Kanamycin and G-Quadruplexes: An Exploration of Binding Interactions

 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Virtual Screening (VS)
2.2. Thermodynamic Analysis of Kanamycin–G4 Complexes
2.3. Characterization of the Dynamic Interactions in G4s–Kanamycin Complexes
2.4. In Vitro Study of Interactions Between Kanamycin and G4s
3. Discussion
4. Materials and Methods
4.1. Database Preparation
4.2. Receptor Preparation and NMR Conformer Selection
4.3. Docking Protocol
4.4. Molecular Dynamics Simulation (MDS) Protocol
4.5. 1D 1H-NMR Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef]
- Parrotta, L.; Ortuso, F.; Moraca, F.; Rocca, R.; Costa, G.; Alcaro, S.; Artese, A. Targeting unimolecular G-quadruplex nucleic acids: A new paradigm for the drug discovery? Exp. Opin. Drug Discov. 2014, 9, 1167–1187. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Webba da Silva, M. Geometric formalism for DNA quadruplex folding. Chemistry 2007, 13, 9738–9745. [Google Scholar] [CrossRef]
- Popenda, M.; Miskiewicz, J.; Sarzynska, J.; Zok, T.; Szachniuk, M. Topology-based classification of tetrads and quadruplex structures. Bioinformatics 2020, 36, 1129–1134. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Rigo, R.; Palumbo, M.; Sissi, C. G-quadruplexes in human promoters: A challenge for therapeutic applications. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Di Porzio, A.; Iaccarino, N.; Riccardi, G.; Di Lorenzo, R.; Laneri, S.; Pagano, B.; Amato, J.; Randazzo, A. G-quadruplexes in cancer-related gene promoters: From identification to therapeutic targeting. Expert. Opin. Ther. Pat. 2023, 33, 745–773. [Google Scholar] [CrossRef]
- Kerkour, A.; Marquevielle, J.; Ivashchenko, S.; Yatsunyk, L.A.; Mergny, J.L.; Salgado, G.F. High-resolution three-dimensional NMR structure of the. J. Biol. Chem. 2017, 292, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Porter, K.I.; Dakup, P.P.; Wang, S.; Robertson, G.P.; Gaddameedhi, S.; Zhu, J. Telomere dysfunction in Tert knockout mice delays Braf. Int. J. Cancer 2024, 154, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Wang, X.D.; Hu, M.H. Novel quinoxaline analogs as telomeric G-quadruplex ligands exert antitumor effects related to enhanced immunomodulation. Eur. J. Med. Chem. 2024, 274, 116536. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, V.; George, S.; Cote, G.M. Molecular Advances in the Treatment of Advanced Gastrointestinal Stromal Tumor. Oncologist 2023, 28, 671–681. [Google Scholar] [CrossRef]
