar] [CrossRef]
- Price, P.S.; Keenan, R.E.; Swartout, J.C. Characterizing interspecies uncertainty using data from studies of anti-neoplastic agents in animals and humans. Toxicol. Appl. Pharmacol. 2008, 233, 64–70. [Google Scholar] [CrossRef]
- Szabó, B.; Lang, Z.; Kövér, S.; Bakonyi, G. The inter-individual variance can provide additional information for the ecotoxicologists beside the mean. Ecotoxicol. Environ. Saf. 2021, 217, 112260. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Zoete, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Dopazo, J. Genomics and transcriptomics in drug discovery. Drug Discovery Today 2014, 19, 126–132. [Google Scholar] [CrossRef]
- Hsieh, J.-H.; Yin, S.; Wang, X.S.; Liu, S.; Dokholyan, N.V.; Tropsha, A. Cheminformatics meets molecular mechanics: A combined application of knowledge-based pose scoring and physical force field-based hit scoring functions improves the accuracy of structure-based virtual screening. J. Chem. Inf. Model. 2012, 52, 16–28. [Google Scholar] [CrossRef][Green Version]
- RDKit. Available online: https://www.rdkit.org/ (accessed on 28 September 2023).
- D3R|D3R Grand Challenge. Available online: https://drugdesigndata.org/about/grand-challenge (accessed on 28 September 2023).
- Ferguson, A.L.; Mueller, T.; Rajasekaran, S.; Reich, B.J. Conference report: 2018 materials and data science hackathon (MATDAT18). Mol. Syst. Des. Eng. 2019, 4, 462–468. [Google Scholar] [CrossRef]
- Drugathon 2023 • BioSolveIT. Available online: https://www.biosolveit.de/drugathon-2023/ (accessed on 28 September 2023).
- Mansouri, K.; Karmaus, A.L.; Fitzpatrick, J.; Patlewicz, G.; Pradeep, P.; Alberga, D.; Alepee, N.; Allen, T.E.; Allen, D.; Alves, V.M.; et al. CATMoS: Collaborative Acute Toxicity Modeling Suite. Environ. Health Perspect. 2021, 129, 047013. [Google Scholar] [CrossRef] [PubMed]
- Kaggle. Available online: https://www.kaggle.com/ (accessed on 28 September 2023).
- Nomad2018 Predicting Transparent Conductors. Available online: https://kaggle.com/competitions/nomad2018-predict-transparent-conductors (accessed on 28 September 2023).
- Novozymes Enzyme Stability Prediction. Available online: https://kaggle.com/competitions/novozymes-enzyme-stability-prediction (accessed on 28 September 2023).
- Predicting Molecular Properties. Available online: https://kaggle.com/competitions/champs-scalar-coupling (accessed on 28 September 2023).
- Bristol-Myers Squibb—Molecular Translation. Available online: https://kaggle.com/competitions/bms-molecular-translation (accessed on 28 September 2023).
- Syntelly Hackathon. Available online: https://syntelly.ru/russianmedia/tpost/g0ainxvja1-obyavleni-pobediteli-hakatona (accessed on 28 September 2023).
- CatBoost. Available online: https://catboost.ai/ (accessed on 28 September 2023).
- Sosnin, S.; Karlov, D.; Tetko, I.V.; Fedorov, M.V. Comparative Study of Multitask Toxicity Modeling on a Broad Chemical Space. J. Chem. Inf. Model. 2019, 59, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Syntelly. Available online: https://app.syntelly.com/login (accessed on 10 May 2023).
- Jiang, J.; Zhang, R.; Ma, J.; Liu, Y.; Yang, E.; Du, S.; Zhao, Z.; Yuan, Y. TranGRU: Focusing on both the local and global information of molecules for molecular property prediction. Appl. Intell. 2023, 53, 15246–15260. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, K.; DeCost, B. Atomistic Line Graph Neural Network for improved materials property predictions. npj Comput. Mater. 2021, 7, 185. [Google Scholar] [CrossRef]
- XGBoost. Available online: https://xgboost.readthedocs.io/en/stable/# (accessed on 28 September 2023).