- Phan, A.T.; Kuryavyi, V.; Burge, S.; Neidle, S.; Patel, D.J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 2007, 129, 4386–4392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Abdihamid, O.; Tan, F.; Zhou, H.; Liu, H.; Li, Z.; Xiao, S.; Li, B. KIT mutations and expression: Current knowledge and new insights for overcoming IM resistance in GIST. Cell Commun. Signal 2024, 22, 153. [Google Scholar] [CrossRef]
- Rankin, S.; Reszka, A.P.; Huppert, J.; Zloh, M.; Parkinson, G.N.; Todd, A.K.; Ladame, S.; Balasubramanian, S.; Neidle, S. Putative DNA quadruplex formation within the human c-kit oncogene. J. Am. Chem. Soc. 2005, 127, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Fernando, H.; Reszka, A.P.; Huppert, J.; Ladame, S.; Rankin, S.; Venkitaraman, A.R.; Neidle, S.; Balasubramanian, S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry 2006, 45, 7854–7860. [Google Scholar] [CrossRef] [PubMed]
- Kotar, A.; Rigo, R.; Sissi, C.; Plavec, J. Two-quartet kit* G-quadruplex is formed via double-stranded pre-folded structure. Nucleic Acids Res. 2019, 47, 2641–2653. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef]
- Neidle, S. A Phenotypic Approach to the Discovery of Potent G-Quadruplex Targeted Drugs. Molecules 2024, 29, 3653. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.; Mondal, S.; Sengupta, P.; Chatterjee, S. In Silico Screening and Binding Characterization of Small Molecules toward a G-Quadruplex Structure Formed in the Promoter Region of. ACS Omega 2017, 2, 4382–4397. [Google Scholar] [CrossRef] [PubMed]
- Kaserer, T.; Rigo, R.; Schuster, P.; Alcaro, S.; Sissi, C.; Schuster, D. Optimized Virtual Screening Workflow for the Identification of Novel G-Quadruplex Ligands. J. Chem. Inf. Model. 2016, 56, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Catalano, R.; Moraca, F.; Amato, J.; Cristofari, C.; Rigo, R.; Via, L.D.; Rocca, R.; Lupia, A.; Maruca, A.; Costa, G.; et al. Targeting multiple G-quadruplex-forming DNA sequences: Design, biophysical and biological evaluations of indolo-naphthyridine scaffold derivatives. Eur. J. Med. Chem. 2019, 182, 111627. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Alcaro, S.; Distinto, S.; Maccioni, E.; Ortuso, F.; Artese, A.; Parrotta, L. Structure-based virtual screening of novel natural alkaloid derivatives as potential binders of h-telo and c-myc DNA G-quadruplex conformations. Molecules 2014, 20, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, S.; Musetti, C.; Distinto, S.; Casatti, M.; Zagotto, G.; Artese, A.; Parrotta, L.; Moraca, F.; Costa, G.; Ortuso, F.; et al. Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure-based virtual screening approaches. J. Med. Chem. 2013, 56, 843–855. [Google Scholar] [CrossRef]
- Artese, A.; Costa, G.; Ortuso, F.; Parrotta, L.; Alcaro, S. Identification of new natural DNA G-quadruplex binders selected by a structure-based virtual screening approach. Molecules 2013, 18, 12051–12070. [Google Scholar] [CrossRef]
- Rocca, R.; Costa, G.; Artese, A.; Parrotta, L.; Ortuso, F.; Maccioni, E.; Pinato, O.; Greco, M.L.; Sissi, C.; Alcaro, S.; et al. Hit Identification of a Novel Dual Binder for h-telo/c-myc G-Quadruplex by a Combination of Pharmacophore Structure-Based Virtual Screening and Docking Refinement. ChemMedChem 2016, 11, 1721–1733. [Google Scholar] [CrossRef]
- Costa, G.; Rocca, R.; Moraca, F.; Talarico, C.; Romeo, I.; Ortuso, F.; Alcaro, S.; Artese, A. A Comparative Docking Strategy to Identify Polyphenolic Derivatives as Promising Antineoplastic Binders of G-quadruplex DNA c-myc and bcl-2 Sequences. Mol. Inform. 2016, 35, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Castillo-González, D.; Pérez-Machado, G.; Guédin, A.; Mergny, J.L.; Cabrera-Pérez, M.A. FDA-approved drugs selected using virtual screening bind specifically to G-quadruplex DNA. Curr. Pharm. Des. 2013, 19, 2164–2173. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Geng, X.; Liu, L.; Gao, J. Identification of Trovafloxacin, Ozanimod, and Ozenoxacin as Potent c-Myc G-Quadruplex Stabilizers to Suppress c-Myc Transcription and Myeloma Growth. Mol. Inform. 2022, 41, e2200011. [Google Scholar] [CrossRef] [PubMed]
- Moraca, F.; Marzano, S.; D’Amico, F.; Lupia, A.; Di Fonzo, S.; Vertecchi, E.; Salvati, E.; Di Porzio, A.; Catalanotti, B.; Randazzo, A.; et al. Ligand-based drug repurposing strategy identified SARS-CoV-2 RNA G-quadruplex binders. Chem. Commun. 2022, 58, 11913–11916. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Yang, H.; Kwan, M.H.; Cheng, Z.; Lee, P.; Bai, L.P.; Jiang, Z.H.; Wong, C.Y.; Fong, W.F.; Leung, C.H.; et al. Structure-based optimization of FDA-approved drug methylene blue as a c-myc G-quadruplex DNA stabilizer. Biochimie 2011, 93, 1055–1064. [Google Scholar] [CrossRef]
- Zidanloo, S.G.; Colagar, A.H.; Ayatollahi, H.; Bagheryan, Z. G-quadruplex forming region within WT1 promoter is selectively targeted by daunorubicin and mitoxantrone: A possible mechanism for anti-leukemic effect of drugs. J. Biosci. 2019, 44, 12. [Google Scholar]
- Ranjan, N.; Arya, D.P. Parallel G-quadruplex recognition by neomycin. Front. Chem. 2023, 11, 1232514. [Google Scholar] [CrossRef]
- Kuryavyi, V.; Phan, A.T.; Patel, D.J. Solution structures of all parallel-stranded monomeric and dimeric G-quadruplex scaffolds of the human c-kit2 promoter. Nucleic Acids Res. 2010, 38, 6757–6773. [Google Scholar] [CrossRef]
- Luu, K.N.; Phan, A.T.; Kuryavyi, V.; Lacroix, L.; Patel, D.J. Structure of the human telomere in K+ solution: An intramolecular (3 + 1) G-quadruplex scaffold. J. Am. Chem. Soc. 2006, 128, 9963–9970. [Google Scholar] [CrossRef]