- Wu, L.; Yan, B.; Han, J.; Li, R.; Xiao, J.; He, S.; Bo, X. TOXRIC: A comprehensive database of toxicological data and benchmarks. Nucleic Acids Res. 2023, 51, D1432–D1445. [Google Scholar] [CrossRef]
- Karim, A.; Lee, M.; Balle, T.; Sattar, A. CardioTox net: A robust predictor for hERG channel blockade based on deep learning meta-feature ensembles. J. Cheminform. 2021, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Blood-Brain Barrier Permeability of Compounds by Machine Learning and Resampling Methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Lim, S.; Kim, Y.; Gu, J.; Lee, S.; Shin, W.; Kim, S. Supervised chemical graph mining improves drug-induced liver injury prediction. iScience 2023, 26, 105677. [Google Scholar] [CrossRef]
- LightGBM. Available online: https://lightgbm.readthedocs.io/en/stable/ (accessed on 28 September 2023).
- Boldini, D.; Grisoni, F.; Kuhn, D.; Friedrich, L.; Sieber, S.A. Practical guidelines for the use of gradient boosting for molecular property prediction. J. Cheminformatics 2023, 15, 73. [Google Scholar] [CrossRef]
- BioScience Database. Available online: https://dbarchive.biosciencedbc.jp/index.html (accessed on 28 September 2023).
- EFSA (European Food Safety Authority). Chemical Hazards Database—OpenFoodTox. Available online: https://www.efsa.europa.eu/en/data-report/chemical-hazards-database-openfoodtox (accessed on 28 September 2023).
- CEBS (Chemical Effects in Biological Systems). Available online: https://cebs.niehs.nih.gov/cebs/ (accessed on 28 September 2023).
- Cosmos, D.B. Available online: https://www.ng.cosmosdb.eu/downloads (accessed on 28 September 2023).
- EPA Ecotox Database. Available online: https://cfpub.epa.gov/ecotox/ (accessed on 28 September 2023).
- OCHEM Database. Available online: https://ochem.eu/home/show.do (accessed on 28 September 2023).
- TensorFlow Datasets. Available online: https://www.tensorflow.org/datasets (accessed on 28 September 2023).
- NCI CACTUS Chemical Identifier Search. Available online: https://cactus.nci.nih.gov/index.html (accessed on 28 September 2023).
- FDA. Drug-Induced Liver Injury Rank (DILIrank) Dataset. Available online: https://fda.gov/science-research/liver-toxicity-knowledge-base-ltkb/drug-induced-liver-injury-rank-dilirank-dataset (accessed on 28 September 2023).
- PubChem Database. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 28 September 2023).
- Richard, A.M.; Huang, R.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem. Res. Toxicol. 2021, 34, 189–216. [Google Scholar] [CrossRef] [PubMed]
- NLM CPDB. Available online: https://www.nlm.nih.gov/index.html (accessed on 28 September 2023).
- Molecules Dataset Collection. Available online: https://github.com/GLambard/Molecules_Dataset_Collection (accessed on 28 September 2023).
- CardioTox. Available online: https://github.com/Abdulk084/CardioTox (accessed on 28 September 2023).
- Wu, K.; Wei, G.-W. Quantitative Toxicity Prediction Using Topology Based Multitask Deep Neural Networks. J. Chem. Inf. Model. 2018, 58, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Filimonov, D.; Zakharov, A.; Xie, W.; Huang, Y.; Zhu, F.; Shen, T.; Yao, J.; Poroikov, V. Computer-Aided Prediction of Rodent Carcinogenicity by PASS and CISOC-PSCT. QSAR Comb. Sci. 2009, 28, 806–810. [Google Scholar] [CrossRef]
- Lee, H.-M.; Yu, M.-S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.H.; Bae, M.A.; Lee, B.H.; Shin, D.S.; Oh, K.S.; Ceong, H.; et al. Computational Determination of hERG-Related Cardiotoxicity of Drug Candidates. BMC Bioinform. 2019, 20, 250. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-Y.; Su, B.-H.; Esposito, E.X.; Hopfinger, A.J.; Tseng, Y.J. A Comprehensive Support Vector Machine Binary hERG Classification Model Based on Extensive but Biased End Point hERG Data Sets. Chem. Res. Toxicol. 2011, 24, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, H.; Liu, H.; Li, D.; Li, Y.; Hou, T. ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol. Pharm. 2016, 13, 2855–2866. [Google Scholar] [CrossRef]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep Learning for Drug-Induced Liver Injury. J. Chem. Inf. Model. 2015, 55, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Mika, S.; Schroeter, T.; Sutter, A.; ter Laak, A.; Steger-Hartmann, T.; Heinrich, N.; Müller, K.-R. Benchmark Data Set for in Silico Prediction of Ames Mutagenicity. J. Chem. Inf. Model. 2009, 49, 2077–2081. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, D.; Wang, J.; Hsieh, C.-Y.; Cao, D.; Hou, T. Mining Toxicity Information from Large Amounts of Toxicity Data. J. Med. Chem. 2021, 64, 6924–6936. [Google Scholar] [CrossRef]
- Braga, R.C.; Alves, V.M.; Silva, M.F.; Muratov, E.; Fourches, D.; Lião, L.M.; Tropsha, A.; Andrade, C.H. Pred-hERG: A Novel Web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform. 2015, 34, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Alexiou, A.; Bilgrami, A.L.; Kamal, M.A.; Ashraf, G.M. DeePred-BBB: A Blood Brain Barrier Permeability Prediction Model with Improved Accuracy. Front. Neurosci. 2022, 16, 858126. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.F.; Teixeira, A.L.; Pinheiro, L.; Falcao, A.O. A Bayesian Approach to in Silico Blood-Brain Barrier Penetration Modeling. J. Chem. Inf. Model. 2012, 52, 1686–1697. [Google Scholar] [CrossRef]
- Tong, X.; Wang, D.; Ding, X.; Tan, X.; Ren, Q.; Chen, G.; Rong, Y.; Xu, T.; Huang, J.; Jiang, H.; et al. Blood–brain Barrier Penetration Prediction Enhanced by Uncertainty Estimation. J. Cheminform. 2022, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, J.; Sivaraman, G.; Picel, K.; Peters, B.; Vázquez-Mayagoitia; Ramanathan, A.; MacDonell, M.; Foster, I.; Yan, E. Uncertainty-Informed Deep Transfer Learning of Perfluoroalkyl and Polyfluoroalkyl Substance Toxicity. J. Chem. Inf. Model. 2021, 61, 5793–5803. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, Y.; Hsieh, C.-Y.; Zhang, S.; Liu, X.; Liu, H.; Song, S.; Yao, X. TrimNet: Learning Molecular Representation from Triplet Messages for Biomedicine. Brief. Bioinform. 2021, 22, bbaa266. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Xi, Y.; Huang, J.; Ayers, P.W. A Curated Diverse Molecular Database of Blood-Brain Barrier Permeability with Chemical Descriptors. Sci. Data. 2021, 8, 289. [Google Scholar] [CrossRef] [PubMed]
- Acute Toxicity Data. Available online: https://www.cerc.usgs.gov/data/acute/acute.html (accessed on 28 September 2023).
- ChEMBL Database. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 28 September 2023).
- BBBP-SMILES Dataset on Kaggle. Available online: https://www.kaggle.com/datasets/priyanagda/bbbp-smiles (accessed on 28 September 2023).
- WeiLab Mathematical Data Library. Available online: https://weilab.math.msu.edu/DataLibrary/2D/ (accessed on 28 September 2023).
- CompTox Chemicals Dashboard. Available online: https://www.epa.gov/chemical-research/comptox-chemicals-dashboard (accessed on 28 September 2023).
- LactMed Database. Available online: https://www.nlm.nih.gov/databases/download/lactmed.html (accessed on 28 September 2023).
- CCRIS Database. Available online: https://www.nlm.nih.gov/databases/download/ccris.html (accessed on 28 September 2023).
- DrugBank Online. Available online: https://go.drugbank.com/ (accessed on 28 September 2023).
- NORMAN Network Data System. Available online: https://www.norman-network.com/nds/SLE/ (accessed on 28 September 2023).