- Costa, G.; Carta, F.; Ambrosio, F.A.; Artese, A.; Ortuso, F.; Moraca, F.; Rocca, R.; Romeo, I.; Lupia, A.; Maruca, A.; et al. A computer-assisted discovery of novel potential anti-obesity compounds as selective carbonic anhydrase VA inhibitors. Eur. J. Med. Chem. 2019, 181, 111565. [Google Scholar] [CrossRef]
- Maruca, A.; Rocca, R.; Catalano, R.; Mesiti, F.; Costa, G.; Lanzillotta, D.; Salatino, A.; Ortuso, F.; Trapasso, F.; Alcaro, S.; et al. Natural Products Extracted from Fungal Species as New Potential Anti-Cancer Drugs: A Structure-Based Drug Repurposing Approach Targeting HDAC7. Molecules 2020, 25, 5524. [Google Scholar] [CrossRef]
- Catalano, R.; Rocca, R.; Juli, G.; Costa, G.; Maruca, A.; Artese, A.; Caracciolo, D.; Tagliaferri, P.; Alcaro, S.; Tassone, P.; et al. A drug repurposing screening reveals a novel epigenetic activity of hydroxychloroquine. Eur. J. Med. Chem. 2019, 183, 111715. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Liu, G.; Sang, X.; Zhu, X.; Fu, X.; Dou, C.; Jian, Y.; Zhang, J.; Zou, S.; Zhang, G.; et al. Targeting RNA G-quadruplex with repurposed drugs blocks SARS-CoV-2 entry. PLoS Pathog. 2023, 19, e1011131. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.S.; Sharma, S.; Rizvi, Z.A.; Sinha, D.; Gupta, D.; Rophina, M.; Sehgal, P.; Sadhu, S.; Tripathy, M.R.; Samal, S.; et al. G4-binding drugs, chlorpromazine and prochlorperazine, repurposed against COVID-19 infection in hamsters. Front. Mol. Biosci. 2023, 10, 1133123. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Mergny, J.L.; Cruz, C. G-quadruplex ligands in cancer therapy: Progress, challenges, and clinical perspectives. Life Sci. 2024, 340, 122481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, Y.; Zhang, W. G-quadruplex structures and their interaction diversity with ligands. ChemMedChem 2014, 9, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Schrödinger. LigPrep; Schrödinger LLC.: New York, NY, USA, 2018. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Schrödinger. Protein Preparation Wizard; Schrödinger, LLC.: New York, NY, USA, 2018. [Google Scholar]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Schrödinger. Glide; Schrödinger LLC.: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger. Canvas; Schrödinger LLC.: New York, NY, USA, 2018. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Cornell, W.D.a.C.P.a.B.C.I.a.K.P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Junmei Wang and Wei Wang and Peter, A.K.a.D.A.C. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Pirota, V.; Platella, C.; Musumeci, D.; Benassi, A.; Amato, J.; Pagano, B.; Colombo, G.; Freccero, M.; Doria, F.; Montesarchio, D. On the binding of naphthalene diimides to a human telomeric G-quadruplex multimer model. Int. J. Biol. Macromol. 2021, 166, 1320–1334. [Google Scholar] [CrossRef]