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial Intelligence Foundation for Therapeutic Science. Nat. Chem. Biol. 2022, 18, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER Database of Drugs and Side Effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Wishart, D.; Arndt, D.; Pon, A.; Sajed, T.; Guo, A.C.; Djoumbou, Y.; Knox, C.; Wilson, M.; Liang, Y.; Grant, J.; et al. T3DB: The Toxic Exposome Database. Nucleic Acids Res. 2015, 43, D928–D934. [Google Scholar] [CrossRef]
- Alves, V.M.; Muratov, E.N.; Capuzzi, S.J.; Politi, R.; Low, Y.; Braga, R.C.; Zakharov, A.V.; Sedykh, A.; Mokshyna, E.; Farag, S.; et al. Alarms about structural alerts. Green. Chem. 2016, 18, 4348–4360. [Google Scholar] [CrossRef] [PubMed]
- SLiang, S.-T.; Chen, C.; Chen, R.-X.; Li, R.; Chen, W.-L.; Jiang, G.-H.; Du, L.-L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharmacol. 2022, 13, 1033003. [Google Scholar]
- Limban, C.; Nuţă, D.C.; Chiriţă, C.; Negreș, S.; Arsene, A.L.; Goumenou, M.; Karakitsios, S.P.; Tsatsakis, A.M.; Sarigiannis, D.A. The use of structural alerts to avoid the toxicity of pharmaceuticals. Toxicol. Rep. 2018, 5, 943–953. [Google Scholar] [CrossRef]
- Prasanna, S.; Doerksen, R.-J. Topological polar surface area: A useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. A widely applicable set of descriptors. J. Mol. Graph Model 2000, 18, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Kier, L.; Hall, L. A Differential Molecular Connectivity Index. Quant. Struct. Act. Relatsh. 1991, 10, 134–140. [Google Scholar] [CrossRef]
- Menchinskaya, E.; Chingizova, E.; Pislyagin, E.; Likhatskaya, G.; Sabutski, Y.; Pelageev, D. europrotective Effect of 1,4-Naphthoquinones in an In Vitro Model of Paraquat and 6-OHDA-Induced Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 9933. [Google Scholar] [CrossRef]





| Toxicity Endpoint | Regression Task | |
|---|---|---|
| RMSEMML | RMSEbenchmark | |
| Mouse oral LD50 | 0.43 | 0.49 |
| Rat oral LD50 | 0.47 | 0.68 |
| Mouse intraperitoneal LD50 | 0.45 | 0.54 |
| Rat intraperitoneal LD50 | 0.58 | 0.64 |
| Mouse intravenous LD50 | 0.46 | 0.52 |
| Rat intravenous LD50 | 0.59 | 0.63 |
| Toxicity Endpoint | Billy QSAR | Smi2Vec-LSTM | Smi2Vec-BiGRU | TranGRU |
|---|---|---|---|---|
| NR.AhR 1 | 0.904 | 0.678 | 0.879 | 0.833 |
| NR.AR 1 | 0.771 | 0.691 | 0.714 | 0.824 |
| NR.AR.LBD 1 | 0.747 | 0.748 | 0.824 | 0.847 |
| NR.Aromatase | 0.802 | 0.496 | 0.699 | 0.784 |
| NR.ER 1 | 0.787 | 0.623 | 0.736 | 0.691 |
| NR.ER.LBD 1 | 0.763 | 0.531 | 0.868 | 0.843 |
| NR.PPAR.gamma 1 | 0.767 | 0.566 | 0.749 | 0.838 |
| SR.ARE 1 | 0.795 | 0.641 | 0.761 | 0.701 |
| SR.ATAD5 1 | 0.806 | 0.5 | 0.763 | 0.727 |
| SR.HSE 1 | 0.796 | 0.612 | 0.785 | 0.736 |
| SR.MMP 1 | 0.951 | 0.743 | 0.86 | 0.816 |
| SR.p53 | 0.818 | 0.518 | 0.732 | 0.81 |
| Target Name | CatBoost Fingerprints | XGBoost Fragments | Benchmark * | n Samples |
|---|---|---|---|---|
| Mouse intraperitoneal LD50 | 0.562 | 0.486 | 0.473 (TOXRIC) | 91,162 |
| Mouse oral LD50 | 0.543 | 0.445 | 0.445 (TOXRIC) | 57,307 |
| Mouse intravenous LD50 | 0.498 | 0.415 | 0.491 (TOXRIC) | 41,630 |
| Rat oral LD50 | 0.589 | 0.466 | 0.592 (Syntelly) | 23,409 |
| Mouse subcutaneous LD50 | 0.696 | 0.652 | 0.55 (Syntelly) | 19,457 |
| Rat intraperitoneal LD50 | 0.71 | 0.618 | 0.61 (Syntelly) | 12,769 |
| Rat intravenous LD50 | 0.894 | 0.853 | 0.644 (TOXRIC) | 7461 |
| Rat subcutaneous LD50 | 0.829 | 0.743 | 0.69 (Syntelly) | 5376 |
| Tetrahymena pyriformis IGC50 40 h | 0.524 | 0.46 | 0.518 (TOXRIC) | 3516 |
| Mouse intraperitoneal LDLo | 0.495 | 0.435 | 0.52 (Syntelly) | 3500 |
| Rabbit skin LD50 | 0.521 | 0.479 | 0.58 (Syntelly) | 3429 |
| Rabbit oral LD50 | 0.626 | 0.588 | 0.588 (Syntelly) | 2969 |
| Guinea pig oral LD50 | 0.703 | 0.659 | 0.69 (Syntelly) | 1778 |
| Fathead minnow LC50 96 h | 0.78 | 0.72 | 0.864 (TOXRIC) | 1739 |
| Rat skin LD50 | 0.665 | 0.63 | 0.62 (Syntelly) | 1673 |
| Rabbit intravenous LD50 | 0.898 | 0.83 | 0.67 (Syntelly) | 1604 |
| Rat intraperitoneal LDLo | 0.512 | 0.482 | 0.63 (Syntelly) | 1568 |
| Mouse intramuscular LD50 | 0.894 | 0.866 | 0.715 (TOXRIC) | 1518 |