- Oliviero, G.; Borbone, N.; Amato, J.; D’Errico, S.; Galeone, A.; Piccialli, G.; Varra, M.; Mayol, L. Synthesis of quadruplex-forming tetra-end-linked oligonucleotides: Effects of the linker size on quadruplex topology and stability. Biopolym. Orig. Res. Biomol. 2009, 91, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, G.; Amato, J.; Borbone, N.; Galeone, A.; Petraccone, L.; Varra, M.; Piccialli, G.; Mayol, L. Synthesis and characterization of monomolecular DNA G-quadruplexes formed by tetra-end-linked oligonucleotides. Bioconjugate Chem. 2006, 17, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Amato, J.; Pagano, A.; Capasso, D.; Di Gaetano, S.; Giustiniano, M.; Novellino, E.; Randazzo, A.; Pagano, B. Targeting the BCL2 Gene Promoter G-Quadruplex with a New Class of Furopyridazinone-Based Molecules. ChemMedChem 2018, 13, 406–410. [Google Scholar] [CrossRef] [PubMed]


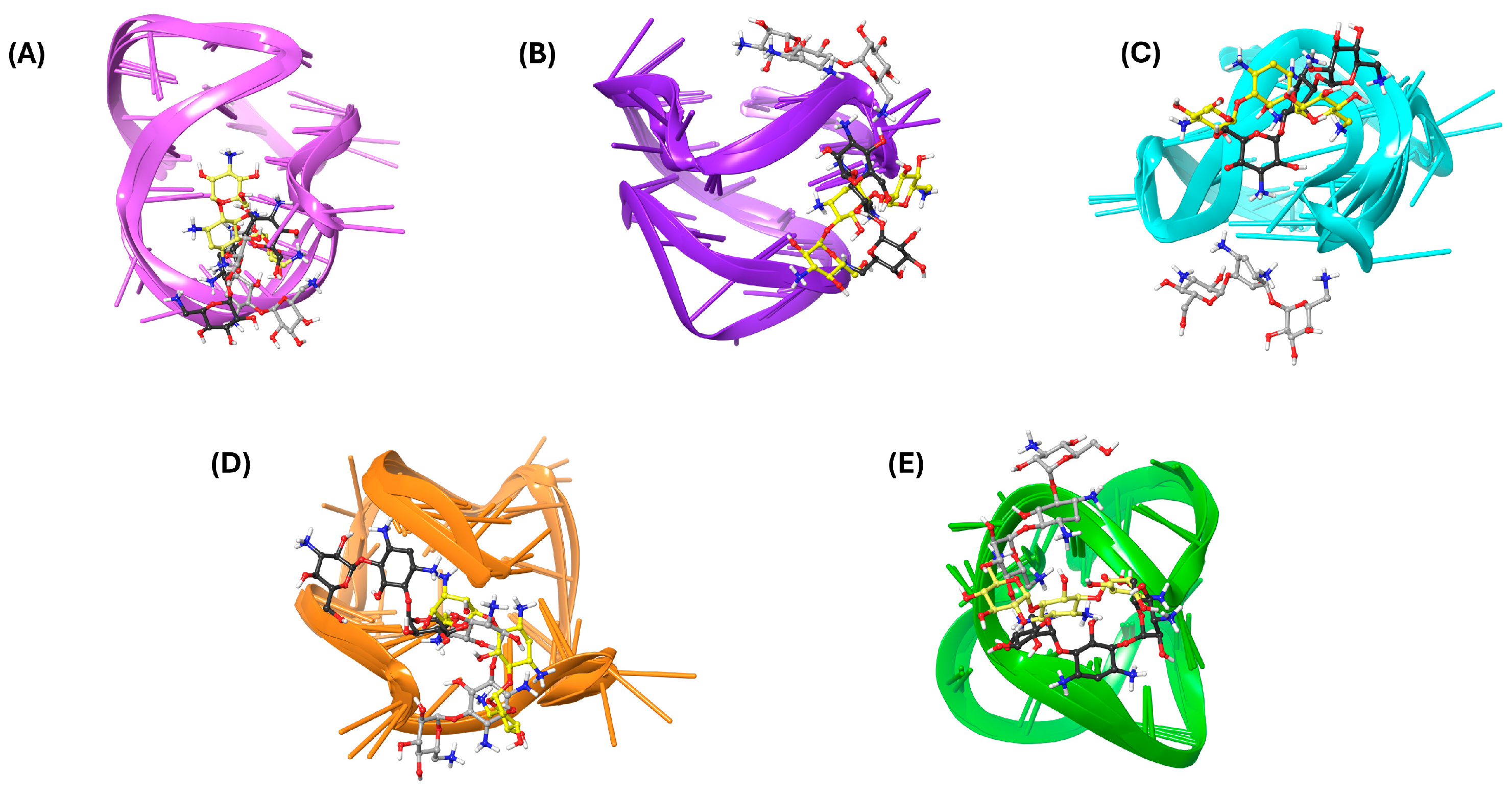

{kind=link}
{kind=link}
{kind=link}
| G4 Model | ΔGbind (kcal/mol) | ΔGcoul (kcal/mol) | ΔGlipo (kcal/mol) | ΔGvdw (kcal/mol) | ΔGsolv (kcal/mol) |
|---|---|---|---|---|---|
| Bcl-2 | −58.89 | −786.47 | −7.86 | −29.44 | 759.36 |
| c-kit1 | −53.94 | −770.08 | −7.82 | −23.74 | 740.36 |
| c-kit2 | −43.81 | −734.77 | −5.75 | −13.77 | 696.23 |
| c-myc | −48.87 | −663.10 | −8.58 | −22.07 | 632.21 |
| kit* | −53.84 | −754.97 | −7.90 | −23.02 | 722.76 |
| mtel24 | −64.71 | −752.54 | −7.86 | −24.36 | 712.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gualtieri, G.; Citriniti, E.L.; Rocca, R.; Arciuolo, V.; Amato, J.; Randazzo, A.; Alcaro, S. Kanamycin and G-Quadruplexes: An Exploration of Binding Interactions. Molecules 2024, 29, 5932. https://doi.org/10.3390/molecules29245932
Gualtieri G, Citriniti EL, Rocca R, Arciuolo V, Amato J, Randazzo A, Alcaro S. Kanamycin and G-Quadruplexes: An Exploration of Binding Interactions. Molecules. 2024; 29(24):5932. https://doi.org/10.3390/molecules29245932
Chicago/Turabian StyleGualtieri, Gianmarco, Emanuele Liborio Citriniti, Roberta Rocca, Valentina Arciuolo, Jussara Amato, Antonio Randazzo, and Stefano Alcaro. 2024. "Kanamycin and G-Quadruplexes: An Exploration of Binding Interactions" Molecules 29, no. 24: 5932. https://doi.org/10.3390/molecules29245932
APA StyleGualtieri, G., Citriniti, E. L., Rocca, R., Arciuolo, V., Amato, J., Randazzo, A., & Alcaro, S. (2024). Kanamycin and G-Quadruplexes: An Exploration of Binding Interactions. Molecules, 29(24), 5932. https://doi.org/10.3390/molecules29245932