| Rat oral LDLo | 0.997 | 0.99 | 0.71 (Syntelly) | 1464 |
| Bioconcentration factor | 0.699 | 0.626 | 0.71 (Syntelly) | 1321 |
| Dog intravenous LD50 | 0.995 | 0.927 | 0.838 (TOXRIC) | 1215 |
| Chicken oral LD50 | 0.906 | 0.941 | 0.916 (TOXRIC) | 743 |
| Quail oral LD50 | 0.816 | 0.852 | 0.817 (TOXRIC) | 735 |
| Dog intravenous LDLo | 0.84 | 0.834 | 0.894 (TOXRIC) | 703 |
| Daphnia magna LC50 | 0.866 | 0.817 | 1.109 (TOXRIC) | 699 |
| Rabbit intravenous LDLo | 0.783 | 0.749 | 1.031 (TOXRIC) | 690 |
| Guinea pig intraperitoneal LD50 | 0.745 | 0.786 | 0.818 (TOXRIC) | 631 |
| Cat intravenous LD50 | 0.877 | 0.851 | 0.836 (TOXRIC) | 542 |
| Mouse skin LD50 | 0.838 | 0.815 | 0.917 (TOXRIC) | 426 |
| Target Name | CatBoost Fingerprints | XGBoost Fragments | Benchmark 1 | n Samples |
|---|---|---|---|---|
| Cardiotoxicity (hERG binary) | 0.888 | 0.926 | 0.930 (CardioTox) | 324,010 |
| Ames test | 0.845 | 0.894 | 0.88 (Syntelly) | 14,168 |
| SR-HSE 2 | 0.839 | 0.836 | 0.736 (TranGRU) | 7281 |
| NR-AR 2 | 0.724 | 0.797 | 0.824 (TranGRU) | 7263 |
| NR-AR-LBD 2 | 0.843 | 0.835 | 0.847 (TranGRU) | 7133 |
| NR-PPAR-gamma 2 | 0.727 | 0.809 | 0.838 (TranGRU) | 6942 |
| NR-aromatase | 0.831 | 0.875 | 0.784 (TranGRU) | 6929 |
| NR-ER-LBD 2 | 0.858 | 0.878 | 0.843 (TranGRU) | 6920 |
| SR-ATAD5 2 | 0.75 | 0.862 | 0.727 (TranGRU) | 6893 |
| SR-ARE 2 | 0.825 | 0.845 | 0.701 (TranGRU) | 6822 |
| SR-p53 | 0.748 | 0.877 | 0.81 (TranGRU) | 6749 |
| NR-ER 2 | 0.852 | 0.866 | 0.691 (TranGRU) | 6585 |
| NR-AhR 2 | 0.816 | 0.871 | 0.833 (TranGRU) | 6446 |
| SR-MMP 2 | 0.853 | 0.896 | 0.816 (TranGRU) | 6361 |
| Eye irritation | 0.977 | 0.98 | 0.966 (TOXRIC) | 5040 |
| Hepatotoxicity | 0.785 | 0.811 | 0.741 (TOXRIC) | 3413 |
| Carcinogenicity | 0.757 | 0.787 | 0.68 (TOXRIC) | 2726 |
| Eye corrosion | 0.993 | 0.99 | 0.948 (TOXRIC) | 2190 |
| Blood–brain barrier penetration | 0.925 | 0.936 | 0.919 (Wang et al.) | 1961 |
| Developmental toxicity | 0.85 | 0.857 | 0.918 (TOXRIC) | 640 |
| DILI 2 | 0.874 | 0.901 | 0.691 (Lim et al.) | 475 |
| Reproductive toxicity | 0.489 | 0.739 | 0.927 (TOXRIC) | 146 |
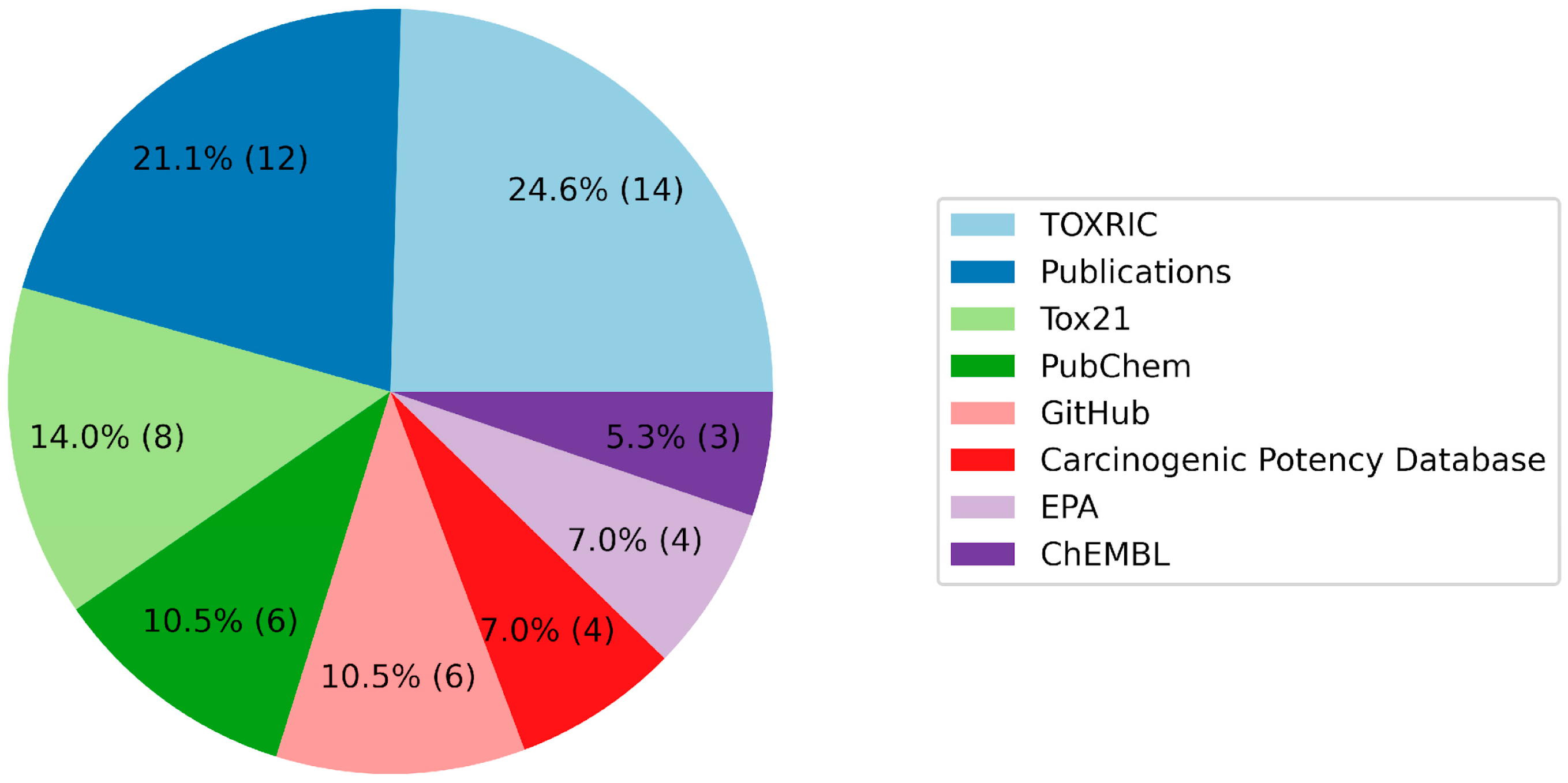
| Name of the Database | References |
|---|---|
| TOXRIC | [48] |
| BioScience DBC | [54] |
| EFSA | [55] |
| National Toxicology Program | [56] |
| COSMOS | [57] |
| EPA | [58] |
| OCHEM | [59] |
| TensorFlow | [60] |
| Cactus NIH | [61] |
| FDA | [62] |
| PubChem | [63] |
| Tox21 | [64] |
| Carcinogenic Potency Database | [65] |
| GitHub | [66,67] |
| Publications | [35,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83] |
| USGS | [84] |
| ChEMBL | [85] |
| Kaggle | [86] |
| PyTDC | [87] |
| WeiLab MSU | [88] |
| CompTox | [89] |
| NLM NIH | [90,91] |
| SIDER | [92] |
| DrugBank | [93] |
| NORMAN | [94] |
| T3DB | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shkil, D.O.; Muhamedzhanova, A.A.; Petrov, P.I.; Skorb, E.V.; Aliev, T.A.; Steshin, I.S.; Tumanov, A.V.; Kislinskiy, A.S.; Fedorov, M.V. Expanding Predictive Capacities in Toxicology: Insights from Hackathon-Enhanced Data and Model Aggregation. Molecules 2024, 29, 1826. https://doi.org/10.3390/molecules29081826
Shkil DO, Muhamedzhanova AA, Petrov PI, Skorb EV, Aliev TA, Steshin IS, Tumanov AV, Kislinskiy AS, Fedorov MV. Expanding Predictive Capacities in Toxicology: Insights from Hackathon-Enhanced Data and Model Aggregation. Molecules. 2024; 29(8):1826. https://doi.org/10.3390/molecules29081826
Chicago/Turabian StyleShkil, Dmitrii O., Alina A. Muhamedzhanova, Philipp I. Petrov, Ekaterina V. Skorb, Timur A. Aliev, Ilya S. Steshin, Alexander V. Tumanov, Alexander S. Kislinskiy, and Maxim V. Fedorov. 2024. "Expanding Predictive Capacities in Toxicology: Insights from Hackathon-Enhanced Data and Model Aggregation" Molecules 29, no. 8: 1826. https://doi.org/10.3390/molecules29081826
APA StyleShkil, D. O., Muhamedzhanova, A. A., Petrov, P. I., Skorb, E. V., Aliev, T. A., Steshin, I. S., Tumanov, A. V., Kislinskiy, A. S., & Fedorov, M. V. (2024). Expanding Predictive Capacities in Toxicology: Insights from Hackathon-Enhanced Data and Model Aggregation. Molecules, 29(8), 1826. https://doi.org/10.3390/molecules29081